


Abstract
Aconitase is an iron–sulfur protein and a major enzyme of the TCA cycle that catalyzes the conversion of citrate to isocitrate under iron-rich conditions. In Escherichia coli, aconitase B (AcnB) is a typical moonlighting protein that can switch to its apo form (apo-AcnB) which favors binding its own mRNA 3′UTR and stabilize it when intracellular iron become scarce. The small regulatory RNA (sRNA) RyhB has previously been shown to promote RNase E-dependent degradation of acnB mRNA when it was expressed from an ectopic arabinose-dependent promoter, independently of intracellular iron levels. In marked contrast, we report here that expression of RyhB under low-iron conditions did not result in acnB mRNA degradation even when RyhB was bound to acnB ribosome binding site (RBS). Genetic and biochemical evidence suggested that, under low-iron conditions, apo-AcnB bound to acnB 3′UTR close to a RNase E cleavage site that is essential for RyhB-induced acnB mRNA degradation. Whereas RyhB can block acnB translation initiation, RNase E-dependent degradation of acnB was prevented by apo-AcnB binding close to the cleavage site. This previously uncharacterized regulation suggests an intricate post-transcriptional mechanism that represses protein expression while insuring mRNA stability.
INTRODUCTION
Aconitases are iron–sulfur proteins and major enzymes of the tricarboxylic acid (TCA) cycle that catalyze the reversible conversion of citrate to isocitrate. The bacterium Escherichia coli possesses two isoforms of aconitases, AcnA and AcnB, whose expression varies according to environmental conditions. For instance, AcnA is expressed during stress and stationary phase whereas AcnB is the major TCA enzyme expressed during exponential growth under aerobic conditions (1). However, AcnB is unstable under certain conditions of environmental stress. For example, AcnB contains a 4Fe–4S group that participates in catalysis but this essential structure becomes disassembled when cells are exposed to oxidative stress or low intracellular iron and that results in loss of activity (2–4).
AcnB is not only sensitive to environmental conditions but it also acts as a moonlighting or bifunctional protein. Whereas AcnB functions as a catalytic enzyme under iron-rich conditions in E. coli, it becomes a regulatory RNA-binding protein when intracellular iron becomes scarce (apo-AcnB). Indeed, under low-iron conditions AcnB can bind its own mRNA 3′UTR, which increases acnB mRNA stability (2–3,5). This creates a dynamic equilibrium between both AcnB states (enzyme/RNA-binding), which depend on the intracellular iron pool (4). This dual function was first described in higher organisms where it was shown that cytoplasmic apo-aconitase could bind mRNAs (6,7). Subsequent investigations in eukaryotes revealed that aconitase mRNAs carried iron-responsive elements (IREs) motifs (8,9). Oxidative stress and iron starvation induce aconitases expression to maintain iron homeostasis, thus classifying aconitases as iron-regulatory proteins (IRPs) (10,11). Acn-IRPs can bind to IREs located in the 5′ or 3′ end of mRNA UTRs to either protect mRNAs from degradation or to prevent translation, respectively. IREs are also found in the 3′UTR of the transferrin receptor mRNA (trf1), where they induce mRNA stabilization by protecting mRNA from cleavage by an uncharacterized endonuclease (12). In contrast, IREs found in the 5′UTR of ferritin lead to translational inhibition of the Fe-storage protein, similarly to many others Fe-metabolism genes (13,14).
The small regulatory RNA (sRNA) RyhB that is strongly expressed in response to low-iron condition has been reported to be an additional factor of regulation of acnB mRNA in E. coli (15). Small RNAs (sRNAs) in bacteria are important regulators of cellular functions by modulating gene expression in response to changes in the environment. More than 80 sRNAs have been identified so far in E. coli (16). A majority of these sRNAs regulate mRNAs by direct base-pairing, which affect either positively or negatively their translation and stability (17,18). In many cases, sRNAs require the RNA chaperone Hfq for optimal regulation by promoting sRNA–mRNA pairing and by stabilizing sRNAs against degradation in vivo (19,20). The chaperone Hfq can also act as a direct translational repressor by competing with initiating 30S ribosomal subunit for accessibility to AU-rich sequences in the vicinity of the ribosome binding site (RBS) (21,22). Such AU-rich sequences have been shown to act as translational enhancers by increasing interactions with ribosomal protein S1 and by stabilizing mRNA (23–28).
RyhB is one of the most extensively studied Hfq-dependent sRNAs. When cells grow under iron-rich conditions, the transcriptional regulator Fur (Fe2+-Fur) represses ryhB promoter. In contrast, Fur becomes inactive and relieves repression of ryhB under conditions of iron starvation (15,29). When iron is depleted, RyhB represses ∼20 different mRNAs, which encode iron-dependent proteins. When bound to those mRNAs, RyhB shuts down translation and promotes their degradation through recruitment of the RNA degradosome, a protein complex that includes endoribonuclease E (RNase E), a 3′-5′ polynucleotide phosphorylase (PNPase), an RNA helicase (RhlB) and an enolase (15,22,30–33). By regulating iron-using proteins, RyhB contributes actively to increase the levels of free intracellular Fe2+ (iron sparing) during iron starvation by reducing the expression of iron-dependent proteins (34).
Previous work from our group has shown that RyhB represses acnB mRNA under defined conditions (31). Here, we extend these observations to show the importance of intracellular iron levels in determining the outcome of RyhB-induced acnB repression. When E. coli is grown under high-iron conditions, RyhB expression results in acnB mRNA degradation. In contrast, low-iron conditions favor translational repression of acnB, leaving acnB mRNA intact. Our data suggest that apo-AcnB protects acnB mRNA from RNase E-mediated cleavage after RyhB pairing. This mechanism highlights a new level of regulation of sRNA-induced mRNA degradation that is linked to the metabolic status of E. coli, which influences the enzyme/RNA-binding properties of AcnB.
MATERIALS AND METHODS
Strains and plasmids
Strains and plasmids used in this study are listed in Supplementary Table S1. The methods used to obtain all constructions are described in Supplemental experimental procedures. Derivatives of E. coli MG1655 were used in all experiments. Escherichia coli expression strain BL21 (DE3) was used for overproduction and purification of AcnB3xFLAG protein. DH5α strain was used for routine cloning procedures. Cells were grown at 37°C in Luria-Bertani (LB) medium with addition of antibiotics depending on plasmids used. In the case of cells carrying pFRΔ, pRS1551, pNM12, pBAD24 and pET-21b, ampicillin (final concentration, 50 μg/ml) was added. Cells harboring pGD3 and pBAD33 plasmids were grown in the presence of chloramphenicol (final concentration, 30 μg/ml).
β-Galactosidase assays
Kinetics assays for β-galactosidase activity were performed as described (35) using a SpectraMax 250 microtitre plate reader (Molecular Devices, Sunnyvale, CA, USA). Briefly, overnight bacterial culture incubated at 37°C were diluted 1000-fold in 50 ml of fresh LB medium and grown under mechanical shaking at 37°C. Fe-starvation was induced by addition of a final concentration of 200 μM of the Fe chelator Dip when cultures reached an OD600 of 0.1. The formula Vmax/OD600 was used to calculate specific β-galactosidase activity. Results reported in this study correspond to a minimum of three independent experiments.
Western blot analysis
Cells were grown in LB medium at 37°C. Appropriate inducer (arabinose or Dip) was added to cells at an OD600 of 0.1. Proteins were harvested by precipitation with trichloroacetic acid (final concentration, 1% (w/v)) as described (36). Proteins were resuspended in protein loading gel electrophoresis buffer before loading on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (7% acrylamide) and transferred on a nitrocellulose membrane. An anti-AcnB polyclonal antibody was used at a dilution of 1:5000 (kind gift of Dr Jeffrey E. Green, National Cancer Institute). The IRDye 800CW-conjugated goat anti-rabbit secondary antibody (Li-Cor Biosciences, Lincoln, NE, USA) was used at a dilution of 1:15 000 and revealed on an Odyssey Infrared Imaging System (Li-Cor Biosciences). Quantifications were carried out using the Odyssey Application Software.
RNA extraction and northern blot analysis
Total RNA was extracted using the hot phenol procedure (37). Cells were grown to an OD600 of 0.5 and 0.1% arabinose or 200 μM of Dip were added. Determination of RNA half-life was performed by addition of 250 μg/ml rifampicin to the culture before total RNA extraction. In the case of acnB northern blots, 40 μg of total RNA were loaded on an agarose gel (1% agarose in 3-(N-Morpholino)propanesulfonic acid (MOPS) buffer). In the case of RyhB sRNA and sodB130-acnB+67 construct northern blots, 5 and 15 μg, respectively of total RNA were loaded on 4% polyacrylamide gel (acrylamide 29:1, 8 M urea). After migration, the RNA was transferred by capillarity (agarose gel) or electro-transferred (acrylamide gel) to a Hybond-XL membrane (Amersham Biosciences) and UV crosslinked. Prehybridization, hybridization and washes were performed as described (36). Membranes were exposed to phosphor storage screens and analyzed using a Typhoon Trio (GE Healthcare) instrument. Quantification was performed using the ImageQuant software (Molecular Dynamics).
RNA immunoprecipitation
Cells carrying plasmids pBAD24-acnB and pBAD24-acnB-3xFLAG (see Supplemental Material for details) were grown in LB medium to an OD600 of 0.1, at which point both AcnB3xFLAG and AcnB were induced by adding arabinose (final concentration of 0.1%). When the culture reached an OD600 of 0.5, Dip (200 μM final) was added to induce RyhB for 10 min. The culture was then chilled on ice for 20 min to stop growth. Cells were then centrifuged and suspended in binding buffer (20 mM Tris–HCl, 0.25 M KCl, 5 mM MgCl2, 10% glycerol, 0.1% Tween 20, pH 8.0) (1 ml). After another centrifugation, cells were suspended in 2 ml of binding buffer and lysed using a French press (800 psi, three times). The lysate was then cleared by centrifugation (17 000 × g, 30 min, 4°C) and proteins were quantified. At this point, 20 μl of soluble fraction were mixed with the same volume of protein sample buffer and 80 μl of the same fraction were used for total RNA extraction (input). The remaining soluble fraction (0.8 ml) was incubated with Anti-FLAG M2 Affinity gel (Sigma-Aldrich, St Louis, MO, USA) for 2 h on a rotating platform (all steps performed at 4°C). The suspension was centrifuged and the beads were washed twice with Tris-buffered saline (TBS) (500 μl each). The beads were then suspended in TBS (100 μl) and FLAG-peptide (Sigma-Aldrich, 15 μg) was added. Eluted RNAs were then subjected to phenol–chloroform extraction, followed by precipitation (2.5 vol. ethanol supplemented by 20 μg of glycogen) of the aqueous phase. Proteins were isolated from the organic phase by acetone precipitation. RNA samples were analyzed by quantitative Real-Time PCR (qRT-PCR) (for acnB2749, acnB2749-Mstem, acnB2749-stem, acnB+20-3′UTR constructs) or by northern blot (endogenous acnB). qRT-PCR reactions were performed four times on a Rotor-Gene 3000 device (Corbett Life Science, Concord, NSW) with Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) (29). Protein samples were analyzed by western blots.
In vitro RNA synthesis and radiolabeling
Oligonucleotides used to generate DNA template for in vitro transcription are listed in Supplementary Table S3. T7 RNA polymerase (Roche Diagnostics GmbH, Mannheim, Germany) was used for radiolabeled probe transcription for northern blot analysis. Antisense transcript of genes of genes of interest were generated with a published method (36). A published protocol (22) was used for transcription of RNA used in secondary structure probing, RNase E degradation assays and toeprinting assays. 32P-pCp 3′ end radiolabeling was carried out as described (33) and 5′-32P-end radiolabeling were done as published (36).
RNA degradosome degradation assays
Experimental procedure for RNase E cleavage sites characterization on 32P-pCp 3′ end radiolabeled RNA transcripts has been performed as described (33). RNase E-FLAG was purified as published (33).
Aconitase B purification
Purified AcnB3xFLAG were prepared as described (33), with some modifications. Escherichia coli (BL21 [DE3] pLysS/pET21b-acnB-3xFLAG) was grown in LB medium (2 l) containing ampicillin (50 μg/ml) and chloramphenicol (30 μg/ml) at 30°C, until it reached an OD600 between 0.5 and 0.6. During growth, Dip was added (final concentration of 200 μM) when the culture had reached an OD600 of 0.1. Expression of AcnB3xFLAG was induced with Isopropyl β-D-1-thiogalactopyranoside (IPTG) (1 mM) for 3 h at 30°C. An additional fast protein liquid chromatography (FPLC) exclusion purification (G200) was performed using modified Ip buffer (Tris–HCl 50 mM, NaCl 150 mM, glycerol 5% and Ethylenediaminetetraacetic acid (EDTA) 1 mM, pH 7.4). Purified apo-AcnB3xFLAG was used in all relevant experiments.
RESULTS
RyhB-induced nucleolytic cleavage of acnB mRNA depends on intracellular Fe levels
The acnB mRNA has been previously shown to be down-regulated by the sRNA RyhB when expressed from the arabinose-inducible promoter of the pBAD-ryhB construct (31). In these experiments, RyhB was induced independently of intracellular iron status and Fur activity, both of which normally control the expression of the endogenous ryhB promoter (38,39). Notably, AcnB has been shown to shift from catalytic (holo-AcnB) to RNA-binding regulatory mode (apo-AcnB) under conditions of iron starvation (4,5). Therefore, we asked whether acnB mRNA could be protected from RyhB-induced mRNA degradation when AcnB switched to binding its own mRNA (apo-form) under conditions of low Fe. We first addressed this question by inducing expression of RyhB under two different modes of induction. The first mode involved cultures in Fe-rich medium (Figure 1A, left panels) of EM1455 (ΔryhB) cells carrying an arabinose-inducible pBAD-ryhB plasmid (0.1% arabinose, Ara). The second mode involved EM1055 (WT) cells grown in Fe-poor medium by adding the iron chelator 2,2′-dipyridyl (Dip) and where RyhB is under the control of its endogenous promoter. Results showed that when RyhB was expressed in cells carrying the pBAD-ryhB plasmid in Fe-rich medium, it triggered a rapid degradation (within 5 min) of acnB mRNA (Figure 1A, lanes 1–5). The mRNA sodB was used as a positive control in these experiments and showed the expected degradation kinetics of a RyhB target (≤5 min). These results were in agreement with our previous observations that RyhB-induced degradation of acnB when expressed under the control of pBAD-ryhB when arabinose was present in the medium (31). In marked contrast, when RyhB sRNA was induced from its endogenous promoter under low Fe conditions, acnB mRNA remained stable even after 10 min of RyhB expression. This result was unexpected given that the fact that RyhB-sensitive sodB mRNA became fully degraded under similar conditions (Figure 1A, lanes 6–10). These results suggested that decreased intracellular iron levels may help to stabilize acnB mRNA although RyhB sRNA was fully expressed and able to induce degradation of control target sodB mRNA. To confirm that these results (Figure 1A, lanes 1–5) were not due to an arabinose-related effect, a third mode of induction was investigated. In this case, EM1455 cells carrying pBAD-ryhB were grown under condition of iron depletion (Dip, 250 μM) and were treated with arabinose to induce RyhB. Induction of RyhB under low iron showed that acnB mRNA remained stable throughout the experiment, although RyhB expression induced sodB degradation within 5 min (Figure 1A, lanes 11–15). These results confirmed that iron-depleted conditions protected acnB mRNA against RyhB-induced degradation.
Figure 1.
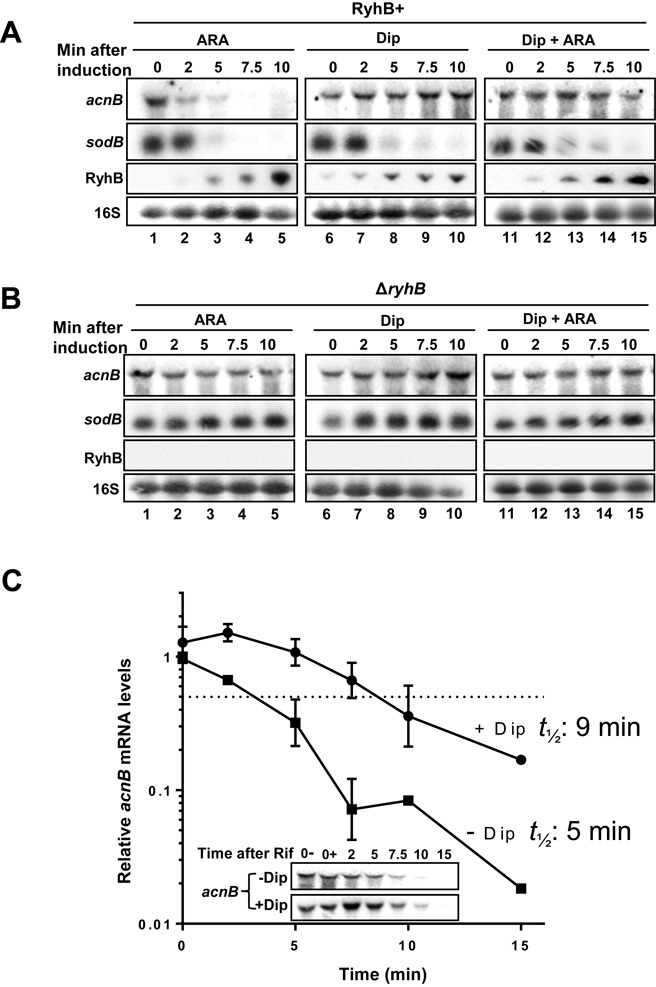
The mRNA acnB is post-transcriptionally regulated as a function of Fe status. (A) Northern blots analysis of RyhB and Fe starvation effects on acnB mRNA levels. Total RNA extracted from strains EM1455 (ΔryhB), carrying pBAD-ryhB, and EM1055 (WT) strains was hybridized with an acnB-specific probe. Cells were grown to an OD600 of 0.5 in LB medium at which point arabinose (0.05% final) (strain EM1455 carrying pBAD-ryhB, lanes 1–5), 2.2′-dipyridyl (Dip) (200 μM final) (strain EM1055, lanes 6–10) or both (strain 1455 carrying pBAD-ryhB, lanes 11–15) were added at time 0. Total RNA was extracted at the indicated time. The well-characterized RyhB target sodB mRNA is shown as a positive control for RyhB-induced degradation under all conditions. 16S rRNA was used as a loading control. (B) Same results as in panel (A) but total RNA extracted from strains EM1455 (ΔryhB) carrying pNM12 and EM1238 (ΔryhB) in which there was no RyhB expression. (C) Decay rates of acnB mRNA in LB medium. Fe chelator Dip (200 μM) was added at time −10 min. Then, rifampicin (Rif) was added (250 μg/ml final concentration) at time 0 before total RNAs were extracted at the indicated time points. These data are representative of four independent biological replicates quantified by densitometry and normalized to time 0 (0+) for both +Dip and −Dip. Mean and SD values experiments are shown. The dashed line corresponds to 50% decrease in mRNA levels. Inset: Northern blots using acnB probe showing acnB mRNA stability. Calculated acnB half-life value is indicated beside respective experimental conditions on the graph.
Control experiments performed in the absence of RyhB (ΔryhB) showed no effect on both acnB and sodB mRNAs (Figure 1B, lanes 1–5). Unexpectedly, addition of Dip (250 μM) induced a 2-fold increase of acnB mRNA levels when cells had been treated with the iron chelator for 10 min (Figure 1B, lanes 6–10). This acnB mRNA stabilization effect was similar in the case of another Fe chelator, diethylene triamine pentaacetic acid (DTPA), which unlike the Fe chelator Dip is not internalized by cells (Supplementary Figure S1A). These results were consistent with the interpretation that DTPA favored the switch to apo-AcnB, which would then bind its own mRNA (4). Taken together, our finding suggested that acnB mRNA was stabilized against RyhB-induced mRNA degradation in a Fe-depleted medium.
To determine whether the promoter of acnB was sensitive to intracellular iron levels, we measured promoter activity in the presence or absence of Dip. Activities of acnB promoter showed that there were no significant differences between these two experimental conditions (Supplementary Figure S1B). This result was in contrast to that observed for the ryhB promoter in which case there was a strong response to iron depletion (Supplementary Figure S1B, ryhB). A possible explanation to account for the fact that the stability of acnB mRNA increased under depleted iron (Figure 1A and B, compare lane 6 with lane 10) was that AcnB switched to its apo form favoring its binding to its own mRNA and increasing its stability. This possibility was tested by measuring acnB mRNA half-life in extracts of ΔryhB cells grown in the presence or absence of Dip, for 15 min. As expected, acnB mRNA stability was significantly increased in the presence of Dip, from 5 to 9 min as compared to growth in the absence of Dip (Figure 1C). These data were consistent with the interpretation of post-transcriptional stabilization of acnB under iron-depleted conditions.
Apo-AcnB binds 3′UTR of acnB mRNA under conditions of iron depletion
Previous in vitro experiments have suggested that apo-AcnB bound to the 3′UTR of acnB mRNA (5). In these experiments, the authors used a 308 nucleotide-long sequence covering the 3′UTR of acnB without giving experimental evidence for the 3′ end of the transcript. We set up experiments to define the 3′ end of acnB by performing 3′RACE experiments, which indicated that the transcript terminated 67 nt after acnB ORF (Figure 2A, sequence and structure of acnB 3′UTR). Next, we performed experiments to identify the apo-AcnB binding site on acnB 3′UTR. Footprinting assays in which radiolabeled acnB 3′UTR was subjected to partial cleavage by RNase T1 (cleaves single-stranded Gs) and RNase TA (cleaves single-stranded As) in the presence of increasing amounts of purified AcnB3xFLAG protein were performed. We found clear protection of the structure surrounding the stem–loop located in 3′UTR (Figure 2B, lanes 4–11). These results suggested that apo-AcnB bound to the 3′UTR of acnB mRNA in the stem–loop structure immediately downstream of the ORF (red nucleotides in Figure 2A).
Figure 2.
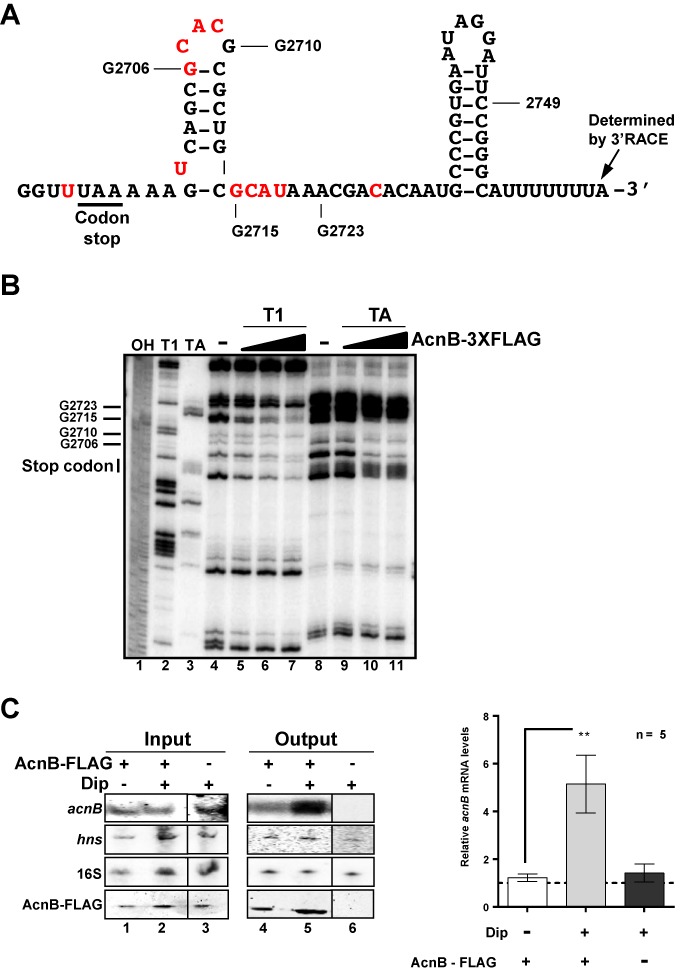
AcnB3xFLAG binds to acnB RNA 3′UTR in vitro and in vivo in Fe-depleted medium. (A) Secondary structure and sequence of the 3′UTR of acnB mRNA. In red, nt protected by apo-AcnB in footprint experiment (see panel B). (B) Footprint analysis of purified AcnB3xFLAG protein showing binding site on acnB 3′UTR RNA. (Lane 1) NaOH ladder. (Lane 2) RNase T1 ladder. (Lane 3) RNase TA ladder. (Lanes 4–7) acnB RNA T1 digestion with addition of increasing amounts of purified AcnB3xFLAG protein (ratio acnB RNA:AcnB3xFLAG, 1:0, 1:1, 1:2 and 1:10, respectively). (Lanes 8–11) TA digestion of acnB RNA with addition of increasing amounts of AcnB3x FLAG protein (same ratio as used for T1 digestion). (C) AcnB3xFLAG binds to acnB mRNA in Fe-depleted medium in vivo. RNA IP performed with pBAD-acnB3xFLAG (JAB146A) (lanes 1, 2, 4 and 5) compared to pBAD-acnB (JAB151) (lanes 3 and 6). Dip (200 μM) was added 10 min before stopping cells growth and proceeding to RNA IP. Northern blots, hybridized with an acnB RNA probe and western blot, using an anti-AcnB antibody, were performed on samples before (input) and after (output) RNA IP. 16S rRNA and hns mRNA were used as negative controls for AcnB3xFLAG enrichment. Northern blot signals were quantified by densitometry and normalized to acnB mRNA levels without Dip. Mean and SD values from five biological replicates are shown. Statistical one-way ANOVA test significance is shown by asterisks (P < 0.002).
Because previous results suggested AcnB bound to acnB 3′UTR in vitro, we next performed AcnB3xFLAG RNA immunoprecipitation (RIP) experiment to determine whether AcnB could bind to acnB mRNA under conditions of iron depletion. Cells were grown in LB medium to an OD of 0.5 after which point cells were left untreated or Dip was added for 15 min. Cells were then lysed and lysate was applied to a FLAG-binding column. After washes and elution, acnB mRNA was detected by northern blot analysis. Densitometry analysis revealed a significant enrichment (4- to 6-fold) in acnB mRNA recovery in the presence of Dip as compared to its absence (Figure 2C, lanes 4 and 5). Control experiments were performed by monitoring acnB mRNA recovery in a strain carrying plasmid pBAD24-acnB without FLAG. Under these control conditions, there was no recovery of acnB mRNA from cultures grown under Fe depletion (Figure 2C, lanes 3 and 6). Furthermore, there were no variations between the various conditions of culture with the non-target control hns mRNA and 16S ribosomal RNA (Figure 2, lanes 1–6). These results gave further support to the interpretation that interaction between AcnB3xFLAG and acnB mRNA depended on depleted intracellular Fe.
Implication of acnB 3′UTR in regulation of its mRNA stability
Results described above (Figure 2) suggested that acnB 3′UTR mediated the stability of acnB mRNA through interaction with AcnB under conditions of iron starvation. To investigate the involvement of acnB 3′UTR in AcnB binding, we engineered a transcriptional lacZ fusion, which carried all but the last 12 nt of acnB mRNA (fused at nt 2749, see Figure 3A). The fusion acnB2749-lacZ reproduced the same pattern of degradation previously observed (Figure 1A and B) 10 min after RyhB expression under control of the Ara-inducible promoter (Fe-rich condition) (Figure 3B, lanes 1 and 2) or in the presence of Dip (Figure 3B, lanes 5 and 6). Furthermore, data indicated (Figure 2B) that the stem–loop located immediately downstream of acnB 3′UTR could bind to AcnB under iron depletion. To investigate the role of this stem–loop in acnB 3′UTR, we constructed an acnB2749Mstem-lacZ fusion that was mutated for 2 nt within the stem structure. This mutation disrupts the stem–loop secondary structure as suggested by Mfold (http://mfold.rit.albany.edu/?q=mfold. Data not shown). Northern blots indicated that acnB2749Mstem-lacZ was completely degraded after 10 min of RyhB expression whether RyhB was expressed from plasmid with Ara (Figure 3C, lanes 1 and 2) or, endogenously, in the presence of Dip (Figure 3C, lanes 5 and 6). To address the question whether the RNA sequence or the stem–loop structure was responsible for the loss of regulation, we constructed a fusion carrying compensatory mutations that reconstituted the stem–loop in acnB 3′UTR (acnB2749-stem, Figure 3A). Northern blots revealed that, under typical growth conditions, the acnB2749-stem construct was degraded when RyhB was expressed from Ara (Figure 3D, lanes 1 and 2) but remained intact when RyhB was expressed in cells cultured in the presence of Dip (Figure 3D, lanes 5 and 6). These results were interpreted to suggest that the stem–loop structure located immediately downstream of acnB ORF played an important role in the stability of acnB mRNA when RyhB is expressed.
Figure 3.
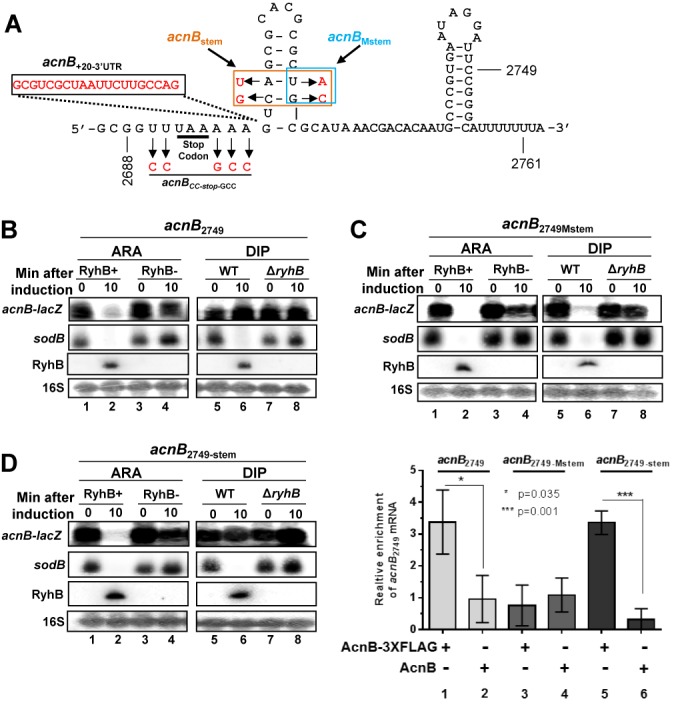
Apo-AcnB binding on acnB mRNA is essential for prevention of sRNA-induced degradation. (A) Scheme of acnB 3′UTR showing acnB2688, acnB2749, acnB2749-Mstem, acnB+20-3’UTR and acnBCC-stop-GCC transcriptional lacZ fusions constructs. The end of three commonly used transcriptional acnB’-lacZ fusion length 2688, 2749 and 2761 are annotated on the scheme. Letters in red corresponds to mutated nt for the indicated construct. (B) Northern blot of acnB2749 transcriptional lacZ fusion, sodB mRNA, RyhB sRNA and 16S rRNA that were hybridized with the corresponding RNA probes (ARA: strain KP1135 carrying pGD3-ryhB (RyhB+) or empty pGD3 plasmid (RyhB−); Dip: strains EM1055 (WT) or EM1238 (ΔryhB)). Total RNA extraction was performed at the indicated time (0 or 10 min), at an OD600 of 0.5. Northern blots showing the effect of RyhB on acnB2749 (B), acnB2749-Mstem (C) and acnB2749-stem (D) transcriptional lacZ fusions. Expression of RyhB was induced at time 0 by addition of arabinose (0.05% final) or Dip (200 μM final). (E) AcnB3xFLAG RNA-IP on acnB2749, on acnB2749-Mstem and on acnB2749-stem constructs were expressed from pBAD33-derived plasmids in JAB292 cells (acnB736::kan). AcnB protein induction (OD600 of 0.1) was performed from pBAD-acnB3xFLAG and from pBAD24 plasmids. Then, when OD600 reached 0.5, RNA IP was performed. qRT-PCR analysis was performed with a probe covering the acnB 3′UTR for determination of acnB constructs enrichment. Mean with SD values from triplicates experiments are shown. Statistical one-way ANOVA test significance is shown by asterisks.
To investigate the possibility that AcnB bound acnB constructs with different affinities, depending on the 3′UTR sequences, we performed RNA IP assays. In these experiments, FLAG-tagged AcnB (AcnB3xFLAG) was used to quantitate binding affinity of the various constructs described above (Figure 3, acnB2749, acnB2749Mstem, acnB2749-stem). Cells were grown to mid-log phase, at which point they were treated with Dip (250 μM, 10 min), followed by lysis and purification (see ‘Materials and Methods’ section for details). Enrichment of the specific construct acnB2749 was then measured by qPCR. Results showed that transcriptional constructs acnB2749 and acnB2749-stem had comparable enrichment with AcnB3xFLAG relative to untagged AcnB (Figure 3E, lanes 1 and 2, and lanes 5 and 6). In contrast, acnB2749Mstem construct showed no enrichment with AcnB3xFLAG (Figure 3E, lanes 3–4). These results established that the stem–loop structure was essential for AcnB binding to acnB 3′UTR under conditions of iron depletion.
The 3′UTR of acnB mRNA is essential for RyhB-induced cleavage
The data described above suggested that binding of AcnB to acnB 3′UTR interfered with RyhB-induced degradation of the mRNA. To add support to this interpretation, we constructed a transcriptional fusion (acnB2688-lacZ) comprising all but the last two codons of acnB ORF (Figure 3A). The reasoning behind the use of this construct was that it would allow RyhB binding to the translation initiation region (Supplementary Figures S2A, S2B and S2C) but would prevent apo-AcnB binding at the 3′UTR of acnB mRNA. The fusion acnB2688-lacZ was tested in cells grown under usual conditions (Figure 1A). Northern blots showed that acnB2688-lacZ remained resistant to the expression of RyhB after 10 min of induction (Figure 4A, lanes 1 and 2).
Figure 4.
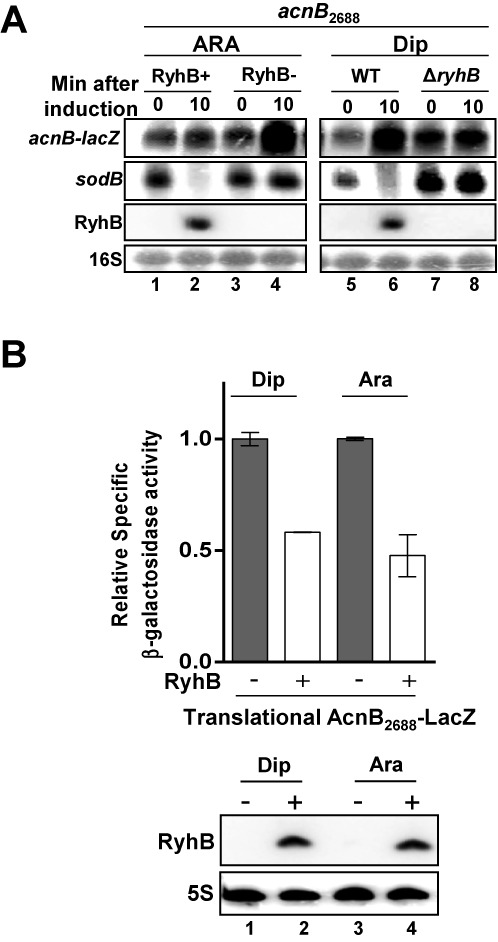
The acnB 3′ end is essential for RyhB-induced mRNA degradation. (A) Northern blot of total RNA of transcriptional acnB2688-lacZ fusion hybridized with acnB RNA probe. RNA was extracted after 10 min of RyhB induction with arabinose (0.05% final) or by Dip (200 μM final) (ARA: strain KP1135 carrying pGD3-ryhB (RyhB+) or empty pGD3 plasmid (RyhB−); Dip: strains EM1055 (WT) or EM1238 (ΔryhB)). RyhB sRNA was hybridized to compare its expression in both conditions. 16S rRNA was used as loading control. (B) β-Galactosidase assays were performed with translational AcnB2688-LacZ fusion, which excluded acnB 3′UTR and its last two codons. Dip (200 μM final) for WT strain (EM1055) or arabinose (0.05% final) for pBAD-ryhB transformed strain (EM1455) were added to induce RyhB expression at an OD600 of 0.1. Specific β-galactosidase activity was measured in triplicates. Mean and SD of relative β-galactosidase activity (normalized to conditions without RyhB) are shown on graph. The empty vector pNM12 (EM1455) or ΔryhB (EM1238) strains were used as a control for AcnB2688-LacZ without RyhB. Northern blot with RyhB probe on total RNA was performed at an OD600 of 0.5 along with β-galactosidase assays. 5S rRNA was used as loading control.
Although these observations suggested that RyhB expression could not induce acnB2688-lacZ degradation, they did not exclude the possibility that RyhB could nevertheless bind the construct. To confirm that RyhB was still able to bind acnB2688 transcript and block translation, we constructed a translational AcnB2688-LacZ fusion. Specific β-galactosidase activities indicated that RyhB significantly reduced expression of translation, regardless of the mode of induction (Dip, Ara) of RyhB (Figure 4B). These results, together with those shown in Figure 4A, strongly suggested that even if RyhB could bind acnB mRNA, the absence of the 3′UTR region prevented sRNA-induced mRNA cleavage. To further investigate the effect of RyhB pairing on 30S ribosomal subunit binding to acnB mRNA translation initiation region (TIR), we performed a toeprint assay in the presence of increasing amount of RyhB. Densitometry analysis revealed a strong inhibition (10-fold) of 30S subunit binding in the presence of added RyhB (Supplementary Figure S3, compare lanes 7 and 11, and 7 and 13). Moreover, the addition of purified Hfq chaperone contributed to block 30S subunit binding on acnB mRNA TIR by an additional 2.3-fold (Supplementary Figure S3, compare lanes 9 and 10, 11 and 12, 13 and 14). Control experiments showed that excess molar ratio of DsrA sRNA had no effect on 30S ribosomal subunit binding on acnB (Supplementary Figure S3, lane 15).
Characterization of RNA degradosome RyhB-induced cleavage site on acnB mRNA
Most trans-acting sRNAs that induce degradation of their target mRNAs usually recruit RNase E through Hfq-mediated interaction (40). To confirm this mechanism in relationship to RyhB and acnB, we performed pulse-expression of RyhB in rne131 background, which carried a deletion of RNase E C-terminus that prevented RNase E recruitment by Hfq. Northern blots confirmed that RNase E was essential for acnB mRNA degradation after a 10 min of RyhB induction with Ara (Figure 5A, lanes 2 and 4). The possibility that acnB 3′UTR carried a cleavage site recognized by RNase E (33) was investigated by degradation assays using purified RNA degradosome in the presence of radiolabeled acnB 3′UTR (from nt 2569 to nt 2767). Degradation assay revealed a strong cleavage signal near the stop codon of acnB ORF (Figure 5B, compare lanes 4 and 5), which mapped between nt 2690 and 2697 (see Figure 3A) close to acnB stop codon. We then mutagenized the cleavage site by modifying the sequence UUUAAAAA to CCUAAGCC (see Figure 3A). The mutant construct was then subjected to RNA degradosome assay. Results showed that CCUAAGCC mutations in the end of acnB ORF prevented cleavage by RNase E (Figure 5B, compare lanes 5 and 10). These results suggested that RNase E required the original AU-rich sequence near the stop codon of acnB mRNA to induce cleavage. To confirm that the RNase E-dependent cleavage site (Figure 5B) was essential, we mutated the acnB2749-lacZ construct to introduce the CCUAAGCC mutation that blocked RNase E cleavage. Cells harboring the construct acnB2749CC-stop-GCC were subjected to RyhB expression and data analyzed by qRT-PCR. The acnB2749CC-stop-GCC construct was completely resistant to RyhB-induced mRNA degradation under Fe-rich condition (Figure 5C, compare acnB2749CC-stop-GCC and sodB panels, columns 1 and 2), highlighting the importance of the RyhB-induced cleavage site that mapped at the end of acnB ORF (Figure 5B, nt 2692–2697).
Figure 5.
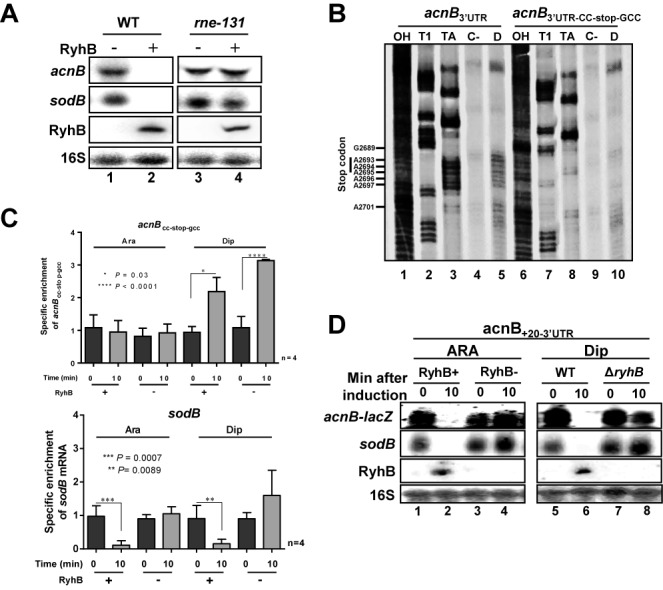
RyhB induces RNase E cleavage in acnB mRNA 3′ end. (A) Northern blot of acnB mRNA were performed on total RNA extracted from strain EM1455 and JF133 (rne131 mutant). Both stains were transformed with plasmid allowing arabinose-dependent RyhB expression (pBAD-ryhB) or with control vector (pNM12). Cells were grown in LB medium to an OD600 of 0.5 and RyhB sRNA was induced at time 0 by addition of arabinose (0.01% final). Total RNA extraction was performed after 10 min of induction. (B) RNA degradosome degradation assay to map RNase E cleavage site on acnB 3′UTR. Effect of purified RNA degradosome (0.5 ng/μl final concentration) on 32P-pCp 3′ end labeling acnB-3′UTR (lanes 4 and 5) and acnB3′UTR-CC-stop-GCC (lanes 9 and 10). (Lanes 1 and 6) NaOH ladder. (Lanes 2 and 7) RNase T1 ladder. (Lanes 3 and 8) RNase TA ladder. (Lanes 4 and 9) Radiolabeled acnB RNA alone (see Figure 3A for cleavage site map). (C) qRT-PCR of acnBCC-stop-gcc transcriptional lacZ fusion from total RNA extraction. RyhB sRNA expression was induced by addition of arabinose (0.05% final) for pGD3-ryhB (KP1135) or by addition of Dip (200 μM final) for wild-type (EM1055). Total RNA extraction was performed at the indicated time, at mid-logarithmic growth phase. Endogenous sodB mRNA levels after RyhB expression are also shown. Mean and SD values of four replicates experiments are shown. Asterisks correspond to statistical significance from one-way ANOVA test. (D) Northern blot using acnB probe showing the effect of RyhB expression on acnB+20-3′UTR-transcriptional lacZ fusion (after 10 min). Before RNA extraction, RyhB sRNA expression was induced by addition of arabinose (0.05% final) for pGD3-ryhB (KP1135) or by addition of Dip (200 μM final) for wild-type (EM1055) at time 0. Empty plasmid pGD3 or ΔryhB strains were used as negatives controls for ARA and Dip panels, respectively.
Next, we asked whether the 3′ end of acnB could function ectopically on sodB, a mRNA that is also a target of RyhB. We used a truncated version of sodB mRNA (sodB130) which has previously been shown to be resistant to RyhB-induced mRNA degradation (33). The construct was fused to the 3′ end of acnB to obtain sodB130-acnB+67 (Supplementary Figure S4A for details). As expected, this construct was sensitive to RyhB sRNA-dependent degradation under Fe-rich conditions (Supplementary Figure S4B, compare lanes 5 and 10). In contrast, induction of RyhB by Dip led to a slower degradation of the sodB130-acnB+67 construct, suggesting that acnB 3′ end can act ectopically to partially stabilize a particular mRNA from RyhB-induced degradation (Supplementary Figure S4).
Our results suggested that apo-AcnB protected acnB mRNA against RyhB-induced mRNA cleavage by occluding RNase E access to the cleavage site. This possibility required that the apo-AcnB binding site must remain proximal to the cleavage site for competitive inhibition. To address this possibility, we initially constructed a lacZ fusion in which a sequence of 20 nt was inserted between RyhB-induced RNase E cleavage site and apo-AcnB binding site (acnB+20-3′UTR-lacZ) (Figure 3A). It must be noted that this 20 nt sequence has previously been reported to be resistant to RNAse E cleavage (41). As expected, northern blot analysis showed that the acnB+20-3′UTR-lacZ construct was sensitive to RyhB when cells were grown under Fe starvation (Figure 5D, lanes 5 and 6). This result could be explained by decreased binding affinity of apo-AcnB for acnB+20-3′UTR-lacZ construct. This possibility was tested by performing an AcnB3xFLAG IP experiment using cells grown in Fe-poor medium. Data of IP experiments revealed acnB mRNA enrichment comparable to acnB2749 WT by apo-AcnB (Supplementary Figure S4C).
Antagonist regulation of acnB mRNA and its effect on protein levels
To evaluate the effect RyhB on AcnB expression, we monitored AcnB levels by western blots and compared the results to native AcnB from WT and ΔryhB cells grown under Fe starvation. Unexpectedly, AcnB levels remained stable over time when RyhB was induced in the presence of Dip (Figure 6A, left panel). In marked contrast, we observed a significant accumulation of AcnB (3.6-fold increase) in ΔryhB cells 2 h after Dip treatment (Figure 6A, right panel). These results suggested that RyhB expression significantly repressed AcnB translation when cells grew under low Fe conditions.
Figure 6.
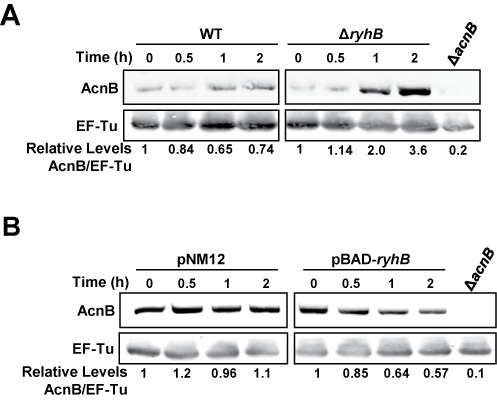
Both apo-AcnB stabilization and RyhB negative regulation affect AcnB levels. (A) Quantitative western blot of AcnB levels in Fe-depleted medium. Fe depletion was induced by addition of Dip (200 μM final) to WT (EM1055) or to ΔryhB strains (EM1238) at time 0. Total proteins were extracted at the indicated time (h). Relative densitometry levels are indicated under the lanes. (B) Quantitative western blot of AcnB levels under Fe-rich conditions when RyhB sRNA was expressed in WT strain (EM1455) from pBAD-ryhB compared with an empty plasmid pNM12 (0.01% arabinose final). Proteins were extracted at the indicated time (h). EF-Tu protein was used as a loading control in both experiments. A polyclonal anti-AcnB antibody was used for hybridization and antibody specificity was determined using a protein extract from ΔacnB strain (strain JAB284 for panel A, JAB154 for panel B). Relative densitometry levels are indicated under the lanes. A IRDye 800CW-conjugated secondary antibody was used for quantification.
We then investigated the effect of RyhB-induced repression of AcnB expression in Fe-rich LB medium (Figure 6B). Western blot analysis revealed an important decrease (43%) of AcnB levels after expression of RyhB sRNA from an arabinose-inducible BAD promoter in ΔryhB cells (Figure 6B, right panel, 2 h). The decrease correlated with the repression observed on translational AcnB2688’-‘LacZ translational fusion (48% decrease) (Figure 4B, Ara). These results added support to the interpretation that RyhB directly blocked acnB mRNA translation initiation, thus significantly affect the levels of AcnB in these cells.
Citrate interferes with acnB mRNA stabilization under low-iron conditions
Low intracellular iron conditions favor the switch of enzymatic (holo) AcnB to its RNA-binding (apo) conformation. We asked whether citrate, the substrate of AcnB, could interfere with this switch. To answer this question, EM1238 (ΔryhB) cells were used to monitor the effect of apo-AcnB alone on acnB mRNA. Thus, cells were grown in iron-rich LB medium until mid-log phase, at which point Dip or a combination of Dip and citrate were added, followed by rifampicin to assess acnB mRNA stability. Densitometry analysis indicated that acnB mRNA half-life was slightly stabilized in the presence of Dip, which correlated with the switch of holo- to apo-AcnB RNA under low iron (Figure 7, top and middle panels). In contrast, the addition of citrate decreased the stability of acnB in a way similar to growth conditions in medium alone (Figure 7). These data were consistent with the interpretation that the presence of citrate prevented the switch of holo- to apo-AcnB even under low Fe conditions.
Figure 7.
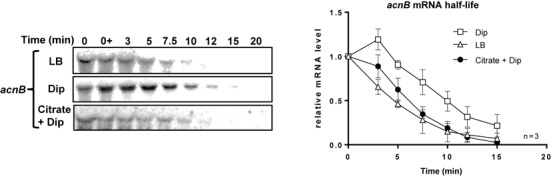
Addition of citrate, the AcnB substrate, abolishes acnB mRNA stabilization. Northern blot of acnB mRNA half-life and corresponding densitometric curves of acnB mRNA are shown. Cells (EM1238) were grown in LB medium to an OD600 of 0.5 at which point citrate (30 mM final) or Dip (200 μM final) were added at indicated time. Thereafter rifampicin (250 μg/ml) was added at time 0 and RNA extraction was performed at the indicated times. Data are representative of two independent experiments performed in triplicates.
DISCUSSION
A common outcome of sRNAs binding to their target mRNAs is rapid RNase E-mediated cleavage of the targets. However, the RyhB-induced acnB mRNA degradation that has been previously reported (15,31) can be prevented when cells are grown under low-iron conditions. Combined with previous studies, data presented here suggested that reduced intracellular iron promoted the switch of holo-AcnB to apo-AcnB with the resulting RNA-binding of acnB mRNA 3′UTR and protection against RyhB-induced mRNA degradation. According to the model proposed here, RyhB still binds to acnB RBS and partially blocks its translation initiation under low-iron conditions. However, our data support the interpretation that bound apo-AcnB seems to interfere with RNase E access to acnB mRNA cleavage site (Figure 8). Accordingly, this mechanism of regulation allows long-term expression homeostasis of AcnB as opposed to a significant stronger expression of AcnB in the absence of RyhB sRNA (Figure 6A). Although RyhB partially blocked acnB translation (Figures 4B and 6A, right), there was a steady production of AcnB (Figure 6A, left). This seemingly paradoxical observation may be explained by the functional nature of AcnB. When cells grow under iron starvation, most RyhB targets become degraded. These degraded targets encode single-function enzymes, whose activity would falter in the absence of sufficient amounts of iron as cofactor. In contrast, bifunctional AcnB switches functions despite low intracellular iron level. When it adopts its catalytic conformation, aconitase has an iron-dependent metabolic function that participates to the TCA cycle. As an RNA-binding protein, apo-AcnB regulates expression of various transcripts such as acnB and ftsH (3) despite environmental variations of iron concentrations and maintains steady expression of acnB that allows sufficient quantities of cellular AcnB to modulate both activities.
Figure 8.
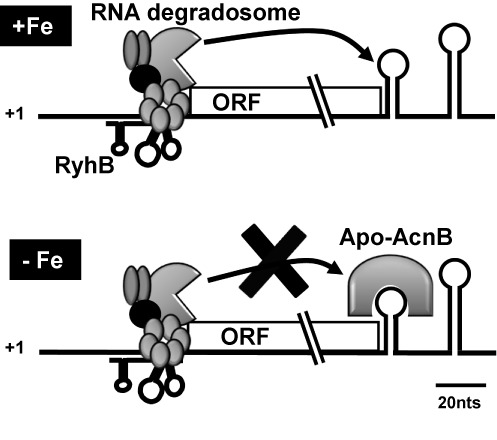
Working model of acnB mRNA double regulation by sRNA RyhB and RNA binding protein apo-AcnB (refer to the text for details).
The question may be raised concerning the reason why two effectors, a sRNA and a RNA-binding protein, would regulate acnB mRNA that results, in any case, in similar expression levels of AcnB? Whereas these findings suggest a robust homeostatic system operating in E. coli, the presence of citrate reduced acnB stability under iron starvation conditions (Figure 7). These data are in agreement with previous observations indicating that citrate, the substrate of AcnB, prevents AcnB from switching to the apo-form (Varghese). This explains why citrate can reduce the expression of acnB even under conditions of low iron. It can be suggested that the sensitivity to citrate may allow the system to sense and respond to metabolic changes. This possibility is especially relevant as citrate can also act as an iron chelator, helping the cell to cope with low-iron conditions (42,43). Thus under growth conditions of low iron but high citrate, AcnB expression would be reduced (less apo-AcnB isoform) and iron acquisition would be increased in the presence of citrate. These growth conditions would favor activity of AcnB. In contrast, when both iron and citrate are low, there would be more AcnB in its apo-AcnB conformation, thus favoring its RNA-binding activity.
Our study suggests a sRNA-induced RNase E mRNA cleavage close to the 3′UTR of acnB mRNA, some 2600 nts from the sRNA binding site. Although we have previously shown a distal sRNA-induced RNase E cleavage on sodB mRNA 350 nt away from the sRNA binding site, this is a considerably longer distance. Such a distal location suggests the importance of regulating acnB mRNA stability accordingly to the RNA binding ability of AcnB. A similar system has been identified with transcripts regulated by microRNA (miRNA) in eukaryotes. In general, miRNAs are post-transcriptional repressors that act together with a protein complex named miRNA-induced-silencing-complex (miRISC). The multimeric complex binds imperfect complementary sequences of 3′UTR mRNA to repress translation. Frequently, RNA degradation follows miRNA pairing by stimulating deadenylation and decapping events (44–46). However, there are some examples where RNA binding proteins antagonize miRNA silencing. One of them occurs in human hepatocarcinoma cells where amino acids starvation leads to HuR protein oligomerization and binding to 3′UTR, impeding miRNA pairing (47,48). Here, we present data that support a similar model in which RyhB-induced degradation is prevented by the RNA binding protein apo-AcnB.
A key point about the mechanism of protection against sRNA-induced acnB cleavage is the close proximity of AcnB binding and RNase E cleavage sites. Evidence for this interpretation was obtained by increasing the distance between both sites by inserting a 20 nt sequence, previously shown to resist RNase E cleavage. Results indicated that increasing the distance abolished the apo-AcnB protection when RyhB was expressed (Figure 5D, acnB+20-3′UTR). This finding was similar to a report in which rpsO stop codon was distanced from a nearby RNase E cleavage site located in the mRNA 3′UTR and that resulted in reduced cleavage protection by ribosomes (41). In spite of these findings, the possibility remained that introduction of a 20 nt sequence in the 3′UTR of acnB could prevent apo-AcnB binding, thus allowing cleavage. However, this possibility was excluded by performing RNA-IP of apo-AcnB. Data showed that similar levels of acnB+20-3′UTR and WT acnB RNA were observed (Supplementary Figure S4C), suggesting that apo-AcnB to acnB exerted steric hindrance to RNase E degrading activity.
Autoregulated proteins such as AcnB are common in bacterial housekeeping genes as observed in the case of many ribosomal proteins and tRNA regulators (49–52). For example, rpmL-rplT transcript is repressed when the second gene product, ribosomal protein L20, is overproduced. The binding of L20 protein to a pseudoknot located in rpmL-rplT 5′UTR blocks 30S ribosomal association with mRNA thereby preventing translation initiation (52,53). A similar case is ribosomal protein RpsO which binds structures in the 5′UTR of its own mRNA when it is overexpressed. Bound RpsO prevents translation initiation of rpsO mRNA, which becomes destabilized by exposed RNase E cleavage sites (41,54). These multiple levels of regulation/autoregulation of the same mRNA seem essential for highly regulated constitutive genes or global regulators.
Cellular adaptation to low iron growth remains a major stress for bacteria. When cells grow under low Fe conditions, many enzymes become virtually inactive in the absence of their iron cofactor. Under these stressful environmental conditions, these iron-dependent enzymes/proteins are repressed through sRNA-induced degradation of their mRNAs whereas in other cases some proteins/enzymes such as AcnB switch to a different functional form.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to Karine Prévost, Phillipe Balthazar and Marie-Claude Carrier for excellent technical assistance. We thank Jeffrey E. Green (National Cancer Institute) for a gift of anti-aconitase B polyclonal antibody. We acknowledge all members of the laboratory for useful comments and discussions.
FUNDING
Canadian Institutes of Health Research [MOP69005 to E.M.]; Natural Sciences and Engineering Research Council of Canada [PhD studentship to J.A.M.B.]; Fonds de Recherche du Québec—Santé [24852 to E.M.]. Funding for open access charge: Canadian Institutes of Health Research [MOP69005 to E.M.]; Natural Sciences and Engineering Research Council of Canada [PhD studentship to J.A.M.B.]; Fonds de Recherche du Québec—Santé [to E.M.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Cunningham L., Gruer M.J., Guest J.R. Transcriptional regulation of the aconitase genes (acnA and acnB) of Escherichia coli. Microbiology. 1997;143:3795–3805. doi: 10.1099/00221287-143-12-3795. [DOI] [PubMed] [Google Scholar]
- 2.Tang Y., Quail M.A., Artymiuk P.J., Guest J.R., Green J. Escherichia coli aconitases and oxidative stress: post-transcriptional regulation of sodA expression. Microbiology. 2002;148:1027–1037. doi: 10.1099/00221287-148-4-1027. [DOI] [PubMed] [Google Scholar]
- 3.Tang Y., Guest J.R., Artymiuk P.J., Read R.C., Green J. Post-transcriptional regulation of bacterial motility by aconitase proteins. Mol. Microbiol. 2004;51:1817–1826. doi: 10.1111/j.1365-2958.2003.03954.x. [DOI] [PubMed] [Google Scholar]
- 4.Varghese S., Tang Y., Imlay J.A. Contrasting sensitivities of Escherichia coli aconitases A and B to oxidation and iron depletion. J. Bacteriol. 2003;185:221–230. doi: 10.1128/JB.185.1.221-230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang Y., Guest J.R. Direct evidence for mRNA binding and post-transcriptional regulation by Escherichia coli aconitases. Microbiology. 1999;145:3069–3079. doi: 10.1099/00221287-145-11-3069. [DOI] [PubMed] [Google Scholar]
- 6.Hentze M.W., Argos P. Homology between IRE-BP, a regulatory RNA-binding protein, aconitase, and isopropylmalate isomerase. Nucleic Acids Res. 1991;19:1739–1740. doi: 10.1093/nar/19.8.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rouault T.A., Stout C.D., Kaptain S., Harford J.B., Klausner R.D. Structural relationship between an iron-regulated RNA-binding protein (IRE-BP) and aconitase: functional implications. Cell. 1991;64:881–883. doi: 10.1016/0092-8674(91)90312-m. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy M.C., Mende-Mueller L., Blondin G.A., Beinert H. Purification and characterization of cytosolic aconitase from beef liver and its relationship to the iron-responsive element binding protein. Proc. Natl. Acad. Sci. U.S.A. 1992;89:11730–11734. doi: 10.1073/pnas.89.24.11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haile D.J., Rouault T.A., Tang C.K., Chin J., Harford J.B., Klausner R.D. Reciprocal control of RNA-binding and aconitase activity in the regulation of the iron-responsive element binding protein: role of the iron-sulfur cluster. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7536–7540. doi: 10.1073/pnas.89.16.7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castro L.A., Robalinho R.L., Cayota A., Meneghini R., Radi R. Nitric oxide and peroxynitrite-dependent aconitase inactivation and iron-regulatory protein-1 activation in mammalian fibroblasts. Arch. Biochem. Biophys. 1998;359:215–224. doi: 10.1006/abbi.1998.0898. [DOI] [PubMed] [Google Scholar]
- 11.Hentze M.W., Kühn L.C. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8175–8182. doi: 10.1073/pnas.93.16.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J., Pantopoulos K. Regulation of cellular iron metabolism. Biochem. J. 2011;434:365–381. doi: 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rouault T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 14.Addess K.J., Basilion J.P., Klausner R.D., Rouault T.A., Pardi A. Structure and dynamics of the iron responsive element RNA: implications for binding of the RNA by iron regulatory binding proteins. J. Mol. Biol. 1997;274:72–83. doi: 10.1006/jmbi.1997.1377. [DOI] [PubMed] [Google Scholar]
- 15.Masse E., Gottesman S. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4620–4625. doi: 10.1073/pnas.032066599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottesman S., Storz G. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harbor Perspect. Biol. 2011;3:a003798. doi: 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waters L.S., Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storz G., Vogel J., Wassarman K.M. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogel J., Luisi B.F. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrade J.M., Pobre V., Matos A.M., Arraiano C.M. The crucial role of PNPase in the degradation of small RNAs that are not associated with Hfq. RNA. 2012;18:844–855. doi: 10.1261/rna.029413.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vytvytska O., Moll I., Kaberdin V.R., von Gabain A., Bläsi U. Hfq (HF1) stimulates ompA mRNA decay by interfering with ribosome binding. Genes Dev. 2000;14:1109–1118. [PMC free article] [PubMed] [Google Scholar]
- 22.Desnoyers G., Massé E. Noncanonical repression of translation initiation through small RNA recruitment of the RNA chaperone Hfq. Genes Dev. 2012;26:726–739. doi: 10.1101/gad.182493.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komarova A.V., Tchufistova L.S., Dreyfus M., Boni I.V. AU-rich sequences within 5′ untranslated leaders enhance translation and stabilize mRNA in Escherichia coli. J. Bacteriol. 2005;187:1344–1349. doi: 10.1128/JB.187.4.1344-1349.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J., Deutscher M.P. A uridine-rich sequence required for translation of prokaryotic mRNA. Proc. Natl. Acad. Sci. U.S.A. 1992;89:2605–2609. doi: 10.1073/pnas.89.7.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ringquist S., Jones T., Snyder E.E., Gibson T., Boni I., Gold L. High-affinity RNA ligands to Escherichia coli ribosomes and ribosomal protein S1: comparison of natural and unnatural binding sites. Biochemistry. 1995;34:3640–3648. doi: 10.1021/bi00011a019. [DOI] [PubMed] [Google Scholar]
- 26.Boni I.V., Musychenko M.L., Tzareva N.V. Ribosome-messenger recognition: mRNA target sites for ribosomal protein S1. Nucleic Acids Res. 1991;19:155–162. doi: 10.1093/nar/19.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hook-Barnard I.G., Brickman T.J., McIntosh M.A. Identification of an AU-rich translational enhancer within the Escherichia coli fepB leader RNA. J. Bacteriol. 2007;189:4028–4037. doi: 10.1128/JB.01924-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duval M., Korepanov A., Fuchsbauer O., Fechter P., Haller A., Fabbretti A., Choulier L., Micura R., Klaholz B.P., Romby P. Escherichia coli ribosomal protein S1 unfolds structured mRNAs onto the ribosome for active translation initiation. PLoS Biol. 2013;11:e1001731. doi: 10.1371/journal.pbio.1001731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salvail H., Lanthier-Bourbonnais P., Sobota J.M., Caza M., Benjamin J.A., Mendieta M.E., Lepine F., Dozois C.M., Imlay J., Masse E. A small RNA promotes siderophore production through transcriptional and metabolic remodeling. Proc. Natl. Acad. Sci. U.S.A. 2010;107:15223–15228. doi: 10.1073/pnas.1007805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masse E., Escorcia F.E., Gottesman S. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev. 2003;17:2374–2383. doi: 10.1101/gad.1127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masse E., Vanderpool C.K., Gottesman S. Effect of RyhB small RNA on global iron use in Escherichia coli. J. Bacteriol. 2005;187:6962–6971. doi: 10.1128/JB.187.20.6962-6971.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carpousis A.J. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 2007;61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- 33.Prevost K., Desnoyers G., Jacques J.F., Lavoie F., Masse E. Small RNA-induced mRNA degradation achieved through both translation block and activated cleavage. Genes Dev. 2011;25:385–396. doi: 10.1101/gad.2001711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacques J.F., Jang S., Prevost K., Desnoyers G., Desmarais M., Imlay J., Masse E. RyhB small RNA modulates the free intracellular iron pool and is essential for normal growth during iron limitation in Escherichia coli. Mol. Microbiol. 2006;62:1181–1190. doi: 10.1111/j.1365-2958.2006.05439.x. [DOI] [PubMed] [Google Scholar]
- 35.Prevost K., Salvail H., Desnoyers G., Jacques J.F., Phaneuf E., Masse E. The small RNA RyhB activates the translation of shiA mRNA encoding a permease of shikimate, a compound involved in siderophore synthesis. Mol. Microbiol. 2007;64:1260–1273. doi: 10.1111/j.1365-2958.2007.05733.x. [DOI] [PubMed] [Google Scholar]
- 36.Desnoyers G., Morissette A., Prevost K., Masse E. Small RNA-induced differential degradation of the polycistronic mRNA iscRSUA. EMBO J. 2009;28:1551–1561. doi: 10.1038/emboj.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aiba H., Adhya S., de Crombrugghe B. Evidence for two functional gal promoters in intact Escherichia coli cells. J. Biol. Chem. 1981;256:11905–11910. [PubMed] [Google Scholar]
- 38.Masse E., Arguin M. Ironing out the problem: new mechanisms of iron homeostasis. Trends Biochem. Sci. 2005;30:462–468. doi: 10.1016/j.tibs.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 39.Salvail H., Massé E. Regulating iron storage and metabolism with RNA: an overview of posttranscriptional controls of intracellular iron homeostasis. Wiley Interdiscip. Rev.: RNA. 2012;3:26–36. doi: 10.1002/wrna.102. [DOI] [PubMed] [Google Scholar]
- 40.Lalaouna D., Simoneau-Roy M., Lafontaine D., Massé E. Regulatory RNAs and target mRNA decay in prokaryotes. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2013;1829:742–747. doi: 10.1016/j.bbagrm.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Braun F., Le Derout J., Régnier P. Ribosomes inhibit an RNase E cleavage which induces the decay of the rpsO mRNA of Escherichia coli. EMBO J. 1998;17:4790–4797. doi: 10.1093/emboj/17.16.4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frost G., Rosenberg H. The inducible citrate-dependent iron transport system in Escherichia coli K12. Biochim. Biophys. Acta (BBA)-Biomembranes. 1973;330:90–101. doi: 10.1016/0005-2736(73)90287-3. [DOI] [PubMed] [Google Scholar]
- 43.Hantke K. Regulation of ferric iron transport in Escherichia coli K12: isolation of a constitutive mutant. Mol. Gen. Genet. 1981;182:288–292. doi: 10.1007/BF00269672. [DOI] [PubMed] [Google Scholar]
- 44.Mauxion F., Chen C.-Y.A., Séraphin B., Shyu A.-B. BTG/TOB factors impact deadenylases. Trends Biochem. Sci. 2009;34:640–647. doi: 10.1016/j.tibs.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen C.-Y.A., Zheng D., Xia Z., Shyu A.-B. Ago-TNRC6 triggers microRNA-mediated decay by promoting two deadenylation steps. Nat. Struct. Mol. Biol. 2009;16:1160–1166. doi: 10.1038/nsmb.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu L., Fan J., Belasco J.G. MicroRNAs direct rapid deadenylation of mRNA. Proc. Natl. Acad. Sci. U.S.A. 2006;103:4034–4039. doi: 10.1073/pnas.0510928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharyya S.N., Habermacher R., Martine U., Closs E.I., Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 48.Kundu P., Fabian M.R., Sonenberg N., Bhattacharyya S.N., Filipowicz W. HuR protein attenuates miRNA-mediated repression by promoting miRISC dissociation from the target RNA. Nucleic Acids Res. 2012;40:5088–5100. doi: 10.1093/nar/gks148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caillet J., Nogueira T., Masquida B., Winter F., Graffe M., Dock-Brégeon A.C., Torres-Larios A., Sankaranarayanan R., Westhof E., Ehresmann B. The modular structure of Escherichia coli threonyl-tRNA synthetase as both an enzyme and a regulator of gene expression. Mol. Microbiol. 2003;47:961–974. doi: 10.1046/j.1365-2958.2003.03364.x. [DOI] [PubMed] [Google Scholar]
- 50.Nomura M., Yates J.L., Dean D., Post L.E. Feedback regulation of ribosomal protein gene expression in Escherichia coli: structural homology of ribosomal RNA and ribosomal protein mRNA. Proc. Natl. Acad. Sci. 1980;77:7084–7088. doi: 10.1073/pnas.77.12.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nomura M., Gourse R., Baughman G. Regulation of the synthesis of ribosomes and ribosomal components. Annu. Rev. Biochem. 1984;53:75–117. doi: 10.1146/annurev.bi.53.070184.000451. [DOI] [PubMed] [Google Scholar]
- 52.Guillier M., Allemand F., Raibaud S., Dardel F., Springer M., Chiaruttini C. Translational feedback regulation of the gene for L35 in Escherichia coli requires binding of ribosomal protein L20 to two sites in its leader mRNA: a possible case of ribosomal RNA-messenger RNA molecular mimicry. RNA. 2002;8:878–889. doi: 10.1017/s1355838202029084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haentjens-Sitri J., Allemand F., Springer M., Chiaruttini C. A competition mechanism regulates the translation of the Escherichia coli operon encoding ribosomal proteins L35 and L20. J. Mol. Biol. 2008;375:612–625. doi: 10.1016/j.jmb.2007.10.058. [DOI] [PubMed] [Google Scholar]
- 54.Philippe C., Eyermann F., Benard L., Portier C., Ehresmann B., Ehresmann C. Ribosomal protein S15 from Escherichia coli modulates its own translation by trapping the ribosome on the mRNA initiation loading site. Proc. Natl. Acad. Sci. U.S.A. 1993;90:4394–4398. doi: 10.1073/pnas.90.10.4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.