


Abstract
eIF5 is the GTPase activating protein (GAP) for the eIF2·GTP·Met-tRNAiMet ternary complex with a critical role in initiation codon selection. Previous work suggested that the eIF5 mutation G31R/SUI5 elevates initiation at UUG codons by increasing GAP function. Subsequent work implicated eIF5 in rearrangement of the preinitiation complex (PIC) from an open, scanning conformation to a closed state at AUG codons, from which Pi is released from eIF2·GDP·Pi. To identify eIF5 functions crucial for accurate initiation, we investigated the consequences of G31R on GTP hydrolysis and Pi release, and the effects of intragenic G31R suppressors on these reactions, and on the partitioning of PICs between open and closed states. eIF5-G31R altered regulation of Pi release, accelerating it at UUG while decreasing it at AUG codons, consistent with its ability to stabilize the closed complex at UUG. Suppressor G62S mitigates both defects of G31R, accounting for its efficient suppression of UUG initiation in G31R,G62S cells; however suppressor M18V impairs GTP hydrolysis with little effect on PIC conformation. The strong defect in GTP hydrolysis conferred by M18V likely explains its broad suppression of Sui− mutations in numerous factors. We conclude that both of eIF5's functions, regulating Pi release and stabilizing the closed PIC conformation, contribute to stringent AUG selection in vivo.
INTRODUCTION
In translation initiation by the scanning mechanism, the small (40S) ribosomal subunit harboring initiator methionyl tRNA (Met-tRNAi) bound to eIF2-GTP in a ternary complex (TC) attaches near the capped 5′ end of the mRNA and scans the leader for an AUG triplet in optimal sequence context (reviewed in (1,2)). According to our current model (Figure 1A), eIF1 and eIF1A promote an ‘open’ conformation of the 40S subunit that is competent for binding the TC in a metastable state (POUT) that allows the Met-tRNAi to sample successive triplets entering the P site for complementarity to the anticodon triplet. The GTPase activating protein (GAP) eIF5 stimulates Guanosine-5'-triphosphate (GTP) hydrolysis by the TC, but completion of the reaction with release of Pi is blocked by eIF1 in the scanning preinitiation complex (PIC). Base-pairing of Met-tRNAi with an AUG triplet evokes a rearrangement of factors in the PIC, including displacement of eIF1 and the C-terminal tail (CTT) of eIF1A from their locations near the P site, and movement of the eIF1A CTT toward the GAP domain of eIF5. These rearrangements enable dissociation of eIF1 from the 40S subunit, evoking a closed, scanning-arrested conformation of the 40S subunit, Pi release from eIF2-GDP, and tighter binding of Met-tRNAi in the P site (PIN state) (reviewed in (2,3)).
Figure 1.
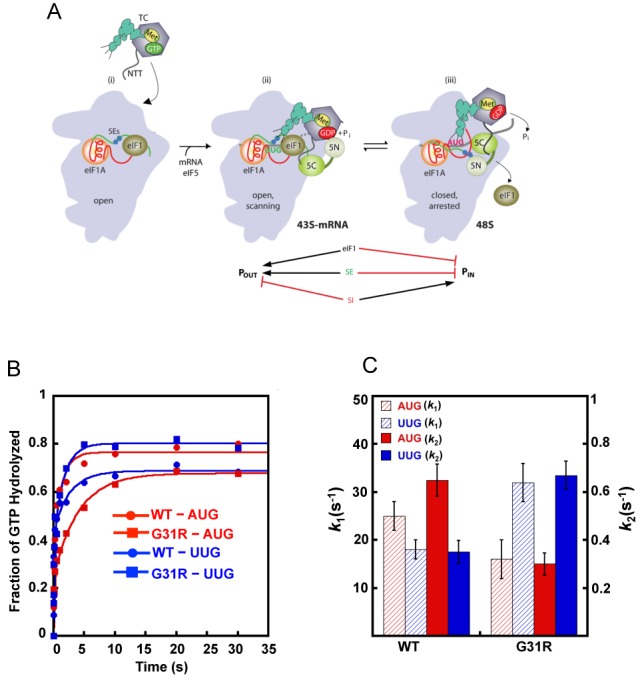
(A) Model describing conformational rearrangements of the PIC during scanning and start codon recognition. (i) eIF1 and the SE elements in the eIF1A CTT stabilize an open 40S conformation to which TC loads rapidly. (ii) The 43S PIC in the open conformation scans for an AUG codon with Met-tRNAi in the POUT state. The GAP domain in the N-terminal domain of eIF5 (5N) stimulates GTP hydrolysis to produce GDP·Pi, but Pi release is blocked. The unstructured NTT of eIF2β interacts with eIF1 to stabilize eIF1·40S association. (iii) On AUG recognition, Met-tRNAi moves from the POUT to PIN state, clashing with eIF1. Movement of eIF1 disrupts its interaction with the eIF2β-NTT, which interacts with the eIF5-CTD instead. eIF1 dissociates from the 40S subunit, and the eIF1A SE elements interact with the eIF5-NTD to facilitate Pi release. (Below) Arrows summarize that eIF1 and eIF1A SE elements promote POUT and block transition to the PIN state, whereas the scanning inhibitor (SI) element in the NTT of eIF1A stabilizes the PIN state. (Adapted from (3,29)). (B) eIF5 Sui− substitution G31R alters start codon regulation of GTP hydrolysis and Pi release from reconstituted 43S·mRNA PICs. The kinetics of GTP hydrolysis and Pi release from 43S PICs was measured after addition of WT eIF5 or G31R eIF5 and mRNAs (AUG or UUG). Aliquots from the reactions were quenched at different times with 100 mM EDTA. γ-32P-GTP and γ-32Pi were then separated using PEI-cellulose TLC and quantified by phosphorimager analysis. The fraction of GTP hydrolyzed versus time was plotted and the data fit with a double exponential rate equation. The fast phase corresponds to GTP hydrolysis and the slower phase to Pi release (10). The curves shown are WT eIF5 and AUG mRNA (red circles); WT eIF5 and UUG mRNA (blue circles); G31R eIF5 and AUG mRNA (red squares); or G31R eIF5 and UUG mRNA (blue squares). (C) Histograms showing the observed rate constants for GTP hydrolysis (k1; left Y-axis) and Pi release (k2; right Y-axis) from (B).
The crystal structure of a 40S·eIF1 complex predicts a clash between eIF1 and Met-tRNAi bound to the P site in the classical P/P state (4). Moreover, a comparison of recent structures of mammalian 40S·eIF1, 40S·eIF1·eIF1A and 40S·eIF1A·mRNA·tRNAi PICs predicts a re-orientation of Met-tRNAi from a position tilted toward the E-site (likened to POUT) in complexes containing eIF1 to a state closer to the canonical P site location, dubbed P/I and likened to PIN, in PICs lacking eIF1; and also that the P/I location clashes with eIF1 (5). These findings support the notion that stable base-pairing of Met-tRNAi with AUG in the P site will displace eIF1 and thereby trigger movement of the eIF5 NTD toward the eIF1A CTT, Pi release from eIF2-GDP-Pi, and rearrangement of the PIC to the closed, scanning-arrested conformation (3) (Figure 1A).
The GAP function of eIF5 is lodged in its N-terminal domain (NTD) (6), and requires Arg-15 in the unstructured N-terminal tail (NTT) (7) to accelerate GTP hydrolysis but not for binding eIF2 (8–10). There is evidence that Arg-15 acts as an ‘Arg finger’ that inserts into the eIF2γ GTP binding pocket to stabilize the transition state for GTP hydrolysis (9); however, GAP function is also highly dependent on PIC assembly (8–10). Thus, whereas yeast eIF5 increases the rate of GTP hydrolysis by free TC by ∼103-fold, the stimulation is ∼106-fold in 43S PICs reconstituted with eIFs -1 and -1A, TC and eIF5 (10). It is not understood how the PIC contributes to eIF5 GAP function.
A previous study indicated that regulating the GAP activity of eIF5 has an important role in accurate selection of AUG start codons, based on analysis of the SUI5 mutation in eIF5, which substitutes Arg for Gly-31 (G31R) in the β1–β2 loop in the structured portion of the NTD. SUI5/G31R elevates the ratio of translation initiation at a UUG versus AUG start codon, conferring the Sui− phenotype in vivo. The Sui− phenotype is scored in strains harboring the his4-301 allele, which in lacking an ATG start codon cannot support growth on medium lacking histidine. Introducing the TIF5-G31R on a plasmid was shown to restore growth on −His medium (dominant His+ phenotype) by increasing initiation at the third, UUG codon present in his4-301 mRNA. It was also reported that the G31R substitution increases eIF5 GAP activity in a model assay containing TC, 40S subunits and AUG triplet, leading to the proposal that G31R provokes GTP hydrolysis, with attendant release of Met-tRNAiMet from eIF2-GDP into the P-site inappropriately at UUG codons (11). It is now understood that Pi release from eIF2·GDP·Pi, rather than GTP hydrolysis, is the reaction most highly stimulated by AUG recognition (10). Accordingly, it was important to reinvestigate the effect of G31R on the rate of Pi release in PICs reconstituted with mRNAs harboring AUG or UUG start codons. As described below, we found that eIF5-G31R dramatically alters the regulation of Pi release by the start codon, accelerating the reaction at UUG codons while suppressing it at AUG start codons.
eIF1A dissociation kinetics is a sensitive probe of the partitioning of PICs between the open and closed conformations, as eIF1A dissociates from PICs more slowly with AUG versus UUG (or other near-cognate triplets) in the P site. We showed previously that eIF5-G31R reverses the opposing effects of AUG and UUG on the eIF1A dissociation rate, and thus appears to enhance closed complex formation at UUG while disfavoring the closed state at AUG (12). Thus, in addition to affecting Pi release, G31R alters regulation of the conformational rearrangement between open and closed states to favor the later at UUG codons. We sought to determine whether eIF5's distinct functions in regulating Pi release and PIC conformation are critically involved in promoting stringent AUG selection in living cells. To this end, we set out to identify intragenic suppressors of the recessive lethality or dominant Sui− phenotype of the G31R mutation, and to characterize their effects on the aberrant activities of the G31R mutant in regulating Pi release and rearrangement to the closed PIC conformation in vitro. In this way, we obtained evidence that both aspects of eIF5 function are crucial to achieve both high efficiency and accuracy of initiation in vivo.
MATERIALS AND METHODS
Plasmids construction
Plasmids used in this study are listed in Table 1. YCpTIF5 (TIF5-FL, LEU2) (13) encoding eIF5 tagged with FLAG epitope at the C-terminus, was modified by replacing the unique Nde I site (CATATG) with (AAGATG), yielding plasmid pAS5-101. TIF5 mutant alleles were constructed by fusion polymerase chain reaction (PCR) using pAS5-101 as template. The fusion PCR products were inserted between the EcoR I and Sal I sites of YCplac111 (sc) or YEplac181 (hc), and the subcloned fragments of all mutant constructs were confirmed by DNA sequencing. TIF11 mutant alleles were constructed by fusion PCR using p3390, containing WT TIF11, as template, as described previously (14). The fusion PCR products were inserted between the EcoR I and Sal I sites of YCplac111 (sc) YEplac181 (hc) or YCplac22, and the subcloned fragments of all mutant constructs were confirmed by DNA sequencing. Plasmids for in vitro expression of eIF5 variants were generated by mutagenesis of pTYB2 eIF5 using the Quik Change II site-directed mutagenesis kit (Agilent Technologies).
Table 1. Plasmids used in this study.
| Plasmid | Description | Source |
|---|---|---|
| YCplac111 | sc LEU2 cloning vector | (30) |
| YCplac22 | sc TRP cloning vector | (30) |
| YEplac181 | hc LEU2 cloning vector | (30) |
| YEplac112 | hc TRP1 cloning vector | (30) |
| pAS5-101 | sc LEU2 TIF5-FL in YCplac111 | This study |
| pAS5-103 | sc LEU2 TIF5-G62S-FL in YCplac111 | This study |
| pAS5-106 | sc LEU2 TIF5-M18V-FL in YCplac111 | This study |
| pAS5-107 | sc LEU2 TIF5-L61A-FL in YCplac111 | This study |
| pAS5-108 | sc LEU2 TIF5-K33E-FL in YCplac111 | This study |
| pAS5-111 | sc LEU2 TIF5-G31R-FL in YCplac111 | This study |
| pAS5-112 | sc LEU2 TIF5-G31R,G62S-FL in YCplac111 | This study |
| pAS5-115 | sc LEU2 TIF5-G31R,M18V-FL in YCplac111 | This study |
| pAS5-116 | sc LEU2 TIF5-G31R,L61A-FL in YCplac111 | This study |
| pAS5-117 | sc LEU2 TIF5-G31R,K33E-FL in YCplac111 | This study |
| pAS5-132 | hc LEU2 TIF5-FL in YEplac181 | This study |
| pAS5-134 | sc TRP1 TIF5-FL in YCplac22 | This study |
| pAS5-135 | hc LEU2 TIF5-T34N-FL in YEplac181 | This study |
| pAS5-136 | hc LEU2 TIF5-G31R,T34N-FL in YCplac181 | This study |
| pAS5-130 | sc TRP1 TIF11-FGFESDE121–127AAAAAAA,FEFGN131–135FAAAA in YCplac22 | This study |
| pAS5-142 | sc TRP1 TIF11 in YCplac22 | This study |
| YCpSUI3-2 | sc TRP1 SUI3-S264Y in YCplac22 | (31) |
| p367 | sc URA3 HIS4(ATG)-lacZ | (32) |
| p391 | sc URA3 HIS4(TTG)-lacZ | (32) |
| pPMB21 | sc TRP1 SUI1 in YCplac22 | (23) |
| pJCB39 | sc TRP1 SUI1-ISQLG93-97ASQAA in YCplac22 | (26) |
| p3342 | sc URA3 TIF5 in YCplac33 | (13) |
Yeast strain construction
Yeast strains used in this study are listed in Table 2. The tif5Δ his4-301 strain ASY100 was constructed as follows. A 1.92 kbp DNA fragment containing a portion of the tif5Δ::kanMX4 allele was amplified by PCR from genomic DNA of a yeast heterozygous tif5Δ::kanMX4/TIF5 diploid strain obtained from Open Biosystems (#YSC1021-672509) using primers F6 (5′-CATCGCGAAGTATGAACGAAAAAAA-3′) and F7 (5′-TACGCATGCGATCGTGGAGCCAAAT-3′). The PCR product was used to transform a derivative of strain H2994 carrying plasmid p3342 (TIF5, URA3) to G418-resistance, replacing chromosomal TIF5 with tif5Δ::kanMX4, generating strain ASY100. The gene replacement was verified by PCR analysis of chromosomal DNA using the appropriate primers. ASY137 was derived from ASY100 by replacing p3342 with TIF5+ TRP1 plasmid pAS5-134 by counter-selection on 5-FOA medium. Strain PMY17, in which TIF5 is under control of the GAL1 promoter, was generated from strain H3582 by the one-step PCR strategy using the kanMX4:PGAL1 cassette (15), selecting for resistance to kanamycin on rich medium containing galactose as carbon source. Integration of the kanMX4:PGAL1 cassette at the correct chromosomal location was verified by PCR analysis of genomic DNA using the appropriate primers.
Table 2. Yeast strains used in this study.
| Strain | Description | Source |
|---|---|---|
| H2994 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) | (31) |
| ASY100 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 p3342 [TIF5, URA3] | This study |
| ASY101 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-101 [TIF5-FL, LEU2] | This study |
| ASY103 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-103 [TIF5-G62S-FL, LEU2] | This study |
| ASY105 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-135 [TIF5-T34N-FL, LEU2] | This study |
| ASY109 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-107 [TIF5-L61A-FL, LEU2] | This study |
| ASY110 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-108 [TIF5-K33E-FL, LEU2] | This study |
| ASY112 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-106 [TIF5-M18V-FL, LEU2] | This study |
| ASY121 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-112 [TIF5-G31R,G62S-FL, LEU2] | This study |
| ASY123 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-136 [TIF5-G31R,T34N-FL, LEU2] | This study |
| ASY124 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-116 [TIF5-G31R,L61A-FL, LEU2] | This study |
| ASY125 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-117 [TIF5-G31R,K33E-FL, LEU2] | This study |
| ASY137 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif5Δ::kanMX4 pAS5-134 [TIF5-FL, TRP1] | This study |
| PMY01 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) sui1Δ::hisG p1200 [SUI1, URA3] PGAL1-TIF5::kanMX6 | (23) |
| ASY230 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) sui1Δ::hisG pJCB39 [SUI1-93-97, TRP1] PGAL1-TIF5::kanMX6 | This study |
| ASY250 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) sui1Δ::hisG pPMB21 [SUI1, TRP1] PGAL1-TIF5::kanMX6 | This study |
| H3582 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif11Δ p3392 [TIF11, URA3] | (33) |
| PMY17 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif11Δ p3392 [TIF11, URA3] PGAL1-TIF5::kanMX6 | This study |
| ASY237 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif11Δ pAS5-142 [TIF11, TRP1] PGAL1-TIF5::kanMX6 | This study |
| ASY238 | MATa ura3-52 trp1Δ63 leu2-3, leu2-112 his4-301(ACG) tif11Δ pAS5-130 [TIF11-FGFESDE121-127AAAAAAA,FEFGN131-135FAAAA, TRP1] PGAL1-TIF5::kanMX6 | This study |
Biochemical assays with yeast extracts
Assays of β-galactosidase activity in whole cell extracts (WCEs) were performed as described previously (16). For western analysis, WCEs were prepared by trichloroacetic acid extraction as described previously (17), and immunoblot analysis was conducted as described (14) using antibodies against FLAG epitope to detect eIF5-FL proteins or against eIF2Bϵ/Gcd6.
Biochemical assays in the reconstituted yeast system
Buffers and reagents
The reconstitution (‘recon’) buffer was composed of 30 mM HEPES-KOH (pH 7.4), 100 mM KOAc (pH 7.4), 3 mM Mg(OAc)2 and 2 mM DTT. The enzyme storage buffer was composed of 20 mM HEPES-KOH (pH 7.4), 2 mM DTT and 10% glycerol. Purification of all components was performed as described previously (18). The model mRNAs used were of the sequence GGAA(UC)7UNNN(CU)10C, where NNN was either AUG or UUG, (referred to as mRNA(AUG) and mRNA(UUG), respectively).
Purification of eIF5 mutants
The eIF5 mutants were expressed in BL21(DE3) Codon Plus cells (Agilent Technologies). WT eIF5 and its mutants were then purified using the IMPACT system (New England Biolabs) as described previously (18).
Fluorescent labeling of WT eIF1A
WT eIF1A was labeled at its C-terminus with Cys-Lys-ϵ-fluorescein dipeptide, using the expressed protein ligation system as previously described (19).
GTP hydrolysis and Pi release kinetics
The kinetics of GTP hydrolysis and phosphate release from 43S complexes in response to start codon recognition was measured using a rapid quench device (Kintek) as described previously (20). TC was formed at 4X concentration: 3.2 μM eIF2, 3.2 μM Met-tRNAi and 250 pM γ[32P]GTP were incubated in 1× recon buffer for 15 min at 26°C. Ribosomal complex was also made at 4× concentration in 1X recon buffer using 800 nM 40S subunits, 3.2 μM eIF1 and 3.2 μM eIF1A. Equal volumes of TC and ribosomal complex were mixed with 2 μM of eIF5 variant, and 20 μM mRNA (AUG or UUG) in a rapid quench. Reactions were quenched at different times with 100 mM ethylenediaminetetraacetic acid (EDTA). The samples were than run on PEI-Cellulose TLC plates using 0.4 M KPO4 buffer, pH 4.0 as the mobile phase, followed by PhosphorImager analysis to quantify the fraction of GTP hydrolyzed over time. The data were fit with a double exponential rate equation. The first phase corresponds to GTP hydrolysis and the second phase corresponds to Pi release, which drives GTP hydrolysis forward (10). All experiments were repeated 2–5 times.
GTP hydrolysis by isolated TC in presence of the eIF5 variants was measured as follows. TC was formed with 800 nM of eIF2, 800 nM Met-tRNAi and 250 pM γ-32P-GTP for 15 min at 26°C. Reactions were initiated by adding the TC to an equal volume of 2 μM of the eIF5 variant and then 2 μl aliquots were removed at various times and stopped by the addition of 20 μl of 100 mM EDTA. The samples were then run on PEI-Cellulose TLC plates using 0.4 M KPO4 buffer, pH 4.0 as the mobile phase, followed by PhosphorImager analysis to quantify the fraction of Pi formed over time. The resulting data were fit with a straight line.
Kinetics of eIF1A dissociation
Kinetic experiments measuring the dissociation of WT eIF1A from 43S complexes were carried out as described previously (12) in reactions with the following concentrations: 15 nM eIF1A-Fl, 1 μM eIF1, 1 μM eIF5 variant, 120 nM 40S, 10 μM mRNA (AUG or UUG), 300 nM eIF2, 150 nM Met-tRNAi and 1 mM GDPNP·Mg2+. Buffer conditions were 30 mM HEPES (pH 7.4), 100 mM potassium acetate, 3 mM Mg(OAc)2and 2 mM dithiothreitol. The fluorophore was excited at 497 nm and fluorescence anisotropy was monitored at 520 nm for up to 3 h, as necessary. Reactions containing fluorescein-labeled WT eIF1A were incubated for 30 min after the addition of TC. Dissociation of eIF1A-Fl was initiated by addition of 15 μM unlabeled eIF1A as a chase. The reaction was carried out on a Fluorolog-3 spectrofluorometer (JobinYvon Horiba). Data were fit using single or double exponential rate equations, as appropriate. All kinetic experiments were repeated 3 times.
RESULTS
The SUI5 mutation in eIF5 alters regulation of GTP hydrolysis and gated Pi release
Because eIF5 is a GAP for eIF2 (8–10), we sought to determine the effect of the eIF5 SUI5/G31R mutation on GTP hydrolysis and subsequent Pi release from eIF2 in the PIC. Our previous results indicated that Pi release is strongly regulated by recognition of the start codon in the mRNA, whereas GTP hydrolysis itself can occur rapidly prior to start codon recognition (10). We assayed eIF5-stimulated GTP hydrolysis and Pi release in reconstituted 43S·mRNA PICs containing 40S subunits, eIF1, eIF1A, TC assembled with [γ-32P]-GTP, eIF5 and a model mRNA containing an AUG or UUG start codon. The PICs were pre-assembled without eIF5 and mRNA, saturating amounts of eIF5 (WT or G31R mutant) and mRNA were added to initiate the reaction, and aliquots were sampled and quenched using a rapid-quench device. Pi formation was monitored by thin-layer chromatography followed by phosphorimaging. When eIF5 binds to the PIC, a fraction of the GTP is rapidly hydrolyzed to GDP·Pi and the Pi from this reaction is observed following the denaturing quench. Upon AUG recognition, eIF1 dissociates from the PIC, triggering Pi release from eIF2, which drives the GTP hydrolysis reaction to completion, producing a second, slower phase with a rate constant of ∼0.6 s−1 (10). This slower, second phase of the reaction corresponds to AUG-dependent Pi release from the PIC. Note that in this experimental set-up GTP hydrolysis and Pi release occur concurrently because eIF5 and mRNA are added together, and the reaction has biphasic kinetics with the rate constants for the fast and slow phases, k1 and k2, corresponding to GTP hydrolysis and Pi release, respectively. By contrast, when eIF5 is added in the absence of a model mRNA containing a start codon, Pi release is dramatically reduced (10).
In accordance with previous findings (10), rapid binding to PICs of a model mRNA with an AUG start codon (mRNA(AUG)) resulted in an observed rate constant for Pi release of ∼0.70 s−1, which was reduced by a factor of ∼2 for complexes assembled with mRNA(UUG) instead (Figures 1B and C and Table 3, WT AUG versus UUG, k2 values). The rate constant for GTP hydrolysis is also somewhat lower for UUG versus AUG complexes (Figures 1B-C & Table 3, WT AUG versus UUG, k1 values). Relative to WT eIF5, the G31R mutant reduced the rate of Pi release in response to mRNA(AUG) by ∼2-fold but increased it by a similar factor in response to mRNA(UUG) (Figures 1B-C and Table 3; WT versus G31R, k2 values). The rate of GTP hydrolysis is also reduced to 1.5-fold and increased nearly 2-fold by the G31R mutation with mRNA(AUG) and mRNA(UUG), respectively (Figures 1B-C and Table 3, WT versus G31R, k1 values). Thus, G31R provokes a switch in the differential between AUG and UUG in the rates of both GTP hydrolysis and Pi release relative to those seen with WT eIF5, which is consistent with the elevated UUG:AUG initiation ratio (Sui− phenotype) of this mutant in vivo. Because Pi release is the rate-limiting step of conversion of eIF2 from its GTP- to GDP-bound states, the differential effects of G31R on the rate contants of Pi release (k2) at UUG versus AUG likely has the greater impact on the fidelity of start codon selection in cells.
Table 3. Kinetic parameters for GTP hydrolysis (k1) and Pi release (k2) from 43S·mRNA complexes with eIF5 mutants.
| AUG | UUG | |||
|---|---|---|---|---|
| eIF5 variant | kobs (s−1) | Amplitude | kobs (s−1) | Amplitude |
| 1. WT | k1 = 25 ± 3.0 | a1 = 0.55 ± 0.02 | k1 = 18.0 ± 2.0 | a1 = 0.60 ± 0.03 |
| k2 = 0.65 ± 0.07 | a2 = 0.45 ± 0.02 | k2 = 0.38 ± 0.05 | a2 = 0.40 ± 0.03 | |
| 2. G31R | k1 = 16 ± 4.0 | a1 = 0.40 ± 0.04 | k1 = 32.0 ± 4.0 | a1 = 0.50 ± 0.02 |
| k2 = 0.32 ± 0.04 | a2 = 0.60 ± 0.04 | k2 = 0.70 ± 0.06 | a2 = 0.50 ± 0.02 | |
| 3. M18V | k1 = 0.4 ± 0.10 | a1 = 0.4 ± 0.05 | k1 = 0.6 ± 0.2 | a1 = 0.3 ± 0.01 |
| k2 = 0.06 ± 0.01 | a2 = 0.6 ± 0.05 | k2 = 0.08 ± 0.02 | a2 = 0.7 ± 0.01 | |
| 4. G31R,M18V | k1 = 0.1 ± 0.02 | a1 = 0.4 ± 0.01 | k1 = 0.13 ± 0.02 | a1 = 0.4 ± 0.01 |
| k2 = 0.028 ± 0.001 | a2 = 0.6 ±0.01 | k2 = 0.03 ± 0.01 | a2 = 0.6 ± 0.01 | |
| 5. G62S | k1 = 28 ± 2.0 | a1 = 0.50 ± 0.03 | k1 = 22 ± 2.0 | a1 = 0.50 ± 0.01 |
| k2 = 0.60 ± 0.08 | a2 = 0.50 ± 0.03 | k2 = 0.40 ± 0.04 | a2 = 0.50 ± 0.01 | |
| 6. G31R,G62S | k1 = 32 ± 4.0 | a1 = 0.35 ± 0.05 | k1 = 24 ± 2.0 | a1 = 0.30 ± 0.02 |
| k2 = 0.60 ± 0.05 | a2 = 0.65 ± 0.05 | k2 = 0.3 ± 0.02 | a2 = 0.70 ± 0.02 | |
· All errors are SE calculated by SD/√n; n = number of experiments.
Genetic characterization of intragenic suppressors of the eIF5-G31R (SUI5) substitution
We reasoned that one way to establish the physiological relevance of the effect of eIF5-G31R on gated Pi release would be to identify intragenic suppressors of the Sui− phenotype of the G31R mutation and determine if they also diminish the effect of G31R on Pi release in vitro. As described above, Sui− mutations can be identified by their ability to restore growth of his4-301 cells on media lacking histidine by increasing initiation at the third, UUG codon in his4-301 mRNA, which lacks an AUG start codon (21). Introducing TIF5-G31R on a plasmid restores histidine prototrophy in his4-301 cells containing WT TIF5 in the chromosome, indicating dominant His+/Sui− phenotypes; however, TIF5-G31R cannot support cell viability as the only source of eIF5 (11). We sought to identify eIF5 intragenic suppressor mutations that would overcome the dominant Sui− phenotype of TIF5-G31R (referred to below as just G31R). To this end, we combined G31R with each of three eIF5 mutations, M18V, T34N and G62S, originally identified by Donahue et al. by their ability to suppress the slow-growth (Slg−) phenotypes conferred by known Sui− mutations in eIF1 (for G62S and M18V) or eIF2β (for T34N) (T. Donahue, unpublished observations). It was shown subsequently that G62S suppresses the His+ phenotype of the Sui− SUI3-2 mutation in eIF2β, consistent with an Ssu− (suppressor of Sui−) hyperaccuracy phenotype (22). The M18V substitution maps in the unstructured NTT of eIF5 only two residues from the key GAP residue Arg-15 (7), whereas T34N and G62S map in the globular portion of the eIF5 NTD. We also combined G31R with the eIF5 mutations, K33E and L61A, as it was proposed that K33 functions as the secondary, stabilizing basic residue described in other GAPs (7) and is adjacent to the residue altered by T34N; and L61 is a highly conserved residue adjacent to the residue altered by suppressor G62S. As described below, the eIF5 mutations we examined fell into three genetic categories: (i) suppression of the His+/Sui− phenotype but not the lethality of G31R, in the case of M18V; (ii) suppression of both the His+/Sui− phenotype and lethality of G31R, for G62S and K33E and (iii) suppression of lethality but not the His+/Sui− phenotype of G31R, for L61A and T34N. These genetic results provided the first indication that distinct biochemical mechanisms underlie the Ssu− phenotypes of different G31R suppressors.
Each of the mutations mentioned above was combined with G31R in a TIF5 allele that also contains the coding sequences for a C-terminal FLAG epitope. The resulting double-mutant alleles were introduced on LEU2 plasmids and transformed into a his4-301 tif5Δ ura3 strain harboring untagged TIF5+ on a URA3 plasmid. We first established that the expression level of all mutant proteins was comparable to FLAG-tagged WT eIF5 by western analysis using FLAG antibody of WCEs prepared from strains either before or after eviction of the TIF5+URA3 plasmid (Supplementary Figure S1). We then evaluated suppression of the dominant His+/Sui− phenotype of G31R by examining growth of the double mutants on −His medium containing only 0.1% of the histidine supplement normally provided in +His medium. Suppression of the lethality of G31R was scored by the ability of the strain to survive eviction of the TIF5+URA3 plasmid by counter-selection on (+His) medium containing 5-fluoro-orotic acid (5-FOA).
M18V strongly suppresses the dominant His+/Sui− phenotype, but not the recessive lethality, conferred by G31R. Thus, transformants harboring tif5-G31R,M18V fail to grow on −His medium in the presence of TIF5+, indicating the Ssu− phenotype, and also cannot grow on 5-FOA medium, indicating inviability of the cells following eviction of TIF5+ (Figure 2A, rows 3–4). The failure to grow on the −His medium does not result from a general growth defect, as the tif5-G31R,M18V strain grows better on +His medium than does the parental His+ tif5-G31R strain, which displays the dominant Slg− phenotype conferred by G31R (Figure 2B, rows 3–4 versus row 2).
Figure 2.
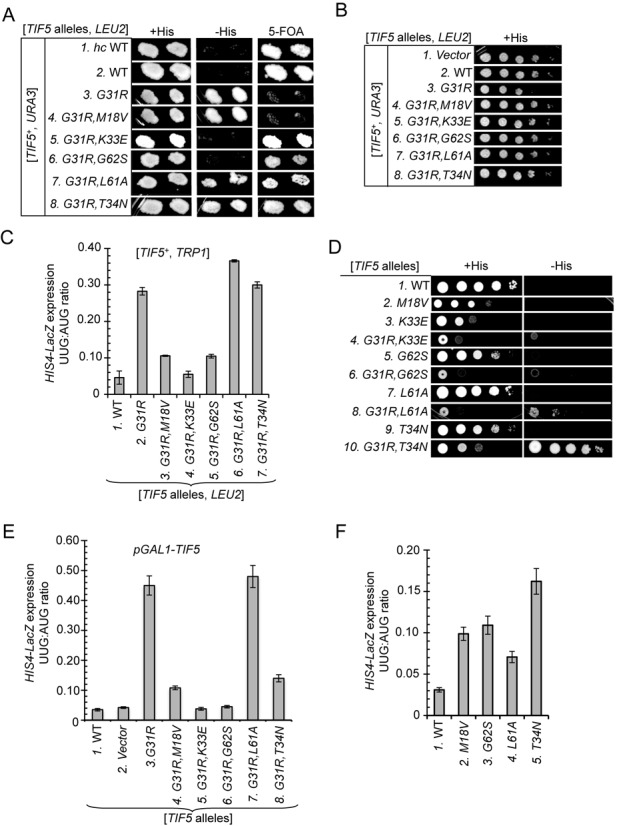
Genetic characterization of intragenic suppressors of TIF5 allele SUI5/G31R. (A) Derivatives of his4-301 tif5Δ strain ASY100 harboring a TIF5+URA3 plasmid and the indicated TIF5-FL alleles on LEU2 plasmids (hc WT on pAS5-132, sc WT on pAS5-101, G31R on pAS5-111, G31R,M18V on pAS5-115, G31R,K33E on pAS5-117, G31R,G62S on pAS5-112, G31R,L61A on pAS5-116 and hc G31R,T34N on pAS5-136) were replica-plated to SC-LU supplemented with either 0.3 mM histidine (-LU) or 0.0003 mM histidine (-LUH), or to SC-L supplemented with 5.2 mM 5-FOA (5-FOA). Cells were incubated for 3d (-LU) or 6d (-LUH and 5-FOA) at 30°C. (B) 10-fold serial dilutions of strains described in (A) were spotted on SC-LU and incubated for 3d at 30°C. (C) Derivatives of tif5Δ strain ASY137 harboring a TIF5+TRP1 plasmid and the indicated TIF5 alleles on LEU2 plasmids were transformed with HIS4-lacZ reporter plasmids with AUG (p367) or UUG (p391) start codons. Cells were cultured in SC lacking leucine, tryptophan and uracil at 30°C and β-galactosidase activities were measured in whole cell extracts (WCEs). Ratios of β-galactosidase expressed from the UUG to AUG reporter were calculated from three independent transformants and mean ratios and and S.E.M.s (error bars) were plotted. (D) Slg− and His+/Sui− phenotypes of the his4-301 tif5Δ strains harboring TIF5 alleles described in (A), and isogenic strains containing M18V on pAS5-106, K33E on pAS5-108, G62S on pAS5-103, L61A on pAS5-107 or hc T34N on pAS5-135, were determined by spotting serial 10-fold dilutions on SC-LU supplemented with 0.3 mM His (+His) or 0.0003 mM His (−His) and incubated for 3d (−His) or 6d (+His) at 30°C. (E) Derivatives of his4-301 sui1Δ strain ASY250 with the chromosomal TIF5 gene under the GAL1 promoter (PGAL-TIF5) harboring SUI1+ on a TRP1 plasmid and the indicated TIF5 alleles on LEU2 plasmids were transformed with the AUG or UUG HIS4-lacZ reporter plasmids. Transformants were cultured in synthetic minimal medium with 2% galactose as carbon source and supplemented with 0.3 mM histidine (SGal+H) and then shifted to synthetic minimal medium with 2% glucose (SD+H) for 16 h. UUG:AUG initiation ratios for the HIS4-lacZ reporters were determined as in (C). (F) HIS4-lacZ UUG:AUG initiation ratios were measured as in (C) for strains described in (D) harboring the indicated plasmid-borne TIF5 alleles. For panels A and D, images have been cropped from results obtained from different plates examined in parallel in the same experiments.
To confirm the Ssu− phenotype of M18V in the G31R,M18V strain, we assayed matched HIS4-lacZ fusions with AUG or UUG start codons in tif5Δ strains harboring TIF5-G31R or TIF5-G31R,M18V on a LEU2 plasmid and also a TRP1 TIF5+ plasmid (provided to rescue growth of the G31R single mutant). As expected, the G31R strain displays an elevated UUG:AUG initiation ratio for these reporters, of ∼0.28, well above that seen in the strain containing only TIF5+ (∼0.05). Importantly, the UUG:AUG ratio drops to ∼0.11 in the G31R,M18V strain (Figure 2C, columns 1–3), indicating that M18V reduces the hypoaccuracy phenotype in the manner expected of an Ssu− mutation, even though it does not overcome the lethality of G31R.
In contrast to the G31R,M18V mutant, the two mutants in which G31R is combined with G62S or K33E can grow on 5-FOA medium (Figure 2A, rows 5 and 6), indicating suppression of the recessive lethality of G31R. As might be expected, G62S and K33E also suppress the dominant Slg− phenotype of G31R on +His medium (Figure 2B, rows 5 and 6 versus row 3). Both double mutants also fail to grow on −His medium (Figure 2A, rows 5 and 6) and, consistent with this, display reduced UUG:AUG initiation ratios compared to the G31R single mutant (Figure 2C, columns 2 versus 4–5). Thus, K33E and G62S suppress the Sui− phenotype of G31R, exhibiting Ssu− phenotypes, in addition to overcoming the lethality of G31R.
T34N and L61A comprise the third class of eIF5 mutations we identified, which suppress the recessive lethality but not the dominant His+/Sui− phenotype of G31R, as the double or triple mutants grow on both 5-FOA and −His medium (Figure 2A, rows 7 and 8 versus 3). Consistent with their His+ phenotypes, when examined in cells harboring TIF5+, these double mutants display no reduction in the UUG:AUG initiation ratio (Figure 2C, columns 6–7 versus 2), indicating that L61A and T34N are Ssu+ rather than Ssu− eIF5 substitutions.
With the exception of the lethal G31R and G31R,M18V alleles, we also characterized the phenotypes of the mutant TIF5 alleles after evicting TIF5+ by 5-FOA selection. These strains showed much the same behavior on −His medium observed prior to TIF5+ eviction described above. Thus, the double mutations combining G31R with T34N or L61A allow growth on –His medium (Figure 2D, rows 1 versus 8 and 10, −His data) indicating a failure to suppress the Sui− phenotype of G31R, even though they have slow-growth (Slg−) phenotypes on +His medium (Figure 2D, rows 1 versus 8 and 10, +His data) that should limit their ability to grow on the −His medium. The Slg− phenotypes of the G31R,T34N and G31R,L61A double mutants are more severe than observed in the corresponding strains harboring the suppressor mutations on their own without G31R (Figure 2D, cf. 7–8 and 9–10, +His), indicating that T34N and L61A do not overcome the growth defects conferred by G31R even though they suppress its lethality.
Following eviction of TIF5+, the two viable double mutants in which G31R is combined with G62S or K33E show little or no growth on −His medium (Figure 2D, rows 4 and 6, −His), consistent with the Ssu− phenotypes observed in the strains harboring TIF5+ (Figure 2A). To confirm this conclusion, we measured the UUG:AUG initiation ratios conferred by the TIF5 suppressor alleles in a strain containing chromosomal TIF5 under the GAL1 promoter (PGAL-TIF5), which expresses WT eIF5 only on galactose medium. The HIS4-lacZ reporters were assayed after incubating cells for 16 h in glucose medium, which is sufficient to deplete the WT eIF5 encoded by PGAL-TIF5 by ∼75% (23). This approach allowed us to compare the UUG:AUG initiation ratios conferred by the G31R and G31R,M18V mutations, which are lethal in the absence of WT eIF5. As shown in Figure 2E, the G31R single mutation confers a UUG:AUG ratio of ∼0.45, considerably larger than it produces in a strain also containing TIF5+ (0.28, Figure 2C), indicating that the Sui− phenotype of G31R is dampened by WT eIF5. As shown above, the M18V substitution greatly reduces, but does not abolish, the elevated UUG:AUG ratio produced by G31R in the G31R,M18V double mutant, which remains ∼2-fold higher than the ratio measured in WT cells (Figure 2E, columns 1, 3–4). The G62S and K33E also strongly suppress the Sui− phenotype of G31R, reducing the UUG:AUG ratio in the relevant double mutants to essentially the WT level (Figure 2E, cf. columns 1 and 5–6). By contrast, L61A does not reduce the UUG:AUG ratio at all in the G31R,L61A double mutant (Figure 2E, column 7 versus 3), in agreement with the results above in cells co-expressing WT eIF5 at native levels (Figure 2C, column 7 versus 2). However, T34N produces a reduction in the UUG:AUG ratio only slightly less than that observed for M18V in the relevant double mutants harboring G31R (Figure 2E, columns 3, 4 and 8). This last result differs from that observed in cells expressing WT eIF5, in which T34N conferred no reduction in UUG:AUG ratio in the G31R,T34N double mutant (Figure 2C, columns 2 and 7). This discrepancy seems to indicate that the ability of T34N to overcome the hypoaccuracy phenotype of G31R in the G31R,T34N double mutant is masked by the presence of WT eIF5.
Because all of the suppressors except L61A conferred a Slg− phenotype when examined as single eIF5 substitutions in the absence of G31R (Figure 2D, rows 2, 3, 5 and 9), we wondered if they might affect AUG selection on their own. In fact, M18V, L61A, G62S and T34N all confer significant increases in the UUG:AUG initiation ratio (Figure 2F). The fact that these strains do not display a His+ phenotype (Figure 2D, rows 2, 5, 7 and 9, −His) probably reflects a combination of their Slg− phenotypes (evident on +His medium) and the relatively moderate increases in UUG initiation they confer (Figure 2F), but it might also involve a defect in another function required for growth under conditions of histidine limitation. Indeed, we found that M18V and T34N diminish the translational induction of the GCN4 gene (data not shown), whose product mediates increased transcription of HIS4 and contributes to the His+ phenotype of many Sui− mutants. In any event, it is interesting that the M18V and G62S substitutions confer moderate Sui− phenotypes on their own (Figure 2F) even though they diminish the Sui− phenotype of the G31R mutation in the corresponding eIF5 double mutants (Figure 2C and E). This finding suggests that the effects of M18V and G62S on eIF5 function are influenced by the presence of the G31R substitution in the same polypeptide.
Distinct effects of G31R suppressors M18V and G62S on the kinetics of GTP hydrolysis and Pi release
The results above indicate that the M18V substitution in eIF5 confers nearly complete suppression of the Sui− phenotype of G31R, without overcoming the lethality of G31R in cells lacking WT eIF5. Given its location only three residues from a key catalytic residue (Arg-15) in the unstructured NTT, we hypothesized that M18V impairs GAP function in the G31R,M18V double mutant. To test this possibility, we assayed eIF5 stimulation of GTP hydrolysis and Pi release from eIF2 in reconstituted PICs. As described above in Figures 1B and C, the G31R single substitution reduces the rates of GTP hydrolysis and Pi release at AUG while increasing them at UUG, reversing the differential effects of AUG and UUG from those seen with WT eIF5 (Figures 3A and B WT versus G31R and Table 3, rows 1–2). In the G31R,M18V double mutant, M18V reduces the rates of Pi release at both AUG and UUG compared to those seen in the G31R single mutant (Figures 3A and B G31R,M18V versus G31R, k2 values), yielding rate constants at AUG and UUG that are 10- and 20-fold, respectively, below those observed with G31R eIF5 (Table 3, row 4 versus 2; k2 values). M18V in the double mutant also dramatically reduces the rates of GTP hydrolysis at AUG and UUG compared to those seen in the G31R single mutant, by factors of >150-fold (Figures 3A and B G31R,M18V versus G31R and Table 3, row 4 versus 2, k1 values), indicating a major defect in the GAP function of the double mutant. These strong defects in both GTP hydrolysis and Pi release at AUG likely account for the inability of M18V to suppress the lethality of G31R in vivo (Figure 2A). Combining M18V with G31R decreases the Pi release rates ∼2-fold more at UUG than at AUG (Figures 3A and B G31R,M18V versus G31R; Table 3, row 4 versus 2, k2 values) to produce a UUG:AUG ratio of Pi release—the rate-limiting step—close to 1 in the double mutant. Although this ratio of release rates is still above that observed with WT eIF5, the trend is consistent with suppression of the Sui− phenotype of G31R by the M18V substitution.
Figure 3.
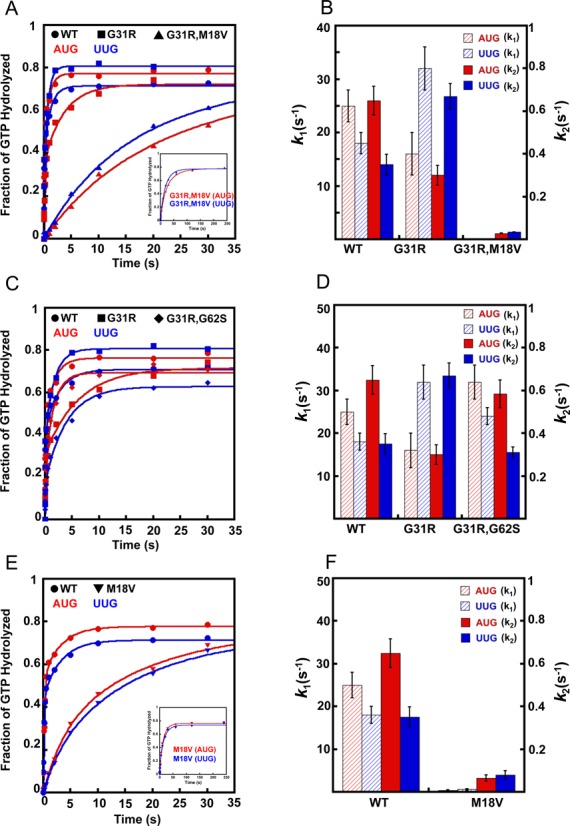
Effects of eIF5 Sui− substitution G31R and its intragenic suppressors M18V and G62S on GTP hydrolysis and Pi release from reconstituted 43S·mRNA PICs. Double mutant derivatives of Sui− G31R eIF5 (G31R,M18V and G31R,G62S) were generated by combining M18V or G62S with G31R in the same recombinant eIF5 variants. The kinetics of Pi formation from the 43S PIC with a model mRNA with an AUG or UUG start codon were measured for these mutants as described in Figure 1B. With WT eIF5, the fast phase of the reaction was shown previously to correspond to GTP hydrolysis and the slow phase to Pi release, which drives GTP hydrolysis to completion (10). (A) GTP hydrolysis and Pi release with WT (circles), G31R (squares) and G31R,M18V (triangles) variants of eIF5 with AUG (red) or UUG (blue) mRNAs. GTP hydrolysis and Pi release was monitored for 4 min in case of G31R,M18V (shown in inset). (B) Observed rate constants for GTP hydrolysis (k1; red and blue striped bars; left Y-axis) and Pi release (k2; red and blue solid bars; right Y-axis) for the cases shown in (A). (C) GTP hydrolysis and Pi release with WT (circles), G31R (squares) and G31R,G62S (diamonds) eIF5 variants with AUG (red) or UUG (blue) mRNAs. (D) Observed rate constants for GTP hydrolysis (k1) and Pi release (k2) for the cases shown in (C). (E) GTP hydrolysis and Pi release with WT (circles) or M18V (inverted triangles) eIF5 variants with AUG (red) or UUG (blue) mRNAs. GTP hydrolysis and Pi release was monitored for 4 min in case of M18V (shown in inset). (F) Observed rate constants for GTP hydrolysis (k1) and Pi release (k2) for the curves shown in (E). Data in (B), (D) and (F) are the averages of three experiments and error bars represent standard errors of the mean.
The M18V substitution on its own results in 60- and 30-fold reductions in the rate constants for GTP hydrolysis with mRNA(AUG) and mRNA(UUG), respectively (Figures 3E and F WT versus M18V; Table 3, row 3 versus 1, k1 values), consistent with the proposed defect in GAP function. The rate constants for Pi release are also reduced, by 10- and 5-fold at AUG and UUG, respectively, compared to the corresponding rates of Pi release observed for WT eIF5, which leads to nearly identical reaction rates at UUG and AUG for the M18V variant (Table 3, row 3 versus 1, k2 values). Although these differences are small, this last observation is consistent with the moderate increase in UUG:AUG initiation ratio (weak Sui− phenotype) observed for the tif5-M18V single mutant in vivo (Figure 2F).
To explore further the effect of the M18V substitution on the GAP activity of eIF5, we measured the slow rate of eIF5-dependent GTP hydrolysis by isolated eIF2·Met-tRNAi·GTP ternary complexes (10) using WT and M18V forms of eIF5. In this assay, pre-formed TCs are mixed with eIF5 to initiate GTP hydrolysis. Aliquots are removed at various times, mixed with a denaturing quench and the amount of Pi formed analyzed by thin-layer chromatography. Even in this context, in which GTP hydrolysis is not influenced by conformational changes in the PIC, the M18V substitution reduces the observed rate of GTP hydrolysis by >20-fold relative to the WT factor (Supplementary Figure S2; 0.0028 versus 0.0003 min−1 with WT and M18V, respectively), supporting the conclusion that M18V diminishes the GAP function of the factor.
The genetic analyses described above in Figure 2 indicated that the G62S substitution is also very effective in suppressing the Sui− phenotype of G31R in vivo. G62S on its own has no significant effect on the GTPase or Pi release rates at AUG versus UUG (Table 3, row 5 versus 1,) or on the rate of GTP hydrolysis by the isolated TC (Supplementary Figure S2). Importantly, however, when combined with G31R, the G62S suppressor essentially reverses the effects of G31R of elevating the rate of Pi release at UUG while depressing it at AUG, to reinstate a nearly WT differential in rates between AUG and UUG (Figures 3C and D G31R,G62S versus G31R and Table 3, row 6 versus 2, k2 values). G62S also restores a WT rate of GTP hydrolysis at AUG codons (Figures 3C and D G31R,G62S versus G31R and Table 3, row 6 versus 2, AUG, k1 values). Note that unlike M18V, which was shown above to slow GTP hydrolysis and Pi release on both AUG and UUG codons in the G31R,M18V double mutant, combining G62S with G31R actually increases the rates of these steps at AUG codons compared to those seen in the G31R single mutant (Figures 3C and D G31R,G62S versus G31R and Table 3 row 6 versus 2, AUG, k1 and k2), consistent with the viability of the G31R,G62S double mutant as the sole version of eIF5 in the cell (Figure 2A).
Although we sought to examine the effects of the eIF5-T34N suppressor on GTP hydrolysis and Pi release, repeated attempts to express and purify both this variant and the eIF5-L61A mutant proteins were unsuccessful.
Distinct effects of G31R suppressors on stabilities of open and closed conformations of the PIC
In addition to affecting the regulation of Pi release, we showed previously that the eIF5 G31R substitution also affects rearrangement of the PIC from the open, scanning conformation to the closed, scanning-arrested state (12). The rate of eIF1A dissociation from the PIC is a sensitive probe of this transition because eIF1A and eIF5 functionally interact on AUG recognition in a way that strengthens eIF1A binding to the PIC. This is manifested as an increase in amplitude or decrease in rate constant (or both) of the slower phase of the biphasic dissociation of eIF1A from the 43S·mRNA complex. The two kinetic phases of eIF1A dissociation are thought to represent the partitioning of PICs between open and closed conformations, with the fast and slow phases corresponding to the fractions of complexes in the open and closed states, respectively. It was demonstrated that eIF5-G31R reverses the differential effects of AUG versus UUG on eIF1A dissociation kinetics, indicating a shift in the balance between the two states of the PIC in favor of the closed complex at UUG codons and against it at AUG codons (12). We considered the possibility that, in addition to influencing Pi release, the G62S and M18V substitutions might reverse the effect of G31R and preferentially destabilize the closed complex at UUG codons, thus contributing to their Ssu− phenotypes. Accordingly, we examined how M18V and G62S influence the effects of G31R on eIF1A dissociation kinetics.
43S·mRNA(AUG) or 43S·mRNA(UUG) complexes were assembled with eIF1A (labeled at its C-terminus with fluorescein) in the presence of eIF5, chased with excess unlabeled eIF1A, and eIF1A dissociation was measured over time as the decrease in fluorescence anisotropy. Dissociation of WT eIF1A from the PIC is biphasic, with rate constants for the fast and slow phases designated k1 and k2, respectively. We defined an apparent equilibrium constant, Kamp, as the ratio of amplitudes of the slow to fast phases (α2/α1). Accordingly, Kamp values >1 indicate that the slow phase dominates the reaction. In accordance with previous findings (12), WT eIF1A dissociates more slowly from AUG versus UUG complexes (Figure 4A), as dissociation from the AUG complexes is dominated by the slow phase (Kamp of 6.0; Table 4, row 1), while replacing AUG with UUG reduces Kamp to 4.0 and also increases k2 by ∼3-fold (row 2). These findings suggest that only approximately one-seventh of the PICs are in the open conformation at AUG (Kamp of 6.0) whereas approximately one-fifth occupy the open conformation at UUG (Kamp of 4.0), and the ∼3-fold higher value of k2 at UUG indicates that eIF1A is bound less stably in the closed conformation at UUG versus AUG. This latter finding could reflect a decreased strength of interaction between eIF1A and eIF5, which we have shown is a key event in mediating a proper response to start codon recognition (3,12).
Figure 4.
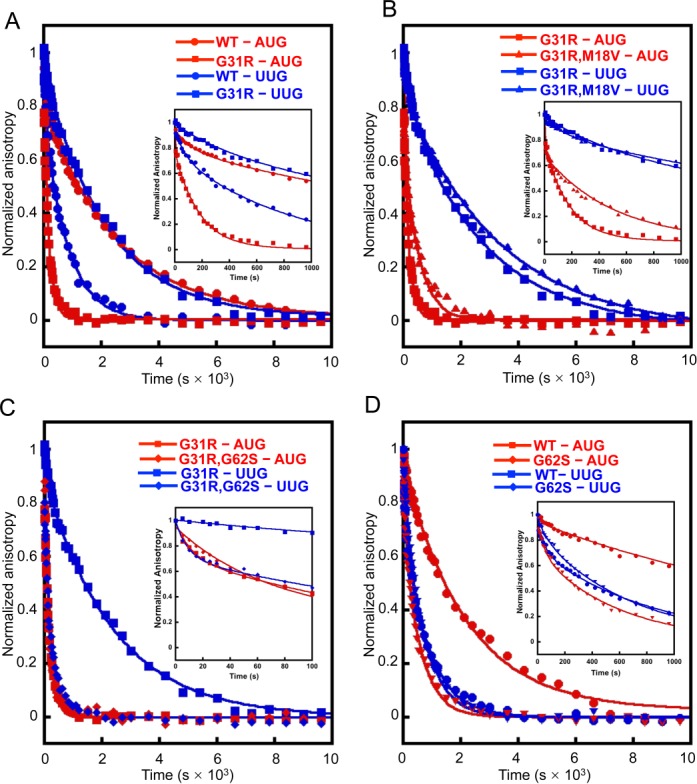
Effects of eIF5 intragenic suppressors M18V and G62S on the kinetics of eIF1A dissociation from reconstituted 43S·mRNA PICs. Dissociation of fluorescein-labeled eIF1A from 43S·mRNA complexes assembled with WT or mutant eIF5 was monitored by following the change in fluorescence anisotropy over time after the addition of a large excess of unlabeled WT eIF1A. The data were fit with a double exponential decay equation. Previous data indicate that the fast phase corresponds to dissociation of eIF1A from the ‘open’ conformation of the PIC and the second phase corresponds to dissociation from the ‘closed’ state. In order to highlight differences in the fast phases, the early time points of the respective eIF1A dissociation curves are shown in insets. The ratio of the amplitudes of the second phase (closed state) to first phase (open state) is defined as Kamp. (A) eIF1A dissociation from PICs assembled with WT (circles) and G31R (squares) eIF5 with mRNA(AUG) (red; Kamp for WT is 6.0 ± 0.7 and for G31R is 3.0 ± 0.2) and mRNA(UUG) (blue; Kamp for WT is 4.0 ± 0.2 and for G31R is 10 ± 0.6). (B) eIF1A dissociation from PICs assembled with G31R (squares) and G31R,M18V (triangles) eIF5 with mRNA(AUG) (red; Kamp is 2.0 ± 0.1 for G31R,M18V) and mRNA(UUG) (blue; Kamp is 9.0 ± 1.0 for G31R,M18V)) mRNA. (C) eIF1A dissociation from PICs assembled with G31R (squares) and G31R,G62S (diamonds) eIF5 with mRNA(AUG) (red; Kamp is 1.1 ± 0.04 for G31R,G62S) and mRNA(UUG) (blue; Kamp is 2.3 ± 0.3 for G31R,G62S). (D) eIF1A dissociation from PICs assembled with WT (circles) and G62S (diamonds) eIF5 with mRNA(AUG) (red; Kamp is 3.0 ± 0.05 for G62S) and mRNA(UUG) (blue; Kamp is 2.3 ± 0.06 for G62S). All experiments were performed at least three times and errors are standard errors of the mean.
Table 4. Rate constants for eIF1A dissociation from 43S·mRNA complexes.
| eIF5 variants | mRNA | k1 (x 10−3 s−1) | k2 (x 10−3 s−1) | α1 | α2 | Kamp (α2/ α1) | k2 (UUG)/k2 (AUG) | Kamp (UUG)/Kamp (AUG) |
|---|---|---|---|---|---|---|---|---|
| 1. WT | AUG | 7.0 ± 1.0 | 0.4 ± 0.01 | 0.14 ± 0.01 | 0.86 ± 0.04 | 6.0 ± 0.7 | 3.0 | 0.7 |
| 2. | UUG | 11 ± 3.0 | 1.1 ± 0.02 | 0.20 ± 0.01 | 0.80 ± 0.01 | 4.0 ± 0.2 | ||
| 3. G31R | AUG | 16 ± 3.0 | 5.0 ± 0.3 | 0.25 ± 0.02 | 0.80 ± 0.06 | 3.0 ± 0.2 | 0.1 | 3.3 |
| 4. | UUG | 8.0 ± 2.0 | 0.5 ± 0.06 | 0.09 ± 0.01 | 0.91 ± 0.06 | 10 ± 0.6 | ||
| 5. M18V | AUG | 3.0 ± 0.1 | 0.3 ± 0.04 | 0.25 ±0.03 | 0.75 ± 0.07 | 3.0 ± 0.2 | 4 | 0.8 |
| 6. | UUG | 11 ± 0.9 | 1.2 ± 0.03 | 0.3 ± 0.01 | 0.70 ± 0.08 | 2.4 ± 0.04 | ||
| 7. G31R,M18V | AUG | 25 ± 2.4 | 2.0 ± 0.20 | 0.30 ± 0.03 | 0.70 ± 0.10 | 2.0 ± 0.1 | 0.15 | 4.5 |
| 8. | UUG | 18 ± 1.0 | 0.3 ± 0.03 | 0.1 ± 0.01 | 0.90 ± 0.10 | 9.0 ± 1.0 | ||
| 9. G62S | AUG | 18 ± 3.5 | 3.0 ± 0.40 | 0.25 ±0.01 | 0.75 ± 0.10 | 3.0 ± 0.05 | 0.7 | 0.8 |
| 10. | UUG | 25 ± 4.0 | 2.0 ± 0.03 | 0.3 ± 0.02 | 0.70 ± 0.04 | 2.3 ± 0.06 | ||
| 11. G31R,G62S | AUG | 18 ± 2.0 | 3.0 ± 0.3 | 0.46 ± 0.04 | 0.54 ± 0.03 | 1.1 ± 0.04 | 1.3 | 2.1 |
| 12. | UUG | 26 ± 3.0 | 4.0 ± 0.07 | 0.30 ± 0.03 | 0.70 ± 0.10 | 2.3 ± 0.30 |
*All errors are SE calculated by SD/√n; n = number of experiments.
As observed previously (12,24), the G31R eIF5 substitution essentially reverses the differential effects of AUG and UUG on eIF1A dissociation kinetics (Figure 4A). Relative to WT eIF5, the G31R mutant displays increased occupancy of the closed state at UUG (Kamp increased >2.5-fold; Table 4, row 4 versus 2) but decreased occupancy of the closed conformation at AUG (Kamp decreased 2-fold; Table 4, row 3 versus 1), increasing the Kamp(UUG):Kamp(AUG) ratio from ∼0.7 (WT) to ∼3.3 (G31R). G31R also stabilizes eIF1A bound in the closed conformation at UUG (k2 decreased by ∼2-fold) while destabilizing it at AUG (k2 increased by ∼12-fold) (Table 4, rows 3–4 versus 1–2). Thus, G31R strongly favors the rearrangement to the closed conformation at UUG and disfavors this transition at AUG, consistent with its Sui− phenotype.
Combining M18V with G31R in the G31R,M18V double mutant does not substantially alter the eIF1A dissociation kinetics (Figure 4B). The Kamp(UUG):Kamp(AUG) ratio (4.5) is relatively unchanged from that observed for the G31R single mutant (3.3) (Table 4, rows 7–8 versus 3–4), indicating that M18V does not reverse the dual effects of G31R of promoting the closed complex at UUG and disfavoring the open complex at AUG. Combining M18V with G31R does appear to destabilize eIF1A binding to the open conformation at UUG (k1 increased by ∼2.0-fold relative to the G31R mutant) and to stabilize its binding to the closed state at AUG (k2 decreased by 2.5-fold relative to G31R) (Table 4, rows 7–8 versus 3–4). These latter effects might contribute to the ability of M18V to lower the UUG:AUG initiation ratio (Ssu− phenotype) in the G31R,M18V double mutant versus the G31R single mutant. On its own in otherwise WT eIF5, with an AUG start codon, M18V decreases the occupancy of the closed complex (Kamp decreased by 2-fold) (Table 4, rows 5–6 versus 1–2), which might contribute to the increased UUG:AUG initiation ratio (Sui− phenotype) observed in vivo for the M18V single mutant (Figure 2F), although there is a similar effect on Kamp at UUG codons.
Comparing the eIF1A dissociation kinetics between the G31R single mutant and the G31R,G62S double mutant indicates that the G62S suppressor exerts a significant effect on the ability of G31R to influence the open-to-closed transition at AUG versus UUG codons (Figure 4C). As noted above, G31R essentially reverses the partitioning of PICs between the open and closed conformations from that observed with WT eIF5 to favor the closed conformation at UUG and disfavor the closed state at AUG. The G62S substitution decreases the occupancy of the closed complex at UUG in the G31R,G62S double mutant relative to its value in the G31R single mutant (Kamp decreased by ∼4-fold), although it has a similar effect of disfavoring the closed state at AUG (Kamp decreased by ∼3-fold) (Table 4, rows 11–12 versus 3–4). Perhaps of greater significance, G62S also strongly destabilizes binding of eIF1A in the closed complex at UUG, increasing k2 by an order of magnitude in the G31R,G62S double mutant versus the G31R single mutant, while actually decreasing somewhat the k2 at AUG under the same circumstances (Table 4, rows 11–12 versus 3–4). Thus, in the presence of G31R, the G62S suppressor has a much larger destabilizing effect on interactions in the closed state at UUG than at AUG. These findings, together with the fact that G62S also mitigates the effects of G31R in promoting Pi release at UUG versus AUG codons (Figure 3C and D), can account for the ability of G62S to efficiently suppress the elevated UUG:AUG ratio conferred by G31R in vivo.
On its own in otherwise WT eIF5, G62S decreases the abundance of the closed complex at AUG (Kamp decreased by ∼2-fold) and also strongly destabilizes eIF1A binding in the closed state at AUG (k2 increased by >10-fold) while producing a smaller reduction in the stability of the open complex at AUG (k1 increased by ∼2.5-fold; Table 4, row 9 versus row 1). It has a similar effect on partitioning between the open and closed states at UUG but destabilizes eIF1A binding in both states to similar extents (k1 and k2 increased by ∼2.5- and ∼2-fold, respectively; Table 4, row 10 versus row 2). The specific destabilization of interactions in the closed state at AUG codons would be expected to elevate the UUG:AUG initiation ratio (Figure 2F) because a preferential reduction in initiation at AUG codons would free up PICs to initiate at UUG codons, increasing UUG initiation by mass action (20).
Intergenic suppression of Sui− mutants of eIF2β, eIF1A and eIF1 by eIF5 suppressors
We sought next to obtain genetic evidence that the Ssu− phenotypes of the M18V and G62S eIF5 substitutions involve distinct molecular mechanisms in vivo. To this end, we asked whether M18V and G62S differ significantly in their ability to suppress Sui− substitutions in other initiation factors, beginning with the dominant Sui− mutation SUI3–2, encoding the S264Y substitution in eIF2β. Plasmid-borne SUI3–2 was introduced into SUI3+ tif5Δ strains also containing TIF5 alleles harboring single suppressor mutations (i.e. without G31R). As expected, in the TIF5+ his4–303 strain, SUI3–2 confers growth on −His medium (Figure 5A, rows 1–2) and elevates the UUG:AUG ratio above ∼0.3 (Figure 5B, columns1–2). The His+/Sui− phenotype of SUI3–2 is suppressed efficiently by the TIF5-M18V and TIF5-K33E alleles (Figure 5A, rows 2–4), with commensurate strong reductions in the UUG:AUG initiation ratio (Figure 5B, columns 2–4). Thus, these two eIF5 mutants are effective suppressors of the Sui− phenotypes of both SUI3–2 and (as shown above) the G31R allele of TIF5. Similarly, the L61A mutation, an ineffective suppressor of G31R/SUI5 (Figure 2) likewise only partially suppresses the His+/Sui− phenotype of SUI3–2 (Figure 5A, row 6 versus 2) and is relatively ineffective in reducing the elevated UUG:AUG ratio conferred by SUI3–2 (Figure 5B, columns 6 versus 2). It is interesting however that G62S, one of the strongest suppressors of G31R (Figure 2C and E), is a relatively inefficient suppressor of SUI3–2, failing to eliminate the His+ phenotype (Figure 5A, row 5) and reducing the UUG:AUG ratio by less than a factor of 2 (Figure 5B, column 6 versus 2). This last finding suggests that the Sui− phenotypes of tif5-G31R and SUI3–2 involve at least partially distinct mechanisms and that the G62S substitution in eIF5 more effectively compensates for the defect(s) conferred by tif5-G31R versus SUI3–2.
Figure 5.
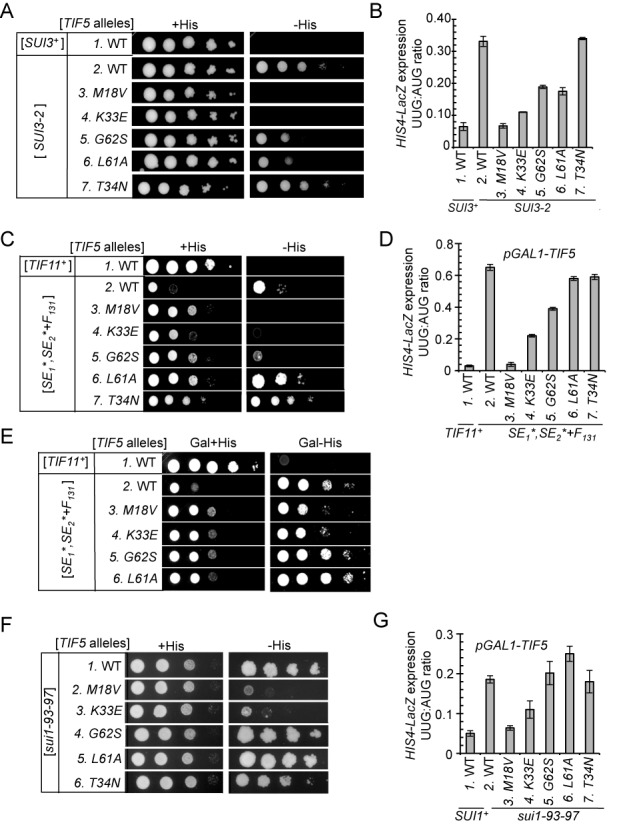
TIF5 suppressors confer allele-specific suppression of Sui− mutations in eIF2β, eIF1A and eIF1. (A) Slg− and His+/Sui− phenotypes of derivatives of his4-301 strain ASY100 harboring the indicated TIF5 alleles on LEU2 plasmid and either YCpSUI3-2 plasmid harboring SUI3-2 (rows 2–7) or empty vector (row 1) were determined by spotting serial 10-fold dilutions on SC medium lacking leucine and tryptophan (SC-LW) supplemented with 0.3 mM His (+His) or 0.0003 mM His (−His) and incubated for 3d (−His) or 6d (+His) at 30°C. (B) Strains described in (A) harboring the AUG or UUG HIS4-lacZ reporters were analyzed as in Figure 2C. (C) Derivatives of his4-301 tif11Δ PGAL-TIF5 strain PMY17 harboring a TRP1 plasmid with WT TIF11 (pAS5-142) (lane 1) or tif11-SE1*,SE2*+F131 (pAS5-130) (Lanes 2–7) were transformed with LEU2 plasmids containing the indicated TIF5 alleles, and Slg− and His+/Sui− phenotypes were determined as in (A). (D) Strains described in (C) harboring the AUG or UUG HIS4-lacZ reporters were analyzed as in (B). (E) Dominant His−/Ssu− and Slg+ phenotypes of the strains in (C) were analyzed by spotting serial 10-fold dilutions on SC medium containing galactose and lacking tryptophan and leucine supplemented with 0.3 mM His (Gal + His) or 0.0003 mM His (Gal −His) and incubating for 3d (Gal + His) or 6d (Gal − His) at 30°C. (F) Slg− and His+/Sui− phenotypes were determined for derivatives of his4-301 sui1Δ PGAL-TIF5 strain PMY01 harboring sui1-93-97 on a TRP1 plasmid and LEU2 plasmids containing the indicated TIF5 alleles were determined as in (C). (G) Strains described in (F) harboring the AUG or UUG HIS4-lacZ reporters were analyzed as described in (D). For panels A, C and F, images have been cropped from results obtained from different plates examined in parallel in the same experiments.
We also examined whether the eIF5 suppressors can mitigate the Sui− phenotype produced by the eIF1A mutation tif11-SE1*,SE2*+F131, containing Ala substitutions in all but one (Phe-131) of the 20 residues that comprise scanning enhancer elements SE1 and SE2 in the eIF1A CTT (25). To this end, we introduced the TIF5 alleles into a his4–303 tif11Δ strain harboring chromosomal PGAL-TIF5 described above, and plasmid-borne tif11-SE1*,SE2*+F131 as the only source of eIF1A. In the TIF5+ strain on glucose medium (where expression of WT eIF5 from PGAL-TIF5 is repressed), SE1*,SE2*+F131 confers a His+ phenotype and elevated UUG:AUG ratio (∼0.65), indicating a Sui− phenotype, and a Slg− phenotype on +His medium (Figure 5C, rows 1–2 and D, columns 1–2), all in agreement with previous results (25). Once again, we found that the tif5-M18V allele confers a dramatic suppression of the strong Sui− phenotype of tif11-SE1*,SE2*+F131, eliminating growth on –His medium (Figure 5C, rows 2–3, −His) and reducing the UUG:AUG ratio to virtually the same low level seen in the isogenic WT TIF11+ strain (Figure 5D, columns 2–3). M18V also partially suppresses the Slg− phenotype on +His medium conferred by SE1*,SE2*+F131 (Figure 5C, rows 2–3, +His). The K33E mutation, shown above to be a potent suppressor of G31R/SUI5 (Figure 2) and SUI3–2 (Figure 5B), also efficiently suppresses the Sui− and Slg− phenotypes of SE1*,SE2*+F131, albeit less completely than does M18V (Figure 5C and D, cf. rows/columns 2–4).
In accordance with their inability to suppress the Sui− phenotypes of tif5-G31R (Figure 2) or SUI3–2 (Figure 5A and B), the L61A and T34N mutations fail to suppress the His+/Sui− phenotype and elevated UUG:AUG ratio conferred by SE1*,SE2*+F131 (Figure 5C, rows 2,6,7, −His; Figure 5D, columns 2,6,7); although they can mitigate the Slg− phenotype of this eIF1A mutation (Figure 5C, rows 2, 4 and 7, +His). Furthermore, the G62S mutation confers only partial suppression of the Sui− phenotype of SE1*,SE2*+F131 (Figure 5C and D, cf. rows/columns 2 and 5), similar to its incomplete suppression of SUI3–2 (Figure 5A and B).
The ability of the M18V and K33E substitutions to suppress the His+/Sui− phenotypes of the SE1*,SE2*+F131 mutation was also observed in the presence of WT eIF5, produced by growing the PGAL-TIF5 strains on galactose medium (Figure 5E, rows 3, 4 versus 2). The fact that the Ssu− phenotypes of M18V and K33E are partially dominant to TIF5+ implies that the mutant proteins compete with WT eIF5 for association with the scanning PIC and mitigate the ability of the eIF1A-SE1*,SE2*+F131 variant to allow aberrant UUG initiation events.
The same trends described above were also observed when we tested the eIF5 mutations for suppression of the Sui− phenotype of sui1–93–97, encoding a cluster of 3 Ala substitutions in helix α2 of eIF1 (26). The TIF5 suppressor alleles were introduced into a sui1Δ PGAL1-TIF5 strain with plasmid-borne sui1–93–97 as the sole source of eIF1. In the TIF5+ strain on glucose medium, the 93–97 mutation in eIF1 confers a His+/Sui− phenotype and increases the UUG:AUG ratio to ∼0.19 (Figure 5F, row 1 and Figure 5G, columns 1–2), consistent with our previous results (26). Similar to observations above for Sui− mutations in other factors, (i) the M18V mutation dramatically suppresses the Sui− phenotype of 93–97; (ii) K33E is a potent suppressor but less effective than M18V and (iii) the T34N and L61A mutations confer no detectable suppression of the Sui− phenotype of 93–97 (Figure 5F and G). Remarkably, G62S is completely ineffective in reducing the Sui− phenotype of 93–97 (Figures 5F, row 4 and 5G, column 5), in sharp contrast to its full suppression of tif5-G31R (Figure 2).
DISCUSSION
eIF5 promotes initiation accuracy by stimulating Pi release and enhancing the closed PIC conformation at AUG codons
In this study, we obtained strong genetic and biochemical evidence that eIF5 controls start codon recognition not merely by functioning as a GAP but also by regulating gated Pi release from eIF2-GDP·Pi. Previous work on the eIF5-G31R variant (SUI5 product) suggested that it enhances UUG initiation by elevating the GAP function of eIF5 (11). We subsequently determined that Pi release is accelerated more strongly than GTP hydrolysis by AUG recognition, and that Pi release is blocked until eIF1 dissociates on start codon recognition (10). In this view, it is less obvious that a simple increase in the rate of GTP hydrolysis evoked by eIF5-G31R would accelerate Pi release preferentially at near-cognates to increase UUG initiation. Indeed, we discovered here that G31R alters the coupling between start codon recognition and Pi release to accelerate release at UUG codons while reducing it at AUG codons. This discovery mirrors our previous findings that eIF5-G31R stabilizes the closed state at UUG while destabilizing it at AUG codons, based on results from an assay (eIF1A dissociation kinetics) in which GTP hydrolysis does not occur (27). Both effects of eIF5-G31R on reconstituted PICs provide a satisfying explanation for its ability to increase the UUG:AUG initiation ratio in vivo (Sui− phenotype). They also suggest that the shift from the open to closed state of the PIC precedes and is required for Pi release, consistent with previous results (3). Interestingly, while this study was underway, we found that eIF5-G31R accelerates eIF1 dissociation from the PIC at UUG codons but does not reduce the eIF1 dissociation rate at AUG codons (28), ostensibly at odds with our findings here on Pi release for PICs reconstituted with mRNA(AUG). Presumably, G31R blocks rapid Pi release at AUG codons following eIF1 dissociation; and one possibility is that it impedes the functional interaction between eIF5-NTD and eIF1A-CTT shown recently to be necessary for Pi release following eIF1 dissociation (3).
Evidence supporting the physiological relevance of the dual effects of eIF5-G31R on Pi release and rearrangement to the closed PIC conformation was obtained by examining second-site suppressors in eIF5 that mitigate the Sui− phenotype of SUI5. We established that two eIF5 substitutions, M18V and G62S, very efficiently suppress the dominant Sui− phenotype of G31R; and that G62S, but not M18V, also suppresses the recessive lethality of G31R. When present as the only substitution in eIF5, M18V also efficiently suppresses the Sui− phenotypes of mutations in eIF2β, eIF1A and eIF1, but this was not true for eIF5-G62S. These marked differences in the genetic properties of M18V and G62S resonate with results of our in vitro analyses, which revealed distinctly different effects of M18V and G62S on GTP hydrolysis/Pi release and the transition from open to closed PIC conformations.
Consistent with the proximity of Met-18 to Arg-15, a key residue of the eIF5 GAP domain, combining M18V with G31R reduces the rates of both GTP hydrolysis and Pi release and partially reverses the aberrant effect of G31R in provoking more rapid Pi release at UUG versus AUG codons. This last effect is consistent with the reduction in the UUG:AUG ratio (Ssu− phenotype) produced by M18V in the eIF5-G31R,M18V double mutant. By contrast, M18V had relatively little impact on the ability of G31R to reverse the differential effects of AUG and UUG on stability of the closed PIC conformation, as revealed by eIF1A dissociation kinetics. Thus, we propose that the principle consequence of M18V is to impair the ability of Arg-15 to function as an arginine finger, reducing GTP hydrolysis and subsequent Pi release. This proposal is supported by our finding that M18V on its own dramatically reduces the rate of the GTP hydrolysis phase of the GTPase reactions at both AUG and UUG codons, as well as the eIF5-dependent stimulation of GTP hydrolysis by isolated TCs. The fact that G31R,M18V is recessive-lethal whereas M18V alone is viable can be explained by proposing that combining the aberrant destabilization of the closed PIC conformation at AUGs conferred by G31R with the impairment of GTP hydrolysis/Pi release by M18V evokes a lethal reduction in bulk translation initiation.
The G62S substitution differs from M18V in suppressing both the lethality and Sui− phenotypes of the G31R mutation in eIF5. The ability of G62S to suppress the Sui− phenotype of G31R can be understood from our biochemical findings that (i) it overcomes the effect of G31R of elevating Pi release at UUG codons and depressing the reaction at AUG codons and (ii) it mitigates the effect of G31R of stabilizing the closed conformation of the PIC at UUG codons. This dual effect of G62S in restoring more nearly WT differential rates of Pi release and closed complex occupancies at both AUG and UUG codons also helps to explain its ability to suppress the lethality of G31R. Thus, compared to the dramatic effect of G31R,M18V in reducing GTP hydrolysis/Pi release rates at both start codons, G62S provokes a more nuanced effect on the kinetics of Pi release in the G31R,G62S variant that moves the system closer to WT behavior. Accordingly, we envision that G62S alters the regulatory mechanism that couples Pi release to the quality of the codon-anticodon helix in the P site, rather than directly altering the catalytic function of the putative Arg finger of the GAP domain.
M18V is the most effective Ssu− mutation described thus far, in completely eliminating the increased UUG:AUG initiation ratio in cells harboring Sui− mutations in eIF1 (sui1–93–97), eIF1A (tif11-SE1*,SE2*+F131) or eIF2β (SUI3–2). The following considerations suggest that the broad Ssu− characteristic of M18V derives from its strong reduction in rate of GTP hydrolysis and Pi release at UUG codons. In addition to eIF1 dissociation, Pi release also requires movement of the eIF5-NTD and eIF1A-CTT closer to one another in the PIC (3). The fact that overexpressing WT eIF1 suppresses UUG initiation in all Sui− mutants tested (2), makes it likely that Pi release at UUGs can be prevented in Sui− mutants by driving the rapid re-association of eIF1 with its 40S binding site in the open PIC conformation after its initial release on UUG recognition. Through a related mechanism, by greatly reducing the rate of GTP hydrolysis/Pi release, eIF5-M18V could increase the probability that scanning can resume at UUG codons by allowing more time for re-association of eIF1 with the PIC. Delaying Pi release in this way would allow eIF5-M18V to suppress any Sui− mutation that shifts the balance toward the closed/PIN state with attendant release of eIF1. Indeed, this property has been described for sui1–93–97 (26), tif11-SE1*,SE2* (25) and SUI3–2 (28), consistent with the strong suppression of all three Sui− mutations by eIF5-M18V. In this view, G62S is inferior to M18V in suppressing these Sui− mutations because it has no intrinsic effect on Pi release and, hence, does not delay this reaction sufficiently to allow eIF1 re-association and resumption of scanning at UUG codons.
It was surprising to find that the T34N and L61A substitutions in eIF5 suppress the recessive lethality of the eIF5-G31R variant but are generally ineffective suppressors of the Sui− phenotypes of eIF5-G31R and the other Sui− mutations we analyzed. These findings likely indicate that the lethality of SUI5/G31R reflects the compound effects of a reduced efficiency of AUG initiation and elevated UUG initiation and that restoring AUG initiation is sufficient to rescue viability. One possibility is that T34N and L61A mitigate the defect in AUG recognition conferred by G31R but produce an equally stimulatory effect on UUG initiation, leaving the ratio of UUG:AUG initiation unaltered from that observed in the G31R/SUI5 single mutant.
Molecular implications of Ssu− mutations for eIF5's multiple functions in start codon recognition
Our finding that the eIF5-M18V substitution strongly impairs GTP hydrolysis and Pi release in the PIC fits with its location only three residues from Arg-15 in the NTT of eIF5 (Figure 6A and B). Arg fingers are usually flanked by hydrophobic residues that stabilize the Arg finger loop and, indeed, R15 is flanked by Phe and Tyr residues: F13Y14R15Y16K17M18 ((7) and references therein). As M18V replaces one flanking hydrophobic residue with another, it would not necessarily perturb hypothetical packing of the Arg finger loop with the core of the GAP domain, but perhaps the Val substitution disturbs the orientation of Arg-15 in a manner that reduces its ability to stimulate GTP hydrolysis.
Figure 6.
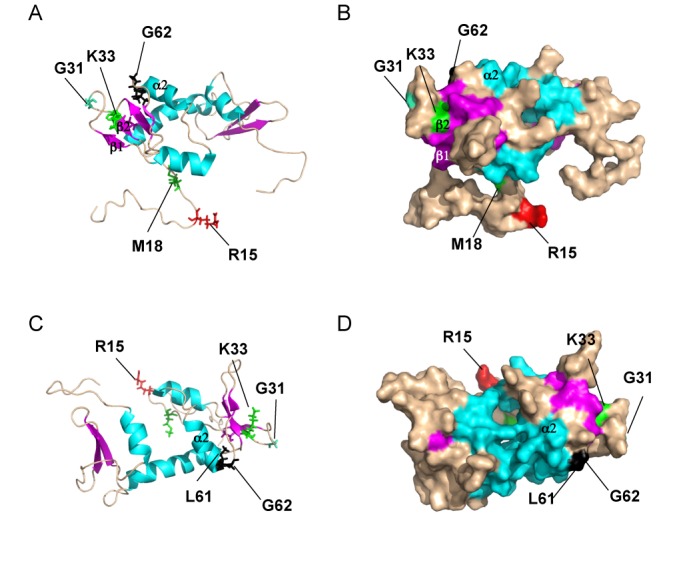
Locations of yeast eIF5 residues altered by Sui− or Ssu− substitutions in the solution structure of the human eIF5-NTD. Ribbon (A and C) and surface (B and D) representations of the human eIF5-NTD were rendered using PyMol from pdb file 2G2K (7) with the locations of residues aligning with the indicated yeast residues colored red, green or black and also shown in stick representation (A and C).
K33E is second only to M18V in its ability to reduce the UUG:AUG initiation ratios conferred by the panel of Sui− mutations we examined in other factors (Figure 5B, D and G), although it differs from M18V in also suppressing the lethality of G31R. Interestingly, the K33A substitution in rat eIF5 impairs its ability to replace endogenous eIF5 in yeast cells and reduces GTP hydrolysis in a model GAP assay (8). It has been suggested that K33 plays the role of the secondary stabilizing Lys or Arg described in other GAPs, which is consistent with its location in the β1–loop–β2 also containing the SUI5/G31R substitution (Figure 6A and B) (7). It seems plausible that K33E reduces GAP function and Pi release, albeit not as severely as M18V, which enables it to suppress multiple Sui− mutations by the mechanism proposed above for M18V, of delaying Pi release to allow time for eIF1 re-association and resumption of scanning at UUG codons. The residue substituted in T34N is immediately adjacent to K33 in β2. We suggested above that T34N stimulates AUG and UUG initiation equally when combined with G31R; but on its own, T34N enhances UUG initiation to a greater extent and confers a Sui− phenotype. These results further implicate the β1–loop–β2, modified by G31R, K33E and T34N, in regulating GTP hydrolysis and Pi release.
We argued above that, when combined with G31R, the G62S substitution perturbs the regulatory mechanism coupling Pi release to the quality of the codon-anticodon helix rather than impairing the catalytic function of Arg-15. G62, and the adjacent residue altered by the suppressor substitution L61, map at the end of helix α2 and beginning of the [α2–β3] loop, and are located on the opposite face of the eIF5 NTD from the NTT and Arg-15 (7) (Figure 6C and D). Considering that G62S also mitigates the effect of G31R in stabilizing the closed complex at UUG, we suggest that α2 and the [α2–β3] loop function in coupling codon-anticodon pairing to stabilization of the closed complex in addition to regulating Pi release. Because eIF1A dissociation kinetics are measured using TC assembled with non-hydrolyzable Guanosine 5′-[β,γ-imido]triphosphate (GDPNP), the effect of G62S in destabilizing the closed conformation of the PIC is unlikely to be secondary to its effects on Pi release; however, the decrease in Pi release could be a consequence of destabilization of the closed complex. An interesting possibility is that eIF5 residues in α2 or the [α2–β3] loop enhance the functional interaction of the eIF5 NTD with the eIF1A CTT required to stabilize the closed/PIN state and trigger Pi release (3).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Tom Donahue for gifts of Ssu− strains and sharing unpublished information, and Tom Dever for many helpful discussions and suggestions.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Intramural Program of the National Institutes of Health [in part to A.G.H. and J.R.L.]; National Institutes of Health Grant [GM62128] [formerly to J.R.L.]; Department of Science and Technology, Government of India Grant [Int/NZ/P-2/13] [to A.K.S.]. Funding for open access charge: Intramural Program of the National Institutes of Health [ to A.G.H. and J.R.L.]; National Institutes of Health Grant [GM62128] [formerly to J.R.L.]; Department of Science and Technology, Government of India Grant [Int/NZ/P-2/13] [to A.K.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Pestova T.V., Lorsch J.R., Hellen C.U.T. In: Translational Control in Biology and Medicine. Mathews M, Sonenberg N, Hershey JWB, editors. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2007. pp. 87–128. [Google Scholar]
- 2.Hinnebusch A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011;75:434–467. doi: 10.1128/MMBR.00008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nanda J.S., Saini A.K., Munoz A.M., Hinnebusch A.G., Lorsch J.R. Coordinated movements of eukaryotic translation initiation factors eIF1, eIF1A, and eIF5 trigger phosphate release from eIF2 in response to start codon recognition by the ribosomal preinitiation complex. J. Biol. Chem. 2013;288:5316–5329. doi: 10.1074/jbc.M112.440693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabl J., Leibundgut M., Ataide S.F., Haag A., Ban N. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science. 2011;331:730–736. doi: 10.1126/science.1198308. [DOI] [PubMed] [Google Scholar]
- 5.Lomakin I.B., Steitz T.A. The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature. 2013;500:307–311. doi: 10.1038/nature12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alone P.V., Dever T.E. Direct binding of translation initiation factor eIF2gamma-G domain to its GTPase-activating and GDP-GTP exchange factors eIF5 and eIF2B epsilon. J. Biol. Chem. 2006;281:12636–12644. doi: 10.1074/jbc.M511700200. [DOI] [PubMed] [Google Scholar]
- 7.Conte M.R., Kelly G., Babon J., Sanfelice D., Youell J., Smerdon S.J., Proud C.G. Structure of the eukaryotic initiation factor (eIF) 5 reveals a fold common to several translation factors. Biochemistry. 2006;45:4550–4558. doi: 10.1021/bi052387u. [DOI] [PubMed] [Google Scholar]
- 8.Das S., Ghosh R., Maitra U. Eukaryotic translation initiation factor 5 functions as a GTPase- activating protein. J. Biol. Chem. 2001;276:6720–6726. doi: 10.1074/jbc.M008863200. [DOI] [PubMed] [Google Scholar]
- 9.Paulin F.E., Campbell L.E., O'Brien K., Loughlin J., Proud C.G. Eukaryotic translation initiation factor 5 (eIF5) acts as a classical GTPase-activator protein. Curr. Biol. 2001;11:55–59. doi: 10.1016/s0960-9822(00)00025-7. [DOI] [PubMed] [Google Scholar]
- 10.Algire M.A., Maag D., Lorsch J.R. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell. 2005;20:251–262. doi: 10.1016/j.molcel.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Huang H., Yoon H., Hannig E.M., Donahue T.F. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 1997;11:2396–2413. doi: 10.1101/gad.11.18.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maag D., Algire M.A., Lorsch J.R. Communication between eukaryotic translation initiation factors 5 and 1A within the ribosomal pre-initiation complex plays a role in start site selection. J. Mol. Biol. 2006;356:724–737. doi: 10.1016/j.jmb.2005.11.083. [DOI] [PubMed] [Google Scholar]
- 13.Asano K., Krishnamoorthy T., Phan L., Pavitt G.D., Hinnebusch A.G. Conserved bipartite motifs in yeast eIF5 and eIF2Be, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J. 1999;18:1673–1688. doi: 10.1093/emboj/18.6.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olsen D.S., Savner E.M., Mathew A., Zhang F., Krishnamoorthy T., Phan L., Hinnebusch A.G. Domains of eIF1A that mediate binding to eIF2, eIF3 and eIF5B and promote ternary complex recruitment in vivo. EMBO J. 2003;22:193–204. doi: 10.1093/emboj/cdg030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Longtine M.S., McKenzie A., III, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R. Additonal modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 16.Moehle C.M., Hinnebusch A.G. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol. Cell Biol. 1991;11:2723–2735. doi: 10.1128/mcb.11.5.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reid G.A., Schatz G. Import of proteins into mitochondria. J. Biol. Chem. 1982;257:13062–13067. [PubMed] [Google Scholar]
- 18.Acker M.G., Kolitz S.E., Mitchell S.F., Nanda J.S., Lorsch J.R. Reconstitution of yeast translation initiation. Methods Enzymol. 2007;430:111–145. doi: 10.1016/S0076-6879(07)30006-2. [DOI] [PubMed] [Google Scholar]
- 19.Maag D., Lorsch J.R. Communication between eukaryotic translation initiation factors 1 and 1A on the yeast small ribosomal subunit. J. Mol. Biol. 2003;330:917–924. doi: 10.1016/s0022-2836(03)00665-x. [DOI] [PubMed] [Google Scholar]
- 20.Nanda J.S., Cheung Y.N., Takacs J.E., Martin-Marcos P., Saini A.K., Hinnebusch A.G., Lorsch J.R. eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J. Mol. Biol. 2009;394:268–285. doi: 10.1016/j.jmb.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donahue T. In: Translational Control of Gene Expression. Sonenberg N., Hershey J.W.B., Mathews M.B., editors. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2000. pp. 487–502. [Google Scholar]
- 22.Singh C.R., Watanabe R., Chowdhury W., Hiraishi H., Murai M.J., Yamamoto Y., Miles D., Ikeda Y., Asano M., Asano K. Sequential eukaryotic translation initiation factor 5 (eIF5) binding to the charged disordered segments of eIF4G and eIF2beta stabilizes the 48S preinitiation complex and promotes its shift to the initiation mode. Mol. Cell Biol. 2012;32:3978–3989. doi: 10.1128/MCB.00376-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin-Marcos P., Cheung Y.N., Hinnebusch A.G. Functional elements in initiation factors 1, 1A, and 2beta discriminate against poor AUG context and non-AUG start codons. Mol. Cell Biol. 2011;31:4814–4831. doi: 10.1128/MCB.05819-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fekete C.A., Mitchell S.F., Cherkasova V.A., Applefield D., Algire M.A., Maag D., Saini A.K., Lorsch J.R., Hinnebusch A.G. N-and C-terminal residues of eIF1A have opposing effects on the fidelity of start codon selection. EMBO J. 2007;26:1602–1614. doi: 10.1038/sj.emboj.7601613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saini A.K., Nanda J.S., Lorsch J.R., Hinnebusch A.G. Regulatory elements in eIF1A control the fidelity of start codon selection by modulating tRNA(i)(Met) binding to the ribosome. Genes Dev. 2010;24:97–110. doi: 10.1101/gad.1871910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheung Y.N., Maag D., Mitchell S.F., Fekete C.A., Algire M.A., Takacs J.E., Shirokikh N., Pestova T., Lorsch J.R., Hinnebusch A.G. Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev. 2007;21:1217–1230. doi: 10.1101/gad.1528307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maag D., Fekete C.A., Gryczynski Z., Lorsch J.R. A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol. Cell. 2005;17:265–275. doi: 10.1016/j.molcel.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 28.Martin-Marcos P., Nanda J.S., Luna R.E., Zhang F., Saini A.K., Cherkasova V.A., Wagner G., Lorsch J.R., Hinnebusch A.G. Enhanced eIF1 binding to the 40S ribosome impedes conformational rearrangements of the preinitiation complex and elevates initiation accuracy. RNA. 2013;20:150–167. doi: 10.1261/rna.042069.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinnebusch A.G., Lorsch J.R. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb. Perspect. Biol. 2012;4:1–25. doi: 10.1101/cshperspect.a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gietz R.D., Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 31.Valasek L., Nielsen K.H., Zhang F., Fekete C.A., Hinnebusch A.G. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell Biol. 2004;24:9437–9455. doi: 10.1128/MCB.24.21.9437-9455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donahue T.F., Cigan A.M. Genetic selection for mutations that reduce or abolish ribosomal recognition of the HIS4 translational initiator region. Mol. Cell Biol. 1988;8:2955–2963. doi: 10.1128/mcb.8.7.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fekete C.A., Applefield D.J., Blakely S.A., Shirokikh N., Pestova T., Lorsch J.R., Hinnebusch A.G. The eIF1A C-terminal domain promotes initiation complex assembly, scanning and AUG selection in vivo. EMBO J. 2005;24:3588–3601. doi: 10.1038/sj.emboj.7600821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.