


Abstract
Nearly all bacteria exhibit a type of phenotypic growth described as persistence that is thought to underlie antibiotic tolerance and recalcitrant chronic infections. The chromosomally encoded high-persistence (Hip) toxin–antitoxin proteins HipASO and HipBSO from Shewanella oneidensis, a proteobacterium with unusual respiratory capacities, constitute a type II toxin–antitoxin protein module. Here we show that phosphorylated HipASO can engage in an unexpected ternary complex with HipBSO and double-stranded operator DNA that is distinct from the prototypical counterpart complex from Escherichia coli. The structure of HipBSO in complex with operator DNA reveals a flexible C-terminus that is sequestered by HipASO in the ternary complex, indicative of its role in binding HipASO to abolish its function in persistence. The structure of HipASO in complex with a non-hydrolyzable ATP analogue shows that HipASO autophosphorylation is coupled to an unusual conformational change of its phosphorylation loop. However, HipASO is unable to phosphorylate the translation factor Elongation factor Tu, contrary to previous reports, but in agreement with more recent findings. Our studies suggest that the phosphorylation state of HipA is an important factor in persistence and that the structural and mechanistic diversity of HipAB modules as regulatory factors in bacterial persistence is broader than previously thought.
INTRODUCTION
To cope with unpredictable environmental fluctuations, bacteria resorted to population diversity as a risk-spreading strategy. By creating a phenotypic or genetically heterogeneous population, bacteria essentially increase the probability that at least a subpopulation is well adapted to the actual environment (1–3). A striking example of such phenotypic diversity is the emergence of a small subpopulation of persister cells that are able to survive against an antibiotic challenge by entering a dormant state with an increased tolerance against a wide variety of stresses (4). Recently, a strong correlation between persister cells and the multidrug tolerance (MDT) of biofilms has been shown. Thus, gaining insights into bacterial persistence could create avenues for the development of new antimicrobial drugs and for combating chronic infections (5,6). While bacterial persistence was discovered almost 70 years ago, its understanding at the molecular level is a relatively recent development and has coincided with the discovery of the hipA gene in Escherichia coli (7–9). The products of hipA and its upstream linked gene hipB form a toxin–antitoxin module, which together are recognized as a major factor involved in persistence (10,11). The toxin HipA is a protein kinase and its activity is essential for growth arrest and MDT in E. coli K12 (12). A first functional annotation of HipA by Schumacher et al. linked HipA to phosphorylation of the elongation factor Tu (EF-Tu) resulting in inhibition of protein biosynthesis. However, more recent studies have demonstrated that not EF-Tu, but glutamyl-tRNA synthetase (GltX) is the phosphorylation target of HipA (13,14). Furthermore, HipA has been shown to trigger growth arrest by controlling RelA-mediated biosynthesis of the alarmone (p)ppGpp (15,16). HipA activity is inhibited upon binding to HipB and through sequestration to the nucleoid, as both HipB and the HipBA complex bind to their corresponding operators, thereby leading to suppression of their own gene transcription (14). However, the molecular mechanisms behind such temporary growth arrest remain unclear and neither is it known what exactly triggers the escape from dormancy.
We previously demonstrated that interruption of the SO0706 gene in Shewanella oneidensis MR-1 (SO) resulted in a 50% decrease of biofilm formation (17). This gene encodes a protein sharing 28% sequence identity with E. coli HipA. S. oneidensis MR-1 is a respiratory diverse model bacterium that attracted interest due to its role in biogeochemistry, its potential for bioremediation of heavy metals and aromatic compounds as well as for its use in microbial fuel cells (18–20). Many of these applications rely on the potential of the organism to form biofilms on inorganic surfaces. Molecular studies on the mechanisms of biofilm formation by S. oneidensis established a role for a type IV pilus system, the presence of extracellular DNA and the mxdABCD gene cluster, encoding a polysaccharide biogenesis system (21). A microarray analysis of the early stage of biofilm formation revealed the involvement of different metabolic processes and cyclic di-GMP dependent pathways, which was recently confirmed by molecular characterization of PdeB, a c-di-GMP phosphodiesterase (22,23). The sequencing of the S. oneidensis MR-1 genome inspired efforts to gain insight into its regulatory and metabolic networks and their relationship to physiology (24,20).
Given the strong correlation between biofilm formation and persistence in bacteria, as well as the potential involvement of SO0706 in this process, we sought to investigate structure–function relationships of the SO0706 product. Here, we provide structural insights into the unexpected participation of the phosphorylated HipA toxin in a ternary assembly with the antitoxin HipB and their operator DNA. We show that phosphorylation of HipA at a serine residue buried deeply in the structure is coupled to conformational ejection of the phosphorylation loop (pLoop). However, our direct observation of phosphorylated HipA in a ternary complex indicates that HipB could rescue growth arrest by directly targeting the phosphorylated HipA population. Furthermore, the C-terminus of HipB undergoes dramatic conformational changes between the unbound form and when in complex with HipA and operator DNA, indicating the possible regulatory role of such interaction via prevention of proteolysis. Furthermore, we find that S. oneidensis MR-1 HipA is neither able to bind to nor can phosphorylate EF-Tu, which is in agreement with recent findings for the E. coli counterpart. Together, our studies provide new insights into the possible involvement of HipA and HipB in bacterial persistence and open new experimental possibilities for interrogating the role of both proteins in the persistence regulatory mechanisms.
MATERIALS AND METHODS
Expression and purification of recombinant proteins and complexes thereof
Expression constructs for S. oneidensis MR-1 HipAso (SO0706) and HipBso (SO0705) were established based on primers MR01, MR02 for HipAso and MR03, MR04 for HipBso (Supplementary Table S1), and were cloned into the pET15b expression vector (Merck Millipore). The D306Q mutant of HipAso was generated via overlap polymerase chain reaction (PCR) using the flanking primer MR01 and internal primer MR06 in one reaction and the flanking primer MR02 and internal primer MR05 in another reaction. The PCR products were gel purified and used in a final overlap PCR round using the flanking primers MR01 and MR02. An expression construct for S. oneidensis MR-1 EF-Tu was cloned into a pET11b vector (Merck Millipore) containing a C-terminal 6xHis-tag sequence with primers MR07 and MR08 from S. oneidensis MR-1 genomic DNA (Supplementary Table S1) and into the pGEX-4T-2 vector (GE Healthcare). S. oneidensis MR-1 HipAso and HipBso were expressed in E. coli BL21 DE3 and EF-Tuso in E. coli C43. Bacterial cultures were induced for 1 h (HipAso) or 4 h (HipBso and EF-Tuso) with 1 mM isopropyl β-D-1-thiogalactopyranoside (Duchefa Biochemie) after growth to an OD600nm of 0.6–0.7 in Lysogeny Broth (LB) medium supplied with Carbenicillin (100 μg/ml) at 37ºC. The cells were harvested by centrifugation at 7000 × g for 10 min at 4°C. After resuspending the cell pellets in 25 mM Na2HPO4 pH 7.5, 500 mM NaCl buffer with ethylenediaminetetraaceticacid-free protease inhibitor cocktail (Roche), the cellular suspension was sonicated and the soluble fraction was clarified by centrifugation at 75 000 × g for 30 min. The clarified lysate was loaded onto a Ni-NTA column (Qiagen) and pre-equilibrated with buffer A (25 mM Na2HPO4 pH 7.5, 500 mM NaCl, 10 mM imidazole), washed with buffer B (25 mM Na2HPO4 pH 7.5, 500 mM NaCl, 50 mM imidazole) and eluted with gradient buffer C (25 mM Na2HPO4 pH 7.5, 500 mM NaCl, 500 mM imidazole). The recombinant proteins were concentrated and subsequently further purified by size exclusion chromatography (SEC) on a Prep-Grade Hiload 16/60 Superdex200 column (GE Healthcare) equilibrated with buffer (10 mM HEPES pH 7.5, 300 mM NaCl and 5% glycerol). EF-Tuso was overexpressed and purified as described in (25), with modification of the buffer (300 mM NaCl, 25 mM Na2HPO4 pH 7.5, 15 μM GDP, 5 mM MgCl2). The N-terminal 6× His-tag of HipB was cleaved with biotinylated thrombin (Novagen) during overnight incubation and repurified by SEC. Pure fractions were pooled and proteins were concentrated for further experiments. To assemble the HipABso:DNA ternary complex, purified HipAso and HipBso were mixed with double-stranded operator DNA obtained by annealing two single primers 5′-ATTAGGTGTACTTATCTACACTTTTT-3′ and 5′-AAAAAGTGTAGATAAGTACACCTAAT-3′ (IDT). The HipABso–DNA complex was further purified by size-exclusion chromatography.
Crystallographic studies of HipABso:DNA complex, HipAso–AMPPNP–Mg2+ and HipBso
All crystallization trial experiments were performed via the vapor diffusion method using a Mosquito robot (TTP LabTech). Crystals of HipAso in complex with Mg2+–AMPPNP grew at 20°C in two days from sitting-drops formed by mixing 200 nl purified recombinant HipAso with 2 mM AMPPNP (Sigma) and 10 mM MgCl2 with an equal volume of reservoir solution (0.2 M sodium chloride, 0.1 M Na HEPES pH 7.0, 25% polyethylene glycol 4000). The crystallization droplets were equilibrated against 40 μl reservoir solution. For X-ray data collection under cryogenic conditions, the crystals were harvested from the drop and frozen in a cryostream using crystallization condition supplemented with 20% glycerol as cryoprotection solvent. Crystals of the HipAso–AMPPNP complex belonged to space group P212121 with a = 60.70 Å, b = 75.83 Å, c = 110.32 Å and α = β = γ = 90° and diffracted to 1.83 Å resolution. Crystals of recombinant HipBso were grown using purified HipBso after cleavage of the N-terminal 6× His-tag, from 0.1 M Na HEPES pH 7.5, 10% v/v 2-propanol, 20% PEG 4000. HipBso crystals diffracted to 1.85 Å and 2.35 Å. and belonged to space group C2221 with a = 53.46 Å, b = 72.95 Å, c = 86.91 Å and α = β = γ = 90°.
Crystals of recombinant HipABso in complex with operator DNA were grown in the crystallization geometries described above from reservoirs containing 0.2 M calcium acetate hydrate pH 7.5, 20% polyethylene glycol 3350. For cryoprotection, the HipABso:DNA crystals were first incubated for several minutes in the crystallization condition supplemented with 20% glycerol. We identified two crystal forms that diffracted to 3.4 Å and 3.8 Å respectively. The first crystal form diffracted to 3.4 Å, and belonged to space group P212121 with a = 57.8 Å, b = 122.5 Å, c = 189.9 Å and α = β = γ = 90°. The second crystal form in space group P21 with a = 72.24 Å, b = 57.33 Å, c = 171.38 Å and α = γ = 90°, β = 95.8° diffracted to 3.8 Å resolution. All X-ray diffraction data were processed with XDS and the XIA2 routine (26,27).
The structure of HipAso–AMPPNP–Mg2+ was determined via the molecular replacement platform BALBES (28), using the S. oneidensis MR-1 HipAso sequence and the crystal structure of E. coli HipA (PDB: 3DNU) structure as input. The structure was extended to completion using Phenix.AutoMR and refined using Phenix.Refine (29). The final HipA–AMPPNP–Mg2+ structure contains one molecule of HipA per asymmetric unit cell including all 433 residues from HipA and 5 residues from the affinity purification tag, 1 AMPPNP molecule, 2 Mg2+ ions, 1 Na+ ion and 486 water molecules.
To determine the structure of HipBso, the S. oneidensis MR-1 HipBso sequence and a subunit from the crystal structure of dimeric E. coli HipB (PDB: 3DNV) were used to create a truncated search model lacking loop regions and non-conserved side chains using CCP4 Chainsaw. The structure was determined using molecular replacement protocols implemented in MOLREP and Phenix.Phaser and was refined using Phenix.Refine (29,30). The final HipBso structure contains one dimer HipBso in the crystal asymmetric unit including residues 24–87 for one subunit and residues 17–87 for the second subunit, 123 water molecules in the dataset of 1.85 Å resolution and includes residues 17–87 for both subunits, and 39 water molecules in the dataset of 2.35 Å resolution.
The HipABso:DNA complex structure was determined by molecular replacement protocols implemented in Phenix.AutoMR using the HipA–AMPPNP monomer and the HipBso dimer as search models. The limited diffraction quality of crystal forms 1 and 2 for the HipABso:DNA complex necessitated the use of weak data at the diffraction limit of the crystals as indicated by half-dataset Pearson correlation coefficient (CC1/2) (31). Both the orthorhombic and monoclinic crystals contain two monomers of HipAso, one dimer of HipBso and double-stranded DNA in the asymmetric unit cell. After initial rounds of refinement, clear electron density could be observed for the DNA duplex that allowed modeling of the complete DNA sequence in COOT (32). The final HipABso:DNA structure contains two molecules of phosphorylated HipAso covering residues 7–433, one dimer of HipBso including residues 20–87 and 95–97, and one molecule of double-stranded DNA with 24 and 25 nucleotides in each strand.
Crystallographic coordinates and structure factors were deposited to the Protein Data Bank with access codes 4PU3, 4PU4, 4PU5, 4PU7 and 4PU8.
Nano liquid chromatography mass spectrometry
Freshly purified HipAso and HipAso treated overnight in vitro with 10 mM ATP, 20 mM MgCl2 were diluted 1:50 with 50 mM ammonium bicarbonate buffer. Sequencing-grade trypsin (Sigma) was added in 1:50 ratio to the diluted protein and digested overnight at 37°C. One picomole HipA digestion from those two samples was applied for LCMS analysis on a nanoACQUITY UPLC coupled to a Synapt mass spectrometer used in the Q-TOF mode (Waters). A nanoACQUITY UPLC@2G-V/M Trap column 108 μm * 20 mm 5 μ symmetry C18 (Waters) was used as the trap column at 15 μl/min for 1 min. A nanoACQUITY UPLC 1.8 μm HSS T3 75 μm * 250 mm column (Waters) was used as the separation column at 0.25 μl/min with a gradient as follows: 3% B to 40% B (0–30 min), 40% B to 85% B (30–31 min), 85% B (31–35 min), 85% B to 3% B (35–35.5 min), 3% B until 65 min using 0.1 formic acid as buffer A and 100% acetonitrile as buffer B. All the Nano LCMS data were processed with the PLGS 2.5 software (Waters).
Isothermal titration calorimetry
Experiments were carried out on a VP-ITC MicroCalorimeter at 25°C and data were analyzed using the origin ITC analysis software package (MicroCal). Purified HipA (WT or D306Q), HipB and DNA were matched into the buffer containing 25 mM Na2HPO4, 300 mM NaCl and 5% glycerol at pH 7.5. The concentrations of HipAso, HipBso and DNA were measured by a Nanodrop spectrophotometer using an extinction coefficient generated from ProtParam (Uniprot Knowledgebase). All samples were degassed extensively prior to the experiments. An initial injection of 2 μl was followed by 10 μl injection spaced 250 s or 350 s apart. The raw data were corrected and fitted to the one binding site model, and apparent molar reaction enthalpy (ΔH), apparent entropy (ΔS), dissociation constant (KD) and stoichiometry of binding (N) were generated.
Small angle X-ray scattering
Small angle X-ray scattering (SAXS) measurements of HipBso, HipBso:DNA and HipABso:DNA were carried out at Beamline SWING in SOLEIL (SOLEIL, Paris, France) with online HPLC separation (Shodex) in 25 mM HEPES pH 7.5, 300 mM NaCl and 5% glycerol buffer. Buffer subtraction and extrapolation were performed with the program FOXTROT or PRIMUS. The Guinier region was evaluated by PRIMUS using the Guinier approximation. The Porod volume was evaluated using PRIMUS; the radius of gyration Rg, forward scattering I0, maximum particle dimension Dmax and the distance distribution function P(r) were evaluated using GNOM. Theoretical scattering patterns from available structures and generated models, together with their respective discrepancy to the experimental data, were calculated using CRYSOL from ATSAS package (33,34). The molecular weight estimation was generated with the protocol described before (35). Modeling of the N- and C-termini of HipBso and HipBso:DNA was performed using crystal structures of HipBso from the 1.85 Å dataset of HipBso and HipBso:DNA from the 3.4 Å dataset of the HipABso:DNA complex. Five CORAL jobs were run for both HipB and HipBso:DNA using different seeds for each run, implying no symmetry (P1) and extending the N- and C-termini with 25 N-terminal and 10 C-terminal dummy residues for HipBso alone and in complex with DNA.
Bacterial growth experiments
Markerless in-frame deletion mutants in S. oneidensis were constructed essentially as previously reported using the suicide vector pNPTS138-R6KT (36,23). The primers used to generate the required N- and C-terminal fragments of the target regions (MR09/10/11/12 for hipBso, MR13/14/15/16 for hipAso, MR09/10/15/17 for hipBAso) are listed in Supplementary Table S1. The mutant alleles were generated by overlap PCR using appropriate primer combinations. Double cross-over mutants were selected as described, and were validated by colony PCR and sequencing of the target regions. Construction of the hipB deletion mutant failed, probably due to the toxicity of HipA. To obtain the overexpression vectors, hipAso, hipBso and hipABso were amplified from genomic DNA of S. oneidensis MR-1 (primers MR18 and MR21 for hipAso, MR19 and MR20 for hipBso and MR19, MR21 for hipABso) (Supplementary Table S1). The obtained PCR products were cloned into the pBAD202/D-TOPO vector (Invitrogen). Subsequently, the thioredoxin sequence was removed by digestion with NcoI, gel purification and re-ligation, producing the pBADHipAso, pBADHipBso and pBADHipABso vectors, respectively. Plasmid DNA was introduced into S. oneidensis MR-1 cells by electroporation using a 2.5 kV pulse. All mutants were confirmed by colony PCR using the appropriate primers. S. oneidensis strains were routinely grown on LB agar plates or shaking (200 rpm) in LB medium at 28°C (media supplemented appropriately with carbenicillin (Cb) 100 μg/ml or kanamycin (Km) 25 μg/ml). For the growth curves, overnight grown precultures were diluted 1:20 to 1:30 to an optical density at 600 nm (OD600nm) of ∼0.3. Aerobic growth was monitored directly, without the need to transfer culture samples into regular cuvettes, using a Shimadzu 1240 Mini Single-Beam UV-Vis spectrophotometer. Each growth curve was derived from three independent experiments for which the mean (±SEM) is plotted.
Radioactive kinase assay to probe HipAso activity
A standard radioactive kinase assay was performed in 11 μl of reaction buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 25 mM β-glycerophosphate, 1 mM Na3VO4, 100 μM ATP and 100 μM MgCl2) in the presence of 10 μCi of [γ-32P]ATP and the purified HipAso and EF-Tuso. The reaction was allowed to incubate for 15 min at room temperature and quenched by the addition of 1/5 volume of sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. The 32P labeled proteins were separated by SDS-PAGE, followed by autoradiography of the gels. As a negative control, a reaction without 32P and a reaction with bovine serum albumin were included.
RT-PCR experiments
Total RNA was isolated from a 0.5 ml late-exponential phase culture (OD600nm = 0.9) of WT S. oneidensis MR-1 using the RNeasy Plus Mini kit in combination with the RNAprotect Bacteria Reagent (Qiagen) (yield: 33 μg total RNA). One microgram of total RNA sample was treated with RNase-free DNase I and Protector RNase Inhibitor (Roche). First strand cDNA was synthesized using random hexamer primers and the Transcriptor First Strand cDNA Synthesis Kit (Roche). Target transcripts were PCR amplified using HotGoldStar polymerase (Eurogentech) (100 ng DNA template; 4 μM primers). PCR products were resolved on an agarose gel.
RESULTS
HipAso and HipBso constitute a toxin–antitoxin module with an atypical operator DNA
Transposon mutagenesis of SO0706 led to a strong reduction of biofilm formation in S. oneidensis MR-1 (17). Sequence analysis revealed that the protein product of SO0706 (HipAso) shows clear homology to E. coli HipA with 28% sequence identity (Supplementary Figure S1). The protein product of the adjacent gene, SO0705 (HipBso), is annotated as a toxin–antitoxin antidote transcriptional repressor (20). Together with the rather low sequence identity between the S. oneidensis and E. coli HipA–HipB sequences, there are also striking differences in the operon structure, which may reflect the functional diversity of such toxin–antitoxin systems. For instance, the SO0705 and SO0706 genes are separated by 3 bp instead of overlapping by 1 bp as seen in the hipAB operon structure in E. coli. Furthermore, a palindromic operator sequence GTGTA(N)6TACAC is repeated four times upstream of the hipABso gene. We note that the random nucleotide sequence within the operator is 2 bp shorter than in the E. coli HipAB system. Interestingly, the fourth operator is located inside the HipBso open reading frame and the spacing between the four operators is different. While in S. oneidensis the operators are equally separated by 34 bp, in E. coli they are separated by 10, 21 and 10 bp, respectively (Supplementary Figure S2a).
To investigate the function of HipAso and HipBso we used S. oneidensis MR-1 cDNA to isolate the genes for HipBso, HipAso, HipABso and its operator DNA. By employing RT-PCR on total RNA using primer pairs designed to span the entire region of HipBso and HipAso, we were able to show that the two genes are transcriptionally coupled (Supplementary Figure S2b). We established complementation and overexpression mutants to study the HipABso physiological function and its role in persistence. To this end, we constructed a series of vectors based on the pBAD vector which was previously shown to allow well-controlled expression to study the toxic effects of HipA (15). Overexpression of HipAso in S. oneidensis MR-1 causes a temporary inhibition of growth in both the wild type and the ΔhipABso strain (Supplementary Figure S2c), an effect that is tempered when also HipBso is overexpressed. However, when HipAso was overexpressed in the ΔhipABso strain growth inhibition was acute, consistent with similar observations in E. coli (10). This strongly suggests that mild overexpression of HipAso only causes a temporary inhibition but not termination of growth (37). Surprisingly, overexpression of the antitoxin HipBso has the same inhibitory effect on growth as expression of toxin HipAso. This could be explained by possible cross talk of HipB with other systems involved in transcriptional repression by analogy to recent findings in E. coli whereby HipB was shown to regulate multiple promoter sequences besides the hipAB module (38).
To enable studies toward obtaining structural and mechanistic insights into the role of HipAso and HipBso in persistence, we overexpressed and purified recombinant HipAso and HipBso. Elution profiles during SEC showed that HipAso could be isolated as a monomer with an apparent molecular weight of 50 kDa, while HipBso could be purified as a dimer with an apparent molecular weight of 30 kDa. Simple mixing of the proteins resulted in the formation of a complex between HipAso and HipBso with an apparent molecular mass of 130 kDa, consistent with 2:2 stoichiometry. Furthermore, the HipABso complex can bind a single operator DNA sequence to form a complex with a distinct chromatographic behavior (Figure 1a). Molecular weights obtained for HipBso, HipBso:DNA HipABso and HipABso:DNA by SAXS correlate well with the apparent sizes estimated from the retention volumes in SEC (Supplementary Table S2).
Figure 1.
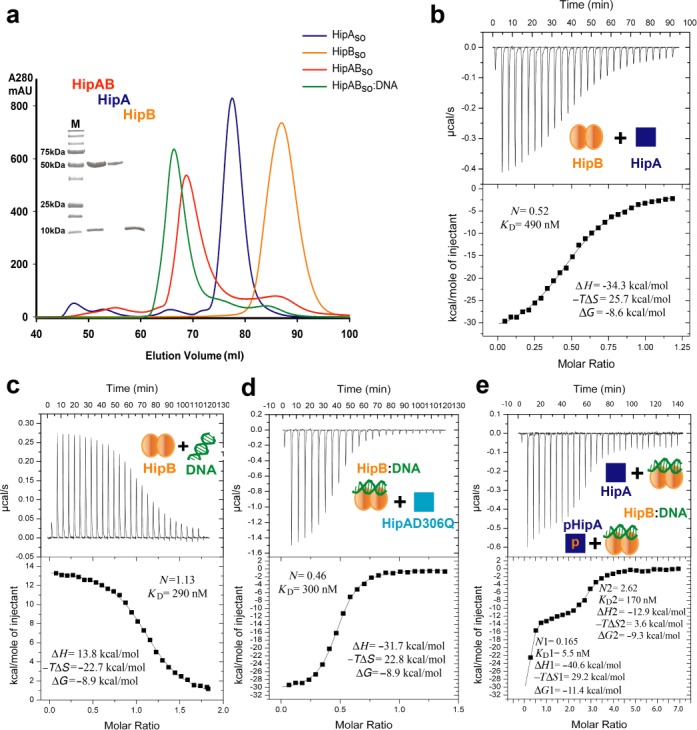
Biochemical reconstitution and characterization of HipBso (SO0705) and HipAso (SO0706) complexes. (a) SEC profiles of HipBso (orange), HipAso (blue), HipABso complex (red) and HipABso:DNA complex (green). SDS-PAGE analysis of peak fraction is accompanied with the SEC profile. (b) Thermodynamic characterization of the interaction between HipAso and HipBso. (c) Thermodynamic characterization of the interaction between HipBso and single hipBASO operator DNA. (d) Thermodynamic characterization of the interaction between the HipBso:DNA complex and the non-phosphorylated active site mutant HipAsoD306Q. (e) Thermodynamic characterization of the interaction between the HipBso:DNA complex and recombinant HipAso as a mixture of phosphorylated (∼10%) and non-phosphorylated HipAso. The derived interaction stoichiometry (N) and equilibrium dissociation constant (KD) are shown together with ΔH, −ΔTS and ΔG.
The interaction between HipAso and HipBso, the binding of HipBso to operator DNA, and the recruitment of HipAso to the HipBso:DNA complex were quantified using Isothermal Titration Calorimetry (ITC) (Figure 1b–e). Two monomers of HipAso bind one dimer of HipBso with an equilibrium dissociation constant (KD) of 490 nM via an enthalpically driven binding event (Figure 1b). On the other hand, the interaction of HipBso with operator DNA (26 bp) is endothermic, resulting in an entropically driven binding event to establish a high-affinity interaction (KD = 290 nM) (Figure 1c). Titration of double operator DNA (76 bp) with HipBso revealed a similar affinity and thermodynamic fingerprint indicating that there is no cooperativity between adjacent operators in HipBso binding (Supplementary Table S3 and Supplementary Figure S3a). Finally, we characterized the recruitment of HipAso to a HipBso:DNA complex. For this study, we employed preformed HipBso:DNA complex and HipAso carrying a D306Q mutation and found that HipAso_D306Q can bind to the HipBso:DNA complex with a KD of 300 nM and a thermodynamic binding profile that is very similar to the HipA:HipB interaction in the absence of DNA (Figure 1d and Supplementary Table S3). The rationale behind employing the HipASO_D306Q mutant for these experiments stemmed from the fact that our preparations of recombinant HipA contained a mixture of non-phosphorylated and phosphorylated HipA, which we estimated by mass spectrometry to be about 10%. However, we were neither able to establish protein expression conditions leading to a stable ratio of non-phosphorylated versus phosphorylated HipA, nor a purification protocol that would allow isolation of the two species for further biochemical and biophysical characterization. Thus, we resorted to the HipAD306Q mutant, which is devoid of autophosphorylation activity, thereby ensuring the homogeneity of our preparations with respect to non-phosphorylated HipA. For comparison purposes and to obtain insights into the possible binding behavior of phosphorylated HipA, we investigated the binding of a freshly prepared HipA to a preassembled HipBso:DNA complex by ITC (Figure 1e). Remarkably, the resultant binding isotherm showed drastically different features when compared to the binding profile of non-phosphorylated HipAD306Q mutant. The data revealed two distinct binding events and could be fitted with a ‘two sets of binding sites’ model. A minority of the pool recombinant HipA molecules consistent with the approximate proportion of phosphorylated HipA bound with high affinity to HipB:DNA (KD = 5.5 nM), while a second pool bound with the expected stoichiometry and a moderately high affinity (KD = 170 nM). Notably, the latter is consistent with the affinity of non-phosphorylated HipAD306Q (KD = 300 nM) for the HipB:DNA complex (Figure 1d). Thus, the two experiments now suggest that phosphorylated HipA binds with a markedly higher affinity to HipB:DNA than non-phosphorylated HipA. In addition, this indicates that the phosphorylation state of HipA bestows high-affinity binding only when HipB is bound to operator DNA, as evidenced by the fact that the thermodynamic binding profile for the HipB:HipA interaction (Figure 1b) closely resembles that for the HipB:HipAD306Q complex (Figure 1d). Together, our data point to the possible functional dichotomy of phosphorylated and non-phosphorylated pools of HipA.
HipAso, HipBso and operator DNA assemble into a distinct ternary complex
The structure of the HipABso:DNA ternary complex was determined in two crystal forms at 3.4 Å and 3.8 Å resolution (Table 1). The S. oneidensis MR-1 HipABso:DNA ternary complex assembles as a compact HipBso dimer bound by a 25 bp duplex operator DNA, with each of the HipBso subunits interacting with a separate HipAso subunit (Figure 2a). Unexpectedly, the overall HipABso:DNA complex assembly is remarkably distinct from the previously characterized E. coli HipABso:DNA ternary complex (PDB:3DNV) (14) (Figure 2b). Notably, E. coli HipA uses both its N- and C-terminal domains to contact the entire side of the HipB dimer, without making any contacts with operator DNA. Thus, viewed from the top of the DNA double helix, HipAso emerges as a toxin that employs dramatically different structural principles to engage in a ternary complex with HipB:DNA compared to the prototypic counterpart in E. coli (Figure 2a and b). We have cross-validated the observed configuration of the HipABso:DNA complex by SAXS experiments of the complex in solution, which revealed that the crystallographically observed complex agrees well with the scattering data in solution. At the same time, a model based on the E. coli HipAB:DNA complex configuration strongly contradicts our SAXS data (Figure 2c). Together this analysis lends orthogonal support to the crystallographically observed HipABso:DNA complex.
Table 1. X-ray data collection and refinement statistics.
| Crystal | HipAB:DNA crystal form 1 | HipAB:DNA crystal form 2 | HipA:AMPPNP:Mg2+ | HipB Dataset 1 | HipB Dataset 2 |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P212121 | P21 | P212121 | C2221 | C2221 |
| a, b, c (Å) | 57.8, 122.5, 189.9 | 72.2, 57.3, 171.4 | 60.7, 75.8, 110.3 | 53.5, 72.9, 86.9 | 53.6, 71.6, 85.8 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 95.8, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 49.4–3.4 (3.5–3.4) | 47.6–3.8 (3.9–3.8) | 44.6–1.8 (1.9–1.8) | 43.1–1.85 (1.95–1.85) | 38.36–2.35(2.43–2.35) |
| Rmeasa | 0.260 (1.112) | 0.281 (1.051) | 0.115 (0.911) | 0.074 (0.761) | 0.147 (0.935) |
| CC(1/2) | 0.989 (0.673) | 0.983 (0.538) | 0.999 (0.904) | 0.998 (0.742) | 0.995 (0.744) |
| I/σ(I) | 6.78 (1.81) | 4.40 (1.21) | 19.96 (3.17) | 11.66 (1.68) | 11.21 (2.15) |
| Completeness (%) | 98.93 (92.72) | 97.03 (84.10) | 99.43 (95.29) | 99.46 (99.20) | 99.71 (97.55) |
| Multiplicity | 6.5 (6.4) | 3.4 (2.9) | 13.1 (12.5) | 4.5 (4.4) | 6.3 (6.3) |
| Wilson B-factor (Å2) | 75.5 | 93.1 | 21.3 | 31.0 | 40.1 |
| Refinement | |||||
| Total reflections | 124924 (11122) | 46748 (3379) | 592535 (53603) | 67170 (6306) | 45147 (4242) |
| Unique reflections | 19211 (1732) | 13858 (1164) | 45100 (4273) | 14778 (1440) | 7125 (676) |
| Rwork/Rfree | 0.256/0.315 | 0.236/0.306 | 0.156/0.175 | 0.186/0.216 | 0.204/0.223 |
| Number of atoms: | |||||
| Protein | 8805 | 8763 | 3466 | 1019 | 1070 |
| Ligand or ion | - | - | 34 | ||
| Water | 486 | 123 | 39 | ||
| Average ADP (Å2) | 46.4 | 80.3 | 27.2 | 33.9 | 49.2 |
| Protein ADP (Å2) | 46.4 | 80.3 | 26.4 | 33.2 | 49.2 |
| Ligand ADP (Å2) | - | - | 11.6 | ||
| Water ADP (Å2) | 33.8 | 39.5 | 47.6 | ||
| Ramachandran Plot | |||||
| Favored/Allowed (%) | 98/2 | 97/3 | 98/2 | 99/1 | 99/1 |
| Root-mean-square deviations: | |||||
| Bond lengths (Å) | 0.003 | 0.005 | 0.005 | 0.004 | 0.004 |
| Bond angles (°) | 0.80 | 0.95 | 0.95 | 0.68 | 0.88 |
aRmeas = ∑h√nh/(nh−1) ∑h∑i|I(h,i)−I(h)>| / ∑h∑iI(h,i), where nh is the multiplicity, I(h,i) is the intensity of the ith measurement of reflection h, and I(h)> is the average value over multiple measurements. Statistics for the highest resolution shell are shown in parentheses.
Figure 2.
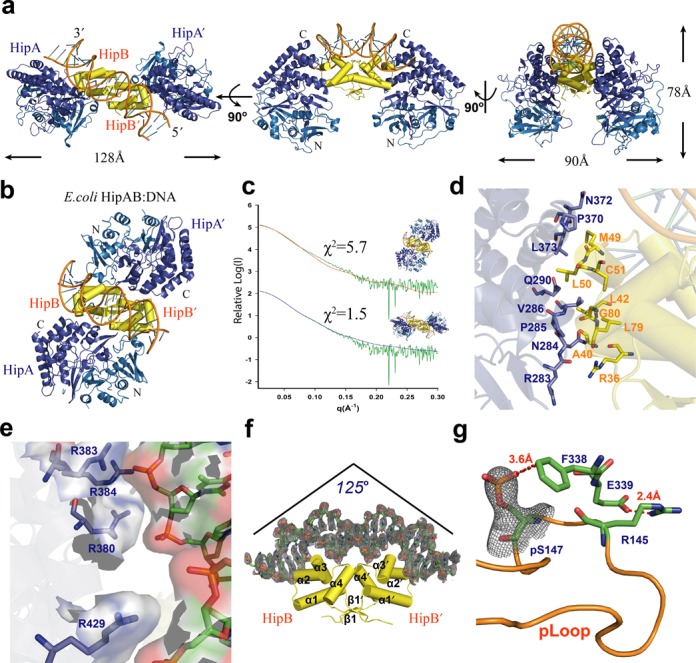
Structural studies of the HipABso:DNA complex. (a) Crystal structure of the HipABso:DNA complex shown in different orientations. A dimer of HipBso (yellow) interacts with the C-terminal helical bundles (dark blue) of two HipAso; the N-terminus (light blue) of HipAso does not take part in the interaction with HipBso. (b) X-ray crystal structure of the E.coli HipAB:DNA complex (PDB: 3DNV) in top view oriented based on a superposition with duplex DNA and HipBso shown in panel (a). (c) SAXS analysis of the HipABso complex and comparison with the crystal structure of HipABso (χ2 = 1.5) and that of E. coli HipAB (χ2 = 5.7). (d) Detail of the residues involved in the interaction between HipAso and HipBso. (e) Detail of the interaction interface between HipBso and single hipABso operator DNA. A positively charged patch on HipAso defined by R380, R383, R384 and R429 forms a complementary interaction site with the negatively charged phosphodiester backbone of the operator DNA. Further interaction details are given in Supplementary Figure S3b. (f) Bending of the operator DNA upon interaction with the helix-turn-helix motif of HipBso. (g) Structural detail of the interactions stabilizing the phosphorylated pLoop of HipAso in the ternary complex.
The HipA–HipB binding interface in the HipABso:DNA ternary complex is mainly hydrophobic and features interactions between the C-terminal helical-bundle domain of HipA and a hydrophobic patch on each HipB subunit creating an interface covering 470 Å2 as determined by the PDB PISA server (39) (Figure 2d). Unexpectedly, in addition to the anticipated extensive interactions of HipB with DNA, HipA also makes specific interactions with the phosphodiester flank of the DNA duplex via a positively charged patch defined by R380, R383 and R384 and R429 covering an interface of ∼150 Å2 (Figure 2e). HipB interacts tightly with its operator DNA using mainly helices α2 and α3 and part of the α4 helix forming a helix-turn-helix DNA binding motif (Figure 2f and Supplementary Figure S3b). Interestingly, binding of duplex DNA to the HipABso complex causes the DNA to bend by about 125° compared to linear DNA, which is 15° more than the bent observed in the interaction of E. coli HipB with operator DNA. A possible reason for this difference may lie in the fact that the inter-palindromic distance between each operators’ binding site is 2 bp shorter than in E. coli (Figure 2f).
Arguably the most surprising feature of the HipABso:DNA complex is the fact that HipAso is phosphorylated at Ser147 (Figure 2g). The phosphate group of pSer147 is observed at full occupancy in both crystal forms of the HipABso:DNA complex and is anchored by an anion-π interaction with F338. This binding mode is further stabilized by a hydrogen bond between main-chain atoms of R145 and F338, and a salt-bridge between E339 and R145 (Figure 2g). This serendipitous structural observation, which may well have been the result of protein crystallization favoring the more stable, albeit much less abundant, ternary complex of phosphorylated HipAso with HipBso:DNA (Figure 1e), offers new functional considerations about the possible interplay between phosphorylated and non-phosphorylated pools of HipAso. Our analysis by SAXS of recombinant HipABso:DNA in solution (Figure 2c), which on average may engage up to 10% phosphorylated HipASO, reveals that the phosphorylated and non-phosphorylated assemblies are overall structurally very similar, and further indicates that the structural basis for the apparent higher affinity of phosphorylated HipAso for HipBso:DNA will have to await further structural characterization of non-phosphorylated HipAso complexes at high resolution.
The flexible C-terminal tail of HipBso becomes ordered upon binding to HipAso
An additional striking difference between E. coli and S. oneidensis HipAB:DNA complexes concerns the structural order of the C-terminal segment of HipB. We posit that this could have functional implications in terms of the post-translational regulation. E. coli HipB also displays structural disorder at its C-terminal end in the HipAB:DNA complex and it has been demonstrated that the C-terminal tryptophan is proteolytically cleaved by the ATP-dependent Lon protease (40). Interestingly, in our structure of HipABso:DNA at 3.8 Å resolution, we can trace the C terminus of HipBso into a hydrophobic pocket of HipAso. Moreover, in our second structure of HipABso:DNA to 3.4 Å resolution, we see clear density in the same pocket for the last three C terminal residues (Gly95, Trp96 and Tyr97) of HipBso (Figure 3a and Supplementary Figure S3c).
Figure 3.
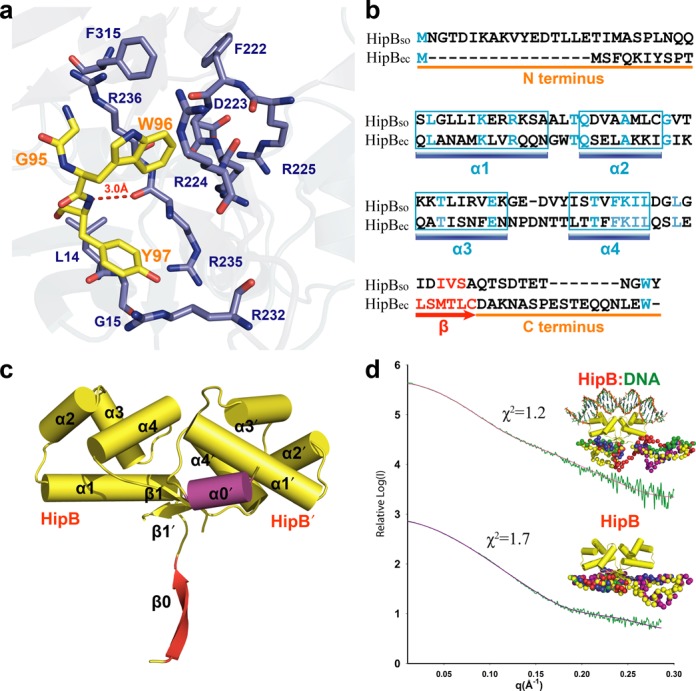
The flexible C-terminal tail of HipBso is accommodated by a hydrophobic pocket in HipAso. (a) Structural detail of the interaction site between HipBso (yellow) and HipAso (blue). (b) Structure-based sequence alignment of S. oneidensis and E. coli HipB (18% identity) showing the conserved W96 residue. (c) Crystal structure (2.35 A) of the HipBso dimer showing the different conformations of the N-termini forming a α0′ helix and β0 sheet, respectively. (d) SAXS modeling showing both HipBso and HipBso:DNA complex have a disordered N- and C-terminus. Five CORAL models, aligned with the N- and C-termini, are colored differently and shown in sphere representation.
As it is widely accepted that the protease sensitivity of the antitoxins provides a mechanism for the regulation of toxin activity (40–42) and given that the C-termimal region of HipBso is drastically different from its counterpart in E. coli (Figure 3b), we pursued structural studies of HipBso outside the context of the ternary complex with HipAso and DNA to obtain insights into the plasticity and structural order of the C-terminus in HipBso. We crystallized HipBso and determined two crystal structures to 1.85 Å and 2.35 Å resolution, respectively (Table 1 and Figure 3c). The electron density for the C-terminal segment in HipBso in both crystal structures did not allow structural modeling, indicative of inherent flexibility in this region in the absence of a binding partner. To obtain additional structural insights into this feature of HipBSO, we measured SAXS data for HipBso alone and HipBso in complex with operator DNA. Modeling of the SAXS data in both cases using the crystal structure of HipBso and dummy atoms for the missing C-terminal residues show that, while HipB retains its compact helical structure, the C-terminus could be modeled as an extended structure protruding outward (Figure 3d). Thus, the C-terminus of HipBso appears to be extended and flexible in the context of HipBso and its binary complex with DNA, while it adopts a more defined structure in the HipABso:DNA complex.
Our modeling of SAXS data for HipBso also provided the opportunity to validate the structural variability of the unique N-terminal region of HipBso (Figure 3b), which adopts two different secondary structures in our structure of HipBso to 2.35 Å resolution, namely a β0 strand in one subunit and α0′ helix in the other (Figure 3c). We note that this region could not be modeled in the ternary complex of HipBso with HipA and DNA due to lack of clear electron density. In solution, however, the N-terminal segment of HipBso could only be modeled as an extended structure analogous to the C-terminal region (Figure 3d). To investigate the possible functional implications of this N-terminal segment and given that it is completely absent in HipBso of E. coli, we produced recombinant HipBsolacking the 16 residues at N-terminus and showed by ITC experiments that its binding behavior to HipAso is indistinguishable from full-length HipBso (Supplementary Figure S3a).
The conformation of HipAso is modulated by autophosphorylation
In E. coli, HipA kinase activity is essential for growth arrest and MDT (12). To further investigate the mechanism of HipAso in S. oneidensis MR-1, we determined the structure of HipAso in complex with the non-hydrolysable ATP analogue AMPPNP and Mg2+ to 1.83 Å resolution (Table 1). This structural analysis at high resolution revealed binding of AMPPNP and Mg2+ at high occupancy and delineation of the protein-nucleotide interaction landscape. The ATP binding site in HipAso is engulfed by the N- and C-terminal domains and most interacting residues are contributed by five loop regions (Figure 4a and Supplementary Figure S4a). All residues involved in Mg2+ and ATP binding are highly conserved in S. oneidensis MR-1 and E. coli (Figure 4a and Supplementary Figure S1). Given that the physiological ATP concentration in the cell is enough to drive the autophosphorylation of HipA, we propose that phosphorylation of HipAso is essential for the formation of the ternary HipABso:DNA complex. The HipAso pLoop consists of residues 129–152 and is ejected out of the pocket compared to the non-phosphorylated form (Figure 4b). Furthermore, the pLoop in autophosphorylated HipAso, as observed in the HipABso:DNA complex, projects to the surface. Such activation mechanism involving ejection of the pLoop upon phosphorylation has also been proposed for E. coli HipA (43). This could be a common feature for the HipA kinase family. To investigate the autophosphorylation capacity of HipAso, we incubated freshly purified HipAso with ATP and Mg2+ overnight and observed a dramatic increase of the corresponding phosphorylated peptide intensity from approximately 5% to almost 45% relative to the signal of the non-phosphorylated peptide in LCMS (Figure 4c).
Figure 4.
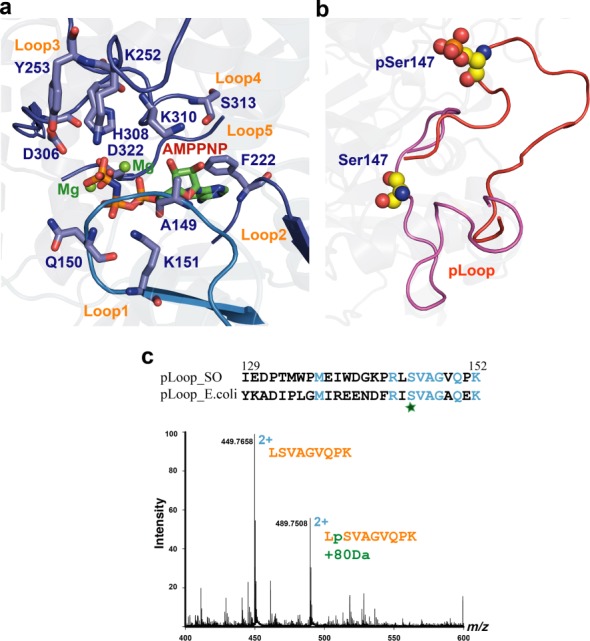
S. oneidensis MR-1 HipAso is regulated conformationally upon autophosphorylation. (a) Structural detail of the AMPPMP (atom colored) and Mg2+ (green spheres) binding site in HipAso (blue). Residues involved are shown in stick representation. (b) Detailed view of the ejection of the pLoop of HipAso upon autophosphorylation. Ser147 and phosphate represented as spheres. (c) Identification of the phosphorylated peptide by nanoLCMS. The peptide LSVAGVQPK was observed at charge state 2+ in two forms differing by 80 Da in molecular weight. Sequence alignment of the pLoop regions in S. oneidensis MR-1 and E. coli.
In E. coli, HipAso is autophosphorylated at Ser150. Similarly, our structural analysis revealed that in S. oneidensis MR-1, HipA can be autophosphorylated at Ser147 which aligns with Ser150 in E. coli HipA. We note that both our crystal forms of the HipABso:DNA complex contain phosphorylated HipA, in sharp contrast to the E. coli HipAB:DNA complex (43,44). Upon superposition of the N-terminal domains of phosphorylated and non-phosphorylated HipAso (derived from the AMPPNP complex) we observed a rigid-body shift of the HipAso C-terminal domain by about 2 Å (Supplementary Figure S4c). Given that this part of HipAso interacts with DNA in the ternary complex with HipBso, we propose that the phosphorylated form of HipAso may be important in the complex assembly and stability.
HipAso neither binds nor phosphorylates EF-Tuso
HipA was the first toxin protein to be linked to the phenomenon of persistence and its overexpression in E. coli resulted in growth arrest, inhibition of protein synthesis, as well as DNA replication and transcription (7,15,12,37). These multiple cellular effects at the molecular level could be traced to the kinase activity of HipA (12). EF-Tu was originally identified as the substrate of the kinase activity of HipA in E. coli, thereby creating a functional link between HipA and the inhibition of translation (14). However, repeated attempts to show that HipAso has an analogous activity failed, leading to the conclusion that EF-Tuso does most likely not constitute a substrate for HipAso (Supplementary Figure S5a). In an in vitro radioactive kinase assay, we could show that HipAso is indeed autophosphorylated, while the active site mutant HipAso D306Q is not. However, no phosphorylation of EF-Tuso could be detected when purified EF-Tuso with C-terminal His-tag or N-terminal glutathione S-transferase (GST) tag was added. Furthermore, ITC and pull-down experiments with GST-tagged EF-Tuso indicated the absence of interaction between these proteins thereby corroborating the functional assay (Supplementary Figure S5b).
DISCUSSION
We here provide structural insights into the toxin–antitoxin system HipA-HipB and its operator DNA assembly in S. oneidensis MR-1, which can now serve as a second example of such complexes next to the previously characterized E. coli HipAB:DNA complex. The S. oneidensis MR-1 HipAB:DNA assembly is sharply distinct from the one in E. coli, providing new insights into the structural and mechanistic diversity of HipAB modules and cautioning against the use of a single model organism for deriving functional generalizations. In E. coli, the phosphorylation of HipA evokes pLoop ejection and is thought to release HipA from HipB in the HipAB:DNA complex (43). However, we have now provided direct structural and biophysical evidence that phosphorylated HipASO can engage in the HipABso:DNA complex form with higher affinity that non-phosphorylated HipASO, and further, that the phosphorylation site is secured in place via anion–π interactions. This suggests that HipB may play an active role in recruiting phosphorylated HipA to avoid growth arrest. We also observed that in the absence of HipA, HipB is structurally destabilized, which may render HipB more susceptible to proteolytic degradation, thereby propagating the effect of HipA-regulated persistence. This raises new questions about the possible molecular trigger for the release of HipA from HipB given the observation that pLoop ejection does not appear to do so.
Understanding MDT and persistence in bacteria is critical for the development of eradication strategies against persisters, and may thus impact therapeutic options for chronic infections. Besides HipBA systems, several other molecular factors have been shown to contribute to bacterial persistence (45,46), and it has been proposed that the level of toxin controls the formation of persister subpopulations in a threshold-based manner (47). In fact, TA modules are tightly regulated based on a complex molecular interplay (48). In particular, a number of type II TA systems exhibit ‘conditional cooperativity’, a term coined to describe the transcriptional repression of the TA system depending on the ratio of toxin to antitoxin (49–51). For instance, conditional cooperativity was reported to stabilize antitoxin levels in rapidly growing cells, preventing the random activation of toxin and promoting fast translational recovery (52), and in Doc-Phd or RelA-RelB toxin-antitoxin systems it is essential for keeping a storage pool of the toxin to generate type I persistence (49,53). However, thus far conditional cooperativity has not been demonstrated for the HipAB module in persister cell formation. Furthermore, the simple threshold-based models are still far from satisfactory because they ignore key aspects of the HipAB autoregulation pathway, such as autophosphorylation of HipA and its possible effects on reverting growth arrest (47,54). In this regard, we envisage that the identification of the effective population of HipA involved in HipB binding will be key to further understand bacterial persistence and to develop bistability models (49,53). Furthermore, it will be crucial to verify the conformational state of HipA involved in the ternary complex with HipB and operator DNA leading to transcriptional suppression. In particular, the involvement of phosphorylated HipA in the HipAB:DNA complex hints that by characterizing the equilibrium between the different conformational states of HipA in a cellular context one may be able to rationalize storage of HipA along the lines of the conditional cooperativity mechanism proposed for other type II toxin–antitoxin systems.
We have further shown that EF-Tu is unlikely to be the substrate of toxin HipA in the case of S. oneidensis, which is in agreement with recent findings in E. coli. While it was originally believed that HipA halted ribosomal activity by phosphorylating EF-Tu (14,43), it was recently established that its natural substrate is actually GltX. By phosphorylating this enzyme at its ATP binding site, uncharged tRNAGlu accumulates, leading to the activation of the stringent response. This induces degradation of the antitoxins by Lon protease, which finally leads to conversion of the cell to the persister state (13,55). However, translational inhibition alone is not sufficient to explain the macromolecular synthesis inhibition triggered by HipA (16).
Mining the S. oneidensis MR-1 genome database and the Toxin-Antitoxin database (TADB) revealed that multiple homologues of the HipAB toxin–antitoxin system exist in S. oneidensis MR-1 (56). Besides SO0705 (HipBso) and SO0706 (HipAso), SO3169, SO3170 and SO0062, SO0063 are also annotated as members of the HipAB toxin–antitoxin system. Interestingly, despite the low sequence identity (15–20%) between the predicted HipA-like proteins SO3170, SO0063 and SO0706, the residues involved in HipA kinase activation, in terms of Mg2+ and ATP binding and the Ser phosphorylation site, are highly conserved (Supplementary Figure S6). Although there is only a low sequence identity between E. coli and S. oneidensis HipA, their structure is highly similar and they share a kinase activation mechanism characterized by a conformational change of the pLoop. Moreover, upstream of the gene encoding for one of these HipA homologues there is a gene encoding for a HipB-like protein. Thus, it appears that there might be functional redundancy of such proteins in S. oneidensis, which may complicate future studies in delineating the exact role of HipA in this organism.
ACCESSION NUMBERS
PDB access codes: 4PU3, 4PU4, 4PU5, 4PU7 and 4PU8.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank the European Synchrotron Radiation Facility (ESRF), SOLEIL, DESY-PETRA3-EMBL, and the Swiss Light Source (SLS) for synchrotron beam time allocation, and the staff of beamlines ID29 (ESRF), Proxima and Swing (SOLEIL), P13 and P14 (PETRA3-EMBL), and PX1 and PX3 (SLS) for technical support.
Footnotes
Present address: Ester Behiels and Jonathan Elegheert, Division of Structural Biology, Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, OX3 7BN Oxford, UK.
FUNDING
Chinese Scholarship Council (CSC) and Ghent University [joint research fellowship to Y.W.]; Research Foundation Flanders, Belgium (FWO) [research fellowships to E.B., J.E., J.F. and B.V.]; FWO; GOA-Ghent University research grant [to S.N.S. and B.D.]; Hercules Foundation grants for research infrastructure [to B.D. and S.N.S.]; Belgian Science Policy office [to B.D., via interuniversity attraction pole project P7/44]. Source of open access funding: GOA-Ghent University research grant.
Conflict of interest statement. None declared.
REFERENCES
- 1.Avery S.V. Microbial cell individuality and the underlying sources of heterogeneity. Nat. Rev. Microbiol. 2006;4:577–587. doi: 10.1038/nrmicro1460. [DOI] [PubMed] [Google Scholar]
- 2.Hersh M.N., Ponder R.G., Hastings P.J., Rosenberg S.M. Adaptive mutation and amplification in Escherichia coli: two pathways of genome adaptation under stress. Res. Microbiol. 2004;155:352–359. doi: 10.1016/j.resmic.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 3.Acar M., Mettetal J.T., van Oudenaarden A. Stochastic switching as a survival strategy in fluctuating environments. Nat. Genet. 2008;40:471–475. doi: 10.1038/ng.110. [DOI] [PubMed] [Google Scholar]
- 4.Lewis K. Persister cells. Annu. Rev. Microbiol. 2010;64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 5.Balaban N.Q., Gerdes K., Lewis K., McKinney J.D. A problem of persistence: still more questions than answers. Nat. Rev. Microbiol. 2013;11:587–591. doi: 10.1038/nrmicro3076. [DOI] [PubMed] [Google Scholar]
- 6.Lewis K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013;12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 7.Moyed H.S., Bertrand K.P. HipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983;155:768–775. doi: 10.1128/jb.155.2.768-775.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bigger J.W. Treatment of staphylococcal infections with penicillin. Lancet. 1944;244:497–500. [Google Scholar]
- 9.Moyed H.S., Broderick S.H. Molecular cloning and expression of hipA, a gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1986;166:399–403. doi: 10.1128/jb.166.2.399-403.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Black D.S., Irwin B., Moyed H.S. Autoregulation of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J. Bacteriol. 1994;176:4081–4091. doi: 10.1128/jb.176.13.4081-4091.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falla T.J., Chopra I. Joint tolerance to beta-lactam and fluoroquinolone antibiotics in Escherichia coli results from overexpression of hipA. Antimicrob. Agents Chemother. 1998;42:3282–3284. doi: 10.1128/aac.42.12.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Correia F.F., D'Onofrio A., Rejtar T., Li L., Karger B.L., Makarova K., Koonin E.V., Lewis K. Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. J. Bacteriol. 2006;188:8360–8367. doi: 10.1128/JB.01237-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Germain E., Castro-Roa D., Zenkin N., Gerdes K. Molecular mechanism of bacterial persistence by HipA. 2013;52:248–254. doi: 10.1016/j.molcel.2013.08.045. [DOI] [PubMed] [Google Scholar]
- 14.Schumacher M.A., Piro K.M., Xu W., Hansen S., Lewis K., Brennan R.G. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science. 2009;323:396–401. doi: 10.1126/science.1163806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korch S.B., Henderson T.a., Hill T.M. Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol. 2003;50:1199–1213. doi: 10.1046/j.1365-2958.2003.03779.x. [DOI] [PubMed] [Google Scholar]
- 16.Bokinsky G., Baidoo E.E.K., Akella S., Burd H., Weaver D., Alonso-Gutierrez J., García-Martín H., Lee T.S., Keasling J.D. HipA-triggered growth arrest and β-lactam tolerance in Escherichia coli is mediated by RelA-dependent ppGpp synthesis. J. Bacteriol. 2013;195:3173–3182. doi: 10.1128/JB.02210-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Theunissen S., De Smet L., Dansercoer A., Motte B., Coenye T., Van Beeumen J.J., Devreese B., Savvides S.N., Vergauwen B. The 285 kDa Bap/RTX hybrid cell surface protein (SO4317) of Shewanella oneidensis MR-1 is a key mediator of biofilm formation. Res. Microbiol. 2010;161:144–152. doi: 10.1016/j.resmic.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Hau H.H., Gralnick J.A. Ecology and biotechnology of the genus Shewanella. Annu. Rev. Microbiol. 2007;61:237–258. doi: 10.1146/annurev.micro.61.080706.093257. [DOI] [PubMed] [Google Scholar]
- 19.Lovley D.R. Electromicrobiology. Annu. Rev. Microbiol. 2012;66:391–409. doi: 10.1146/annurev-micro-092611-150104. [DOI] [PubMed] [Google Scholar]
- 20.Heidelberg J.F., Paulsen I.T., Nelson K.E., Gaidos E.J., Nelson W.C., Read T.D., Eisen J.A., Seshadri R., Ward N., Methe B., et al. Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat. Biotechnol. 2002;20:1118–1123. doi: 10.1038/nbt749. [DOI] [PubMed] [Google Scholar]
- 21.Thormann K.M., Saville R.M., Shukla S., Dale A., Spormann A.M., Saville M., Pelletier D.A. Initial phases of biofilm formation in Shewanella oneidensis MR-1. J. Bacteriol. 2004;186:8096–8104. doi: 10.1128/JB.186.23.8096-8104.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chao L., Rakshe S., Leff M., Spormann A.M. PdeB, a cyclic Di-GMP-specific phosphodiesterase that regulates Shewanella oneidensis MR-1 motility and biofilm formation. J. Bacteriol. 2013;195:3827–3833. doi: 10.1128/JB.00498-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thormann K.M., Duttler S., Saville R.M., Hyodo M., Shukla S., Hayakawa Y., Spormann A.M. Control of formation and cellular detachment from Shewanella oneidensis MR-1 biofilms by cyclic di-GMP. J. Bacteriol. 2006;188:2681–2691. doi: 10.1128/JB.188.7.2681-2691.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fredrickson J.K., Romine M.F., Beliaev A.S., Auchtung J.M., Driscoll M.E., Gardner T.S., Nealson K.H., Osterman A.L., Pinchuk G., Reed J.L., et al. Towards environmental systems biology of Shewanella. Nat. Rev. Microbiol. 2008;6:592–603. doi: 10.1038/nrmicro1947. [DOI] [PubMed] [Google Scholar]
- 25.Jonák J. Bacterial elongation factors EF-Tu, their mutants, chimeric forms, and domains: isolation and purification. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2007;849:141–153. doi: 10.1016/j.jchromb.2006.11.053. [DOI] [PubMed] [Google Scholar]
- 26.Kabsch W. XDS. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winter G. Xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010;43:186–190. [Google Scholar]
- 28.Long F., Vagin A.A., Young P., Murshudov G.N. BALBES: a molecular-replacement pipeline. Acta Crystallogr. D. Biol. Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams P.D., Afonine P. V, Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vagin a., Teplyakov a. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 31.Karplus P.A., Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P. V., Svergun D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Svergun D., Barberato C., Koch M.H.J. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. 1995;28:768–773. [Google Scholar]
- 35.Rambo R.P., Tainer J.a. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lassak J., Henche A.-L., Binnenkade L., Thormann K.M. ArcS, the cognate sensor kinase in an atypical Arc system of Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 2010;76:3263–3274. doi: 10.1128/AEM.00512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korch S.B., Hill T.M. Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: effects on macromolecular synthesis and persister formation. J. Bacteriol. 2006;188:3826–3836. doi: 10.1128/JB.01740-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin C.-Y., Awano N., Masuda H., Park J.-H., Inouye M. Transcriptional repressor HipB regulates the multiple promoters in Escherichia coli. J. Mol. Microbiol. Biotechnol. 2013;23:440–447. doi: 10.1159/000354311. [DOI] [PubMed] [Google Scholar]
- 39.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 40.Hansen S., Vulić M., Min J., Yen T.-J., Schumacher M.a., Brennan R.G., Lewis K. Regulation of the Escherichia coli HipBA toxin-antitoxin system by proteolysis. PLoS One. 2012;7:e39185. doi: 10.1371/journal.pone.0039185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donegan N.P., Thompson E.T., Fu Z., Cheung A.L. Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 2010;192:1416–1422. doi: 10.1128/JB.00233-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brzozowska I., Zielenkiewicz U. Regulation of toxin-antitoxin systems by proteolysis. Plasmid. 2013;70:33–41. doi: 10.1016/j.plasmid.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 43.Schumacher M.A., Min J., Link T.M., Guan Z., Xu W., Ahn Y.-H., Soderblom E.J., Kurie J.M., Evdokimov A., Moseley M.A., et al. Role of unusual P loop ejection and autophosphorylation in HipA-mediated persistence and multidrug tolerance. Cell Rep. 2012;2:518–525. doi: 10.1016/j.celrep.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evdokimov A., Voznesensky I., Fennell K., Anderson M., Smith J.F., Fisher D.A. New kinase regulation mechanism found in HipBA: a bacterial persistence switch. Acta Crystallogr. D. Biol. Crystallogr. 2009;65:875–879. doi: 10.1107/S0907444909018800. [DOI] [PubMed] [Google Scholar]
- 45.Maisonneuve E., Castro-Camargo M., Gerdes K. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell. 2013;154:1140–1150. doi: 10.1016/j.cell.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 46.Maisonneuve E., Shakespeare L.J., Girke M., Gerdes K. Bacterial persistence by RNA endonucleases. PNAS. 2011;108:13206–13211. doi: 10.1073/pnas.1100186108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Rotem E., Loinger A., Ronin I., Levin-Reisman I., Gabay C., Shoresh N., Biham O., Balaban N.Q. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl. Acad. Sci. U.S.A. 2010;107:12541–12546. doi: 10.1073/pnas.1004333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayes F., Van Melderen L. Toxins-antitoxins: diversity, evolution and function. Crit. Rev. Biochem. Mol. Biol. 2011;46:386–408. doi: 10.3109/10409238.2011.600437. [DOI] [PubMed] [Google Scholar]
- 49.Gelens L., Hill L., Vandervelde A., Danckaert J., Loris R. A general model for toxin-antitoxin module dynamics can explain persister cell formation in E. coli. PLoS Comput. Biol. 2013;9:e1003190. doi: 10.1371/journal.pcbi.1003190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Pino A., Balasubramanian S., Wyns L., Gazit E., De Greve H., Magnuson R.D., Charlier D., van Nuland N.A.J., Loris R. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell. 2010;142:101–111. doi: 10.1016/j.cell.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 51.Overgaard M., Borch J., Jørgensen M.G., Gerdes K. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 2008;69:841–857. doi: 10.1111/j.1365-2958.2008.06313.x. [DOI] [PubMed] [Google Scholar]
- 52.Cataudella I., Trusina A., Sneppen K., Gerdes K., Mitarai N. Conditional cooperativity in toxin-antitoxin regulation prevents random toxin activation and promotes fast translational recovery. Nucleic Acids Res. 2012;40:6424–6434. doi: 10.1093/nar/gks297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cataudella I., Sneppen K., Gerdes K., Mitarai N. Conditional cooperativity of toxin–antitoxin regulation can mediate bistability between growth and dormancy. PLoS Comput. Biol. 2013;9:e1003174. doi: 10.1371/journal.pcbi.1003174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feng J., Kessler D.A., Ben-Jacob E., Levine H. Growth feedback as a basis for persister bistability. Proc. Natl. Acad. Sci. U. S. A. 2014;111:544–549. doi: 10.1073/pnas.1320396110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaspy I., Rotem E., Weiss N., Ronin I., Balaban N.Q., Glaser G. HipA-mediated antibiotic persistence via phosphorylation of the glutamyl-tRNA-synthetase. Nat. Commun. 2013;4:1–7. doi: 10.1038/ncomms4001. [DOI] [PubMed] [Google Scholar]
- 56.Shao Y., Harrison E.M., Bi D., Tai C., He X., Ou H.-Y., Rajakumar K., Deng Z. TADB: a web-based resource for type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011;39:D606–D611. doi: 10.1093/nar/gkq908. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.