


Abstract
Noncoding RNAs (ncRNAs) and RNA-binding proteins are potent post-transcriptional regulators of gene expression. The ncRNA 7SL is upregulated in cancer cells, but its impact upon the phenotype of cancer cells is unknown. Here, we present evidence that 7SL forms a partial hybrid with the 3′-untranslated region (UTR) of TP53 mRNA, which encodes the tumor suppressor p53. The interaction of 7SL with TP53 mRNA reduced p53 translation, as determined by analyzing p53 expression levels, nascent p53 translation and TP53 mRNA association with polysomes. Silencing 7SL led to increased binding of HuR to TP53 mRNA, an interaction that led to the promotion of p53 translation and increased p53 abundance. We propose that the competition between 7SL and HuR for binding to TP53 3′UTR contributes to determining the magnitude of p53 translation, in turn affecting p53 levels and the growth-suppressive function of p53. Our findings suggest that targeting 7SL may be effective in the treatment of cancers with reduced p53 levels.
INTRODUCTION
Long noncoding RNAs (lncRNAs; >200 nucleotides) have emerged as potent regulators of gene expression at different levels, including chromatin remodeling, transcriptional and post-transcriptional control and protein metabolism (1,2). At the post-transcriptional level, lncRNAs can modulate mRNA decay and translation working jointly with microRNAs (miRNAs) and with RNA-binding proteins (RBPs) such as HuR and Staufen 1 [reviewed in (2)]. For instance, the lncRNAs 1/2-sbsRNAs (1/2-Staufen 1-binding site) interact with target mRNAs containing Alu elements in the 3′ untranslated region (UTR) by imperfect base-pairing and recruitment of the RBP Staufen 1 to promote decay (3). In contrast, the lncRNA BACE1AS (β amyloid-cleaving enzyme 1 antisense) forms a region of perfect base-pairing with BACE1 mRNA, which encodes an enzyme involved in cleaving the protein APP (amyloid precursor protein), and protects BACE1 mRNA from RNase-mediated degradation (4); this effect was attributed, at least in part, to the fact that the region of complementarity prevents binding of miR-485-5p to the BACE1 3′UTR (5). Examples of the impact of lncRNAs on mRNA translation have also been reported. For example, Uchl1 mRNA (encoding ubiquitin carboxyterminal hydrolase) was recently found to interact with antisense (AS)Uchl1 lncRNA via short interspersed element (SINE) sequences, leading to enhanced mRNA translation (6). Among the lncRNAs that inhibit translation, lincRNA-p21 repressed the translation of JUNB and CTNNB mRNAs by forming partial hybrids and recruiting translation repressors Rck and Fmrp (7).
RBPs are broadly involved in regulating gene expression at all post-transcriptional levels, including pre-mRNA splicing, transport, stability and translation (8–10). The RBP HuR has three RNA recognition motifs through which it binds numerous mRNAs to enhance their translation or stability, as well as many pre-mRNAs and noncoding RNAs (both miRNAs and lncRNAs) (7,11). Through its impact on target RNAs, HuR has been implicated in several cellular processes, including cell division, survival, senescence, immune activation and the stress response, as well as in pathological conditions such as cancer (12,13).
HuR function is regulated by three major mechanisms. HuR abundance is negatively regulated by miRNAs (miR-519, miR-125), ubiquitination and caspase-mediated cleavage in response to apoptotic stress [reviewed in (13)]. HuR subcellular localization is governed via phosphorylation by Cdk1, PKC and JAK3, and via methylation (13–15). HuR binding to mRNAs is controlled through phosphorylation by Chk2, PKC and JAK3 (13–15), and is competed by other RBPs (e.g. AUF1) or by miRNAs (e.g. miR-122, miR-494) (16–18). However, whether lncRNAs modulate HuR function is unknown.
The 300-nt long ncRNA 7SL (NR_002715; gene name RN7SL1) forms a ribonucleoprotein complex (RNP) with six signal-recognition proteins (SRPs). The RNP binds signal sequences of secretory and transmembrane proteins to facilitate their targeting to the membrane translocation apparatus in the endoplasmic reticulum (19,20). Chen et al. previously reported increased expression of the BC200 (related to 7SL) in cancer cells [(21), reviewed by White (22)], but the impact of this enhanced expression on cancer cell fate is not known. Here, we report that 7SL is highly expressed in cancer tissues and its silencing resulted in a significant reduction in cell proliferation. We identified an interaction between 7SL and the 3′UTR of TP53 mRNA, encoding the tumor suppressor p53, that led to the suppression of p53 translation. HuR also binds the TP53 3′UTR and promotes p53 translation, but binds immediately outside of the 7SL binding sites (9,23). Interestingly, 7SL silencing led to increased HuR binding to TP53 mRNA and promoted p53 translation and p53 accumulation, indicating that 7SL functionally prevented HuR binding to TP53 mRNA and suggesting that 7SL and HuR might compete for binding TP53 mRNA. We further discovered that 7SL promotes cancer cell growth by repressing p53 production, as p53-deficient cells were refractory to the growth-inhibitory effects of silencing 7SL. The enhanced expression of p53 in 7SL-silenced cells promoted cell cycle arrest, senescence and autophagy. We conclude that the proliferative effects of the ncRNA 7SL are mediated in part by displacement of HuR from TP53 mRNA leading to repression of p53 translation and p53 accumulation in cancer cells.
MATERIALS AND METHODS
Tumor and normal tissues, and cell culture, transfection and proliferation
Total RNAs from normal and adjacent cancer tissues were purchased from BioChain Institute, Inc. (liver, lung, breast, stomach) or obtained from the University of Maryland Tissue Bank samples (kidney, ovary, breast). Human cervical carcinoma HeLa cells and human pancreatic adenocarcinoma MiaPaCa cells were cultured in Dulbecco's modified Eagle's medium containing 5% fetal bovine serum (FBS), human colorectal carcinoma RKO cells in minimum essential medium (+10% FBS), and colon cancer HCT116 cells were in McCoy's 5A (+10% FBS). All cultures were supplemented with antibiotics. Transfection of cells (∼50% confluence) was carried out using lipofectamine 2000 (Invitrogen). The siRNAs used (from Qiagen) were Ctrl siRNA (AATTCTCCGAACGTGTCACGT), HuR siRNAs (AAACCAACAAGTCCCACAAAT, AATCTTAAGTTTCGTAAGTTA and TTCCTTTAAGATATATATTAA) and 7SL siRNA (AAGCACTAAGTTCGGCATCAA); SRP proteins were silenced using siRNAs from Santa Cruz Biotechnology directed to SRP68 [sc-94109], SRP19 [sc-44117] and SRP54 [sc-106810].
Plasmid pcDNA3-7SL was prepared by cloning polymerase chain reaction (PCR)-amplified 7SL. Luciferase reporter constructs were pmirGlo (control), pmirGlo-p53 (3′UTR) and pmirGlo-p53 (3′UTRΔ), the latter lacking 140 nt (2161–2300) and thus missing the region of interaction with 7SL. Forty-eight hours after siRNA transfection, DNA replication was assessed by measuring [3H]-thymidine incorporation, and cell cycle distribution was assessed using propidium iodide-stained, ethanol-fixed cells (analyzed using Multicycle software) as described (24). To assay luciferase reporter activity, cells were lysed and firefly and Renilla luciferase activities were measured using Dual-Luciferase Assay system (Promega) and a 96-well Microplate Luminometer BD PharmingenTM Monolight 3096. The ratios of firefly luciferase (FL) to Renilla luciferase (RL) activities were determined, and all values were normalized to those of control cells, transfected with an empty vector.
Protein analysis
Whole-cell lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer, resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes. After incubations with primary mouse monoclonal antibodies recognizing HuR, p53, β-tubulin, p21, SRP68, SRP54, HSP90 (all Santa Cruz Biotechnology), LC3-II (Abcam) and β-actin (Abcam), blots were incubated with the appropriate secondary antibodies (Amersham) and signals detected using enhanced luminescence (Amersham).
Nascent (de novo) translation of p53 and β-tubulin was studied as described (24). Briefly, HeLa cells were incubated with 1 mCi L-[35S]methionine and L-[35S]cysteine (Easy Tag TMEXPRESS, NEN/Perkin Elmer) per 60-mm plate for 15 min. Cells were lysed in RIPA buffer [10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 0.1% SDS and 1 mM DTT] and immunoprecipitation (IP) reactions carried out in TNN buffer [50 mM Tris-HCl (pH 7.5), 250 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40] for 16 h at 4°C using anti-IgG1 (BD PharMingen), anti-β-tubulin and anti-p53 antibodies. Following extensive washes in TNN buffer, the IP samples were resolved by SDS/PAGE, transferred onto PVDF filters and visualized with a Phosphor-Imager (Molecular Dynamics).
RNA analysis
Total cellular RNA was prepared using TRIzol (Invitrogen) or was isolated after RIP analysis [IP of cellular RNP (ribonucleoprotein) complexes using anti-HuR or IgG as described (25). After reverse transcription (RT) using random hexamers and Maxima reverse transcriptase (Fermentas), quantitative PCR (qPCR) analysis was performed using gene-specific primer pairs and SYBR Green PCR master mix (Kapa Biosystems). Specific forward and reverse primers to detect endogenous RNAs using RT-qPCR analysis were: CAAAACTCCCGTGCTGATCA and GGCTGGAGTGCAGTGGCTAT for 7SL, ATGGAAATCCCATCACCATCTT and CGCCCCACTTGATTTTGG for GAPDH, GAAGGAAATGGCCAAACAGA and ACGCTTTCTCCAGGTCTTCA for NCL, CAGGTTGCGGGAATCCAAAG and GCTGGGCACCTAGGACATCG for SIRT1, AGAAGGTGGTGGCATTTTTG and CAGTCTTCCCAAAGCAGGAG for VHL, AGGCCTTGGAACTCAAGGAT and TGAGTCAGGCCCTTCTGTCT for TP53, TCCTGAAACTCCTCTTTGTTTAACTG and CACCAACTCTCCCACTAGGCTATAA for MIAT, TGGCAAAATTGAACCAACAA and TTGAATCCCATCATGCCTTT for SRP54, CAGGCAGCTACCATGAGTGA and TCAAACAGGCGCTCCTTAGT for SRP68, and GAAGGCGAATCCCCATAAGT and CCGGACTCTGCCTCTGTATT for SRP19.
Antisense oligonucleotides (ASOs), biotinylated at the 3′ end, were as follows: AATTCTCCGAACGTGTCACGT (Ctrl-biot-ASO); TTTCACAGATATGGGCCTTGAAGTTAG and AGATGAAATCCTCCAGGGTGTGGGA (TP53-biot-ASO); CACCAACTCTCCCACTAGGCTATAA (MIAT-biot-ASO); GGGTGTGTAGAAGAAGCCACGCTCCCC and TTGCAGTAGTTCTCCAGCTGGTAGAG (INS-biot-ASO), and TGTTTGATGGATAGTTCATGTCTGT and TGAAACATTTCCAAAGCATTTATTT (SIRT1-biot-ASO). RNA and protein in sucrose and glycerol gradients were analyzed as described (24).
Northern blot analysis was performed using standard methods; 7SL and 18S were detected after end-labeling the oligonucleotides GTGATTCAAGCCGTAGTTATACCACTGGAG and CCAATGGATCCTCGTTAAAGGATTT, respectively (25).
Sucrose and glycerol gradient analysis
For the analysis of sucrose gradients, HeLa cells were incubated with cycloheximide (Calbiochem; 100 μg/ml, 15 min), and cytoplasmic lysates (500 μl) were prepared in polysome extraction buffer (PEB), fractionated by centrifugation through 10–50% linear sucrose gradients and divided into 10 fractions for RT-qPCR analysis, as described (24).
For glycerol gradient analysis, HeLa cells were lysed in a buffer containing 20 mM Tris-HCl, pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.5% (v/v) Nonidet P-40, RNAse-OUT and 1X protease inhibitor cocktail. After centrifugation (39 000 rpm, 18 h, 4°C) of the lysates through glycerol gradients (10–40%), 12 fractions (1 ml each) were collected; 0.3 ml was used for preparing RNA using TRIzol (Invitrogen) followed by RT-qPCR to measure 7SL and 18S rRNA levels, and 0.7 ml was precipitated using trichloroacetic acid (TCA) and used for western blot analysis to detect SRP proteins.
Pulldown of endogenous and biotinylated RNA
HeLa cells were lysed with PEB containing protease inhibitors (Roche) and RNase inhibitor (Thermo Fisher). Lysates were incubated with 100 pmoles of biotin-labeled oligomers complementary to TP53 mRNA (TP53-biot-ASO) or MIAT (MIAT-biot-ASO) for 30 min at RT as described (7). Dynabeads M-280 Streptavidin (Invitrogen) (100 μl) were added and further incubated (1 h, 4°C with rotation). Beads were washed with NT2 buffer, RNA was isolated using TRIzol, and the enrichment of TP53mRNA and 7SL bound to biotinylated ASOs was measured by RT-qPCR.
Biotinylated and non-biotinylated RNA was prepared as explained below. To assess binding of 7SL to the TP53 3′UTR, 25 ng of non-biotinylated 7SL was incubated with biot-TP53(3′) or with biot-TP53(3′Δ) (30 min, 37°C) in 1X TENT buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA [pH 8.0], 250 mM NaCl, 0.5% [v/v] Triton X-100), and protease and RNase inhibitors. After incubation with Dynabeads M-280 Streptavidin (30 min, 25°C), the beads were washed with phosphate buffered saline (PBS), the RNA isolated using TRIzol, and 7SL levels in the beads analyzed using RT-qPCR.
To measure HuR binding to TP53(3′) and TP53(3′Δ), the biotinylated RNAs (25 ng) were incubated for 30 min at 37°C in 1X TENT buffer with protease and RNase inhibitors, in the presence of 0.5 μg GST or GST-HuR. In competition experiments, unlabeled 7SL or GAPDHRNAs (25 ng each) was incubated along with biot-TP53(3′) and GST-HuR. After incubation with streptavidin dynabeads (30 min, 25°C) and washes with PBS, western blot analysis was used to detect GST-HuR.
Longer biotinylated RNAs were prepared in vitro using T7 and using primers (forward and reverse in each case; lowercase letters representing the T7 RNA polymerase promoter sequence): ttctaatacgactcactatagggCTTGTTTTATGCTCAGGGTC and CACCCCTCAGACACACAGGTGGCA for TP53 3′UTR, and ttctaatacgactcactatagggGCCGGGCGCGGTGGCGCGTG and AGAGACGGGGTCTCGCTATGTTG for 7SL.
RESULTS
7SL is selectively elevated in cancer tissues and is required for cell growth
The ncRNA 7SL, as well as the related ncRNA BC200, were shown to be widely upregulated in various cancer types (21,22). In agreement with this finding, RT followed by real-time qPCR analysis revealed higher 7SL levels in various tumor (T) tissues (liver, lung, breast, stomach) than in normal (N) adjacent tissues (Figure 1A; similar results were obtained by northern blot analysis of other N and T pairs, Supplementary Figure S1). To study the impact of 7SL on cancer cells, we tested the effect of knocking down 7SL levels in different cancer cell lines. As shown (Figure 1B), 48 h after transfecting HeLa cells (cervical carcinoma) with 7SL siRNA, 7SL levels were reduced by ∼60% compared to control cells. Silencing 7SL significantly reduced cell number in a variety of cultured cancer cell types, including HeLa cells, pancreatic carcinoma (Mia PaCa-2) and colon cancer (HCT116 and RKO) cell lines, and repressed HeLa cell growth (Figure 1C and D). Together, these data indicate that 7SL is highly expressed in cancer cells and enhanced cancer cell growth.
Figure 1.
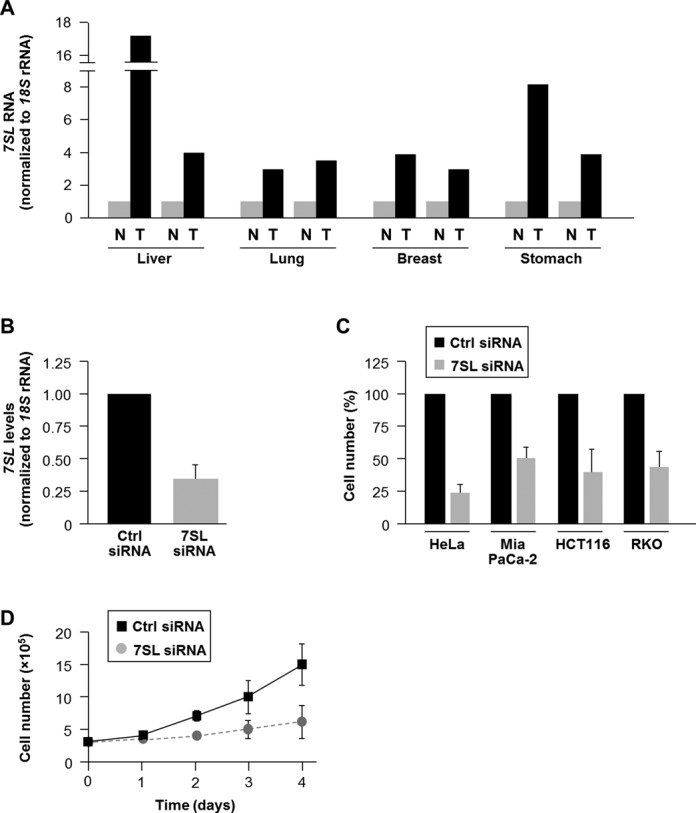
7SL is highly expressed in cancer tissues and is required for cell growth. (A) The abundance of 7SL in the tumor tissues (T) indicated (liver, lung, breast, stomach) and adjacent normal tissues (N) was measured by RT-qPCR analysis and normalized to 18S rRNA levels. (B) RT-qPCR analysis of 7SL levels 48 h after transfection of HeLa cells with Ctrl siRNA or 7SL siRNA. (C) Forty-eight hours after siRNA transfection of HeLa, Mia PaCa-2, HCT116 or RKO cells, cell numbers were measured using a hemocytometer and represented as % of cells relative to the Ctrl group. (D) Growth kinetics of HeLa cells transfected with Ctrl siRNA or 7SL siRNA. Cells were counted daily for 4 days. Data in (B)–(D) are the means ± S.D. from three independent experiments.
7SL interacts with TP53 mRNA and lowers p53 abundance
Long ncRNAs can enhance or suppress gene expression by interacting directly with mRNAs (2). A BLAST survey to identify possible interactions between 7SL and regulators of cell growth revealed that 7SL was predicted to form extensive regions of sense–antisense interaction with the 3′UTR of the TP53 mRNA (nucleotides 2209–2367; Figure 2A, Supplementary Figure S2); sequence identities were 85–93%. Other putative interaction partners of 7SL are listed (Supplementary Table S1). To verify if endogenous 7SL and TP53 mRNAs formed hybrid RNA interactions, we designed biotinylated AS oligomers complementary to TP53 mRNA (TP53-biot-ASO), to a negative control (the lncRNA MIAT, MIAT-biot-ASO), and to mRNAs encoding insulin (INS-biot-ASO) and Sirtuin-1 (SIRT1-biot-ASO). Incubation of lysates prepared from HeLa cells with biot-ASOs revealed that 7SL was enriched in TP53-biot-ASO pulldown samples, but not in the other biot-ASO pulldowns, indicating that 7SL selectively associated with TP53 mRNA (Figure 2B). The enrichment of GAPDH mRNA in the pulldown samples was measured to assess background RNA binding to biot-ASOs.
Figure 2.
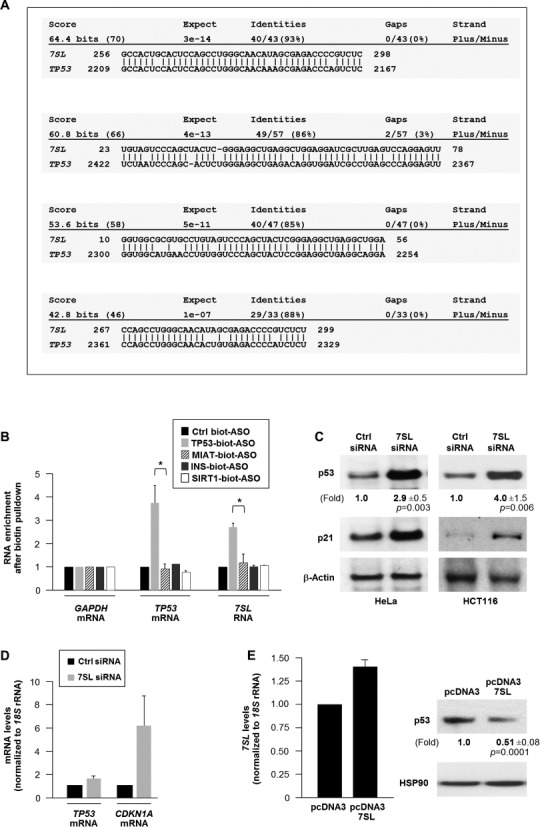
7SL binds TP53 mRNA and lowers p53 abundance. (A) Sequence alignment of 7SL and TP53 mRNA using BLAST shows potential sense–antisense interactions. (B) TP53 mRNA pulldown using the biotin-ASOs shown (see the Materials and Methods section) was followed by RT-qPCR analysis to quantify TP53 mRNA and 7SL levels; GAPDH mRNA was used for normalization. (C) HeLa cells were transfected with the indicated siRNAs; 48 h later, the levels of p53, p21 and loading control β-actin were assessed by western blot analysis. (D) The levels of TP53 and CDKN1A mRNA (encoding p21) and normalization control 18S rRNA were quantified by RT-qPCR. (E) Forty-eight hours after transfecting plasmids pcDNA3 or pcDNA3-7SL, total cellular 7SL levels were measured by RT-qPCR analysis (left) and the levels of p53 and loading control HSP90 by western blot analysis (right). Data in (B) and (E) are the means + S.D. from three independent experiments. In (C) and (E), the means ± standard deviation of p53 signals are indicated and the p values are shown.
Next we asked if 7SL could influence p53 expression. By 48 h after transfection, silencing 7SL elevated p53 protein levels in both HeLa and HCT116 cells (Figure 2C) and increased the levels of p21 protein and CDKN1A mRNA [encoding p21 (Figure 2C and D), a transcriptional target of p53 (26)], but it did not increase TP53 mRNA levels (Figure 2D). The increase in p53 expression was not likely due to cell toxicity derived from lowering 7SL (Supplementary Figure S3). Strong overexpression of 7SL could not be attained in HeLa cells, but a moderate increase in 7SL from a plasmid vector was tolerated and led to a reduction in p53 protein levels (Figure 2E); despite higher levels of 7SL in tumor samples (T), TP53 mRNA abundance did not consistently differ from that in normal samples (Supplementary Figure S1B). Likewise, silencing 7SL in the cultured tumor lines only affected TP53 mRNA modestly, but led to a robust increase in p53 protein levels (Supplementary Figure S1C–E).
Since 7SL is the RNA component of the signal recognition particle (SRP), we studied if the reduction in p53 protein levels was affected by other SRP components. Silencing protein components of the SRP (SRP19, SRP54 or SRP68; Figure 3A) lowered p53 abundance (Figure 3B) but did not affect the levels of TP53 mRNA (Figure 3B) or 7SL (Figure 3C). In addition, silencing SRP proteins did not significantly affect cell viability by 48 h after silencing SRP proteins (Figure 3D). These findings suggest that silencing SRP proteins does not recapitulate the increase in p53 elicited by silencing 7SL in HeLa cells. To test this possibility further, glycerol gradients were studied in order to study if 7SL is present in fractions devoid of SRP proteins. As shown in Figure 3E–G, SRP54 and SRP68 overlapped substantially with 7SL (particularly in fractions 5 through 9 of the gradient), but a sizeable amount of 7SL was found outside of these fractions. In fact, >30% of 7SL was detected in fractions that did not have SRP54 or SRP68. These results support the notion that a subset of 7SL exists outside of the SRPs and further indicate that the influence of 7SL on p53 expression is likely independent of its function as part of the SRP.
Figure 3.
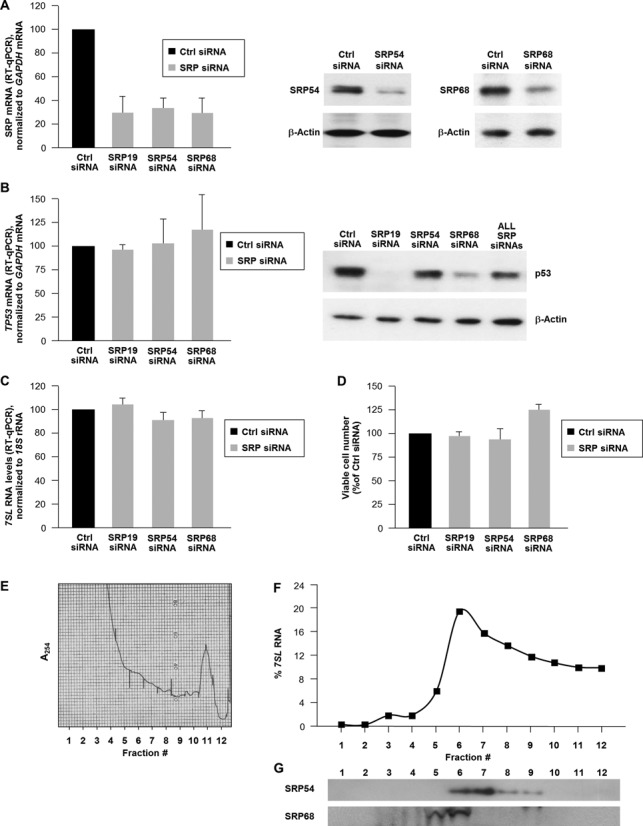
(A) HeLa cells were transfected with siRNAs directed to SRP proteins and 48 h later the levels of the mRNAs encoding each SRP protein were measured by RT-qPCR (left) and the levels of protein were assessed by western blot analysis (right; the SRP19 antibody was not adequate for this analysis, not shown) and loading was monitored by detecting the housekeeping control protein β-actin. (B) In cells processed as in (A), the levels of TP53 mRNA were measured by RT-qPCR (left) and the levels of p53 protein (as well as loading control β-actin) were assessed by western blot analysis (right). This group included analysis of cells in which SRP68, SRP54 and SRP19 were silenced simultaneously (‘ALL SRP siRNAs’). (C) In cells processed as in (A), the levels of 7SL were measured by RT-qPCR analysis. (D) Forty-eight hours after transfection as in (A), cells were counted using a hemocytometer. In (A)–(D), data are shown as the means + S.D. from three independent experiments. (E)–(G) From each of 12 fractions from glycerol gradients shown in the global RNA profile (10-40% glycerol) (E), RNA was isolated and used for RT-qPCR analysis of 7SL levels (F), and protein was precipitated and used for western blot analysis of SRP54 and SRP68. Data are representative of three independent experiments.
In sum, these data indicate that 7SL interacts with TP53 mRNA and lowers p53 protein levels. Since TP53 mRNA levels were not affected, we set out to investigate if 7SL reduced p53 translation.
7SL and HuR compete for binding to TP53 mRNA
The RBP HuR binds the 3′UTR of TP53 mRNA and promotes its translation following irradiation with ultraviolet light (23). Recent HuR PAR-CLIP data revealed that the sites where HuR binds within TP53 3′UTR (11) were adjacent to the site of interaction with 7SL (Figure 4A; yellow, HuR interaction sites; green, putative 7SL sites). Since HuR enhances p53 abundance, 7SL lowers it, and both HuR and 7SL bind TP53 3′UTR, we hypothesized that HuR and 7SL specifically compete for binding to TP53 mRNA. To test this possibility, we first silenced 7SL and studied if this intervention affected HuR binding to target mRNAs (15,18,23). As shown, silencing 7SL selectively enhanced HuR binding to TP53 mRNA, but not to VHL, SIRT1 or NCL mRNAs (Figure 4B).
Figure 4.
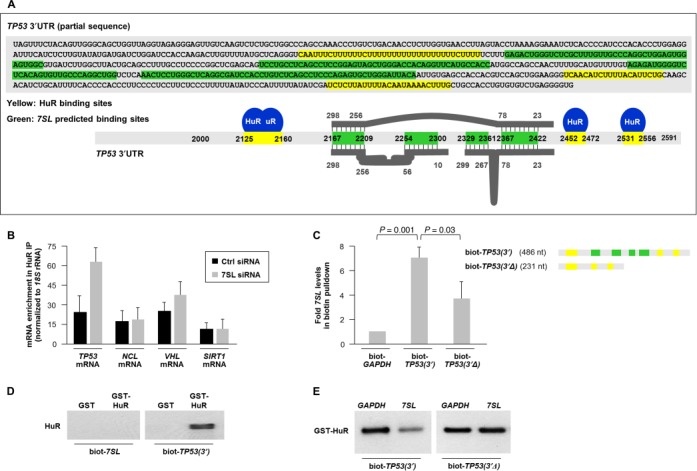
Competitive regulation of p53 expression by 7SL and HuR. (A) Schematic of TP53 mRNA depicting potential binding sites for 7SL (green) and HuR (yellow). The thick grey lines and numbers above and below TP53 3′UTR depict regions of complementarity with 7SL. (B) HeLa cells were transfected with 7SL siRNA; 48 h later, HuR association with the indicated mRNAs was quantified by RIP analysis. Data were normalized to the levels of GAPDH mRNA in each IP sample and represented as the enrichment of each mRNA relative to the levels in IgG IP. (C) Top, partial in vitro TP53 3′UTR biotinylated transcripts bearing the sites of interaction with HuR and 7SL [biot-TP53(3′)] or bearing only the sites of interaction with HuR [biot-TP53(3′Δ)]. Bottom, biotinylated RNAs were incubated with non-biotinylated 7SL and the levels of 7SL in the pulldown were measured by RT-qPCR analysis. (D) GST or GST-HuR were incubated in vitro with the biotinylated RNAs shown; following pulldown, western blot analysis was carried out to detect HuR levels. (E) GST-HuR was incubated in vitro with the biotinylated and non-biotinylated RNAs shown and its presence in the pulldown material was assessed by western blot analysis. Data in (B) are the means + S.D. from three independent experiments; data in (C) and (E) are average of two repeats showing similar results; data in (D) are representative of three repeats.
To gain more direct evidence that 7SL associated with TP53 3′UTR, we transcribed in vitro a biotinylated (biot) TP53 3′UTR RNA bearing the putative 7SL RNA complementarity sites [TP53(3′), 486 nt long], a biot-TP53 3′UTR RNA lacking the 7SL RNA interaction sites [TP53(3′Δ), 231 nt long], 7SL, and biot-7SL. RNAs biot-TP53(3′) and biot-TP53(3′Δ) were incubated with 7SL; 30 min later, RNA complexes were pulled down using streptavidin beads, extracted, and 7SL levels were analyzed by RT-qPCR. As shown (Figure 4C), 7SL was strongly enriched in biot-TP53(3′) pulldown samples, indicating that the two RNAs interacted; far less 7SL was found in biot-TP53(3′Δ) pulldown samples. Other binding sites on TP53 mRNA may exist in the regions surrounding the mapped hybrid sites on the TP53 3′UTR (green regions), since biot-TP53(3′Δ) shows residual binding to 7SL above the background levels seen with the non-target control transcript biot-GAPDH (Figure 4C). In keeping with the lack of HuR PAR-CLIP tags in 7SL, only biot-TP53(3′) (not biot-7SL) pulled down GST-HuR, as detected by western blot analysis (Figure 4D).
It was also important to assess directly if HuR binding to TP53 3′UTR was influenced by 7SL. To this end, we incubated biot-TP53(3′) with GST-HuR in the presence of 7SL or a control non-target transcript (GAPDH RNA), pulled down the complex with streptavidin beads, and measured GST-HuR levels in the complex. As shown (Figure 4E), adding 7SL reduced the binding of GST-HuR to TP53(3′), indicating that 7SL could function as a competitor and reduced GST-HuR interaction with biot-TP53(3′). Importantly, GST-HuR binding to biot-TP53(3′Δ), an RNA that binds HuR but lacks the 7SL complementarity region, was not competed by 7SL (Figure 4E). Together, these findings indicate that TP53 3′UTR can interact with 7SL in cells and in vitro, and that 7SL competed with HuR for binding to TP53 3′UTR via the regions of 7SL homology with TP53 3′UTR.
Antagonistic effects of 7SL and HuR on p53 translation
Consistent with earlier evidence (23), HuR promoted p53 expression (Figure 5A), but the enhancement of p53 levels after silencing 7SL was inhibited when HuR was silenced (Figure 5A). These data suggested that HuR and 7SL competed for binding to TP53 mRNA and that HuR was required for enhancing p53 expression in 7SL-silenced cells. Since modulating 7SL or HuR did not alter TP53 mRNA levels [Figure 2D; (23)], we hypothesized that 7SL RNA and HuR may regulate p53 mRNA translation by binding to the TP53 3′UTR. To test this hypothesis, we cloned the TP53 3′UTR [nucleotides 1421–2629 (23)] into a dual luciferase construct [pmirGlo-p53(3′UTR)] and prepared a TP53 3′UTR reporter plasmid lacking the 7SL interaction region (nucleotides 2161–2300) [pmirGlo-p53(3′UTRΔ). In cells transfected with pmirGlo-p53(3′UTR), luciferase activity [FL/RL] increased in 7SL-silenced cells and decreased in HuR-silenced cells (Figure 5B). Importantly, the induction in luciferase activity by 7SL silencing was attenuated in cells with both 7SL and HuR silenced and in cells expressing pmirGlo-p53(3′UTRΔ). Moreover, HuR binding to TP53 3′UTR, as determined by ribonucleoprotein immunoprecipitation (RIP) analysis of HuR binding to FL RNA expressed from the reporter plasmids in Figure 5B, revealed that removing the 7SL site increased the interaction of HuR with the TP53 3′UTR. Since the precise TP53 3′UTR nucleotides within the HuR PAR-CLIP sites with which HuR interacts are not known, we were unable to test the impact of introducing point mutations upon 7SL binding to TP53 3′UTR. Collectively, these results indicate that 7SL repressed reporter activity by interacting with the TP53 3′UTR region complementary to 7SL and that derepression by silencing 7SL required the presence of HuR.
Figure 5.
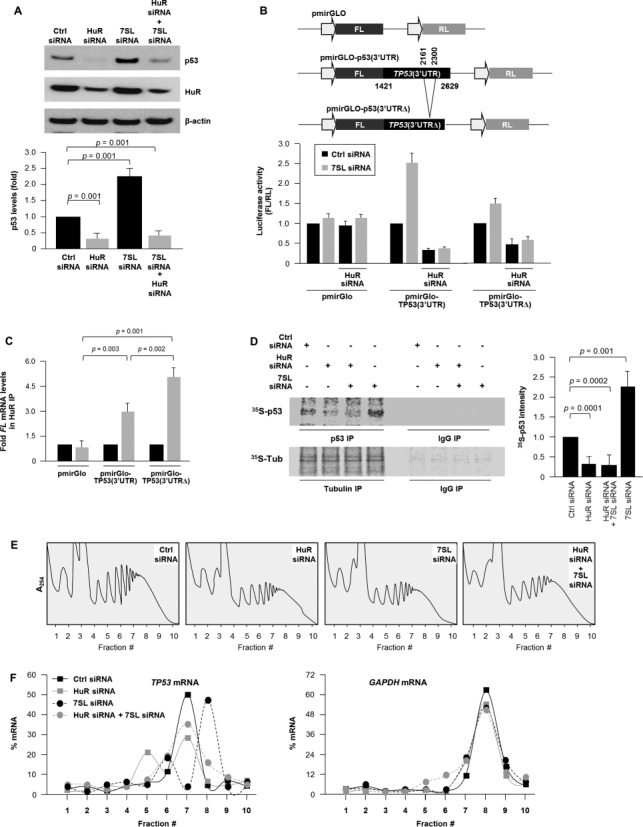
Opposite regulation of p53 translation by 7SL and HuR. (A) HeLa cells were transfected with the indicated siRNAs; 48 h later, the levels of p53, HuR and loading control β-actin were assessed by western blot analysis and p53 signals were quantified by densitometry and plotted. (B) Top, luciferase reporter constructs pmirGLO-p53(3′UTR), bearing partial sequence of TP53 3′UTR (nucleotides 1421–2629) that includes 7SL and HuR binding sites, and pmirGLO-p53(3′UTRΔ) that lacks the 7SL interaction site (nucleotides 2161–2300). Thirty-six hours after transfection of the indicated siRNAs, luciferase plasmids were transfected for 6 h and followed by luciferase assay 12 h thereafter. (C) RIP analysis of HuR interaction with the FL mRNAs expressed from the reporters in panel (B); data represent enrichment levels relative to FL mRNA abundance in IgG IP, using GAPDH mRNA for normalization. (D) De novo translation of p53 as well as housekeeping control protein β-Tubulin (Tub), as assessed by 35S-p53 IP and 35S-Tub IP 48 h after transfection of the indicated siRNAs (details in Materials and Methods section); ‘Fold’ quantified 35S-p53 signals, relative to signals in the Ctrl siRNA group. (E) and (F) Cells were transfected as described in (A) followed by fractionation through sucrose gradients. Global RNA profile for these transfection conditions are shown (E). The relative distribution of TP53 mRNA (and housekeeping GAPDH mRNA) was studied by RT-qPCR analysis of RNA in each of 10 gradient fractions (F). Data in (A)–(C) represent the means and S.D. from three independent experiments; data in (D)–(F) are representative of three independent experiments; P values are shown.
To study if these effects were also observed with the endogenous TP53 mRNA, we performed two sets of experiments. First, nascent p53 translation was measured by a brief (15 min long) incubation of HeLa cells with L-[35S]methionine and L-[35S]cysteine, after which p53 and housekeeping control protein β-Tubulin (β-Tub) were subjected to IP and the incorporation of radiolabeled amino acids visualized by autoradiography; this assay routinely yields faint signals because the time of incorporation of radiolabeled amino acids is very brief (15 min) to ensure that the signal reflects only de novo translation. As shown, relative to control cells, de novo p53 translation was lower in HuR siRNA-transfected cells and was higher in 7SL siRNA-transfected cells (Figure 5D). Consistent with the pattern of luciferase activity (Figure 5B), the 7SL silencing-induced p53 translation was lost in cells in which 7SL and HuR were silenced (Figure 5D).
Second, we investigated whether the levels of 7SL and HuR affected p53 translation by monitoring the association of TP53 mRNA with the translational machinery in each of the four transfection groups. Cytoplasmic extracts were fractionated on sucrose gradients and the relative abundance of TP53 mRNA in each fraction was calculated to measure the association of TP53 mRNA with the translational apparatus. As shown in Figure 5E, TP53 mRNA levels were very low in non-translating and low-translating fractions of the gradient (fractions 1–6). In control cells, TP53 mRNA was found in the actively translating fractions of the gradient (fractions 7–9); in this section of the gradient, TP53 mRNA in 7SL-silenced cells showed a rightward shift, indicating that TP53 mRNA associated with larger polysomes and further suggesting that 7SL suppressed the initiation of TP53 mRNA translation (Figure 5F). By contrast, in HuR-silenced cells, the peak of TP53 mRNA decreased compared to the peak in control cells, even after silencing 7SL, indicating that a smaller number of TP53 mRNAs were engaged in translation (Figure 5F). These differences were not seen when testing the distribution of a control transcript (GAPDH mRNA) that encodes the housekeeping protein GAPDH in the four groups (Figure 5F). In summary, these data indicated that 7SL suppresses TP53 mRNA translation while HuR is required for efficient translation even in the absence of 7SL RNA.
7SL deficiency promotes cell cycle arrest, senescence and autophagy
Several studies underscore the role of HuR in orchestrating anti-apoptotic and pro-survival programs (13,27). In agreement with this influence, HuR silencing decreased cell numbers and silencing both HuR and 7SL decreased cell numbers even further (Supplementary Figure S4A). Therefore, we sought to study in more detail the cellular response to lowering 7SL levels. The 7SL silencing-triggered decrease in cell numbers was specifically dependent on p53, since p53-deficient (p53KO) HCT116 cells were refractory to the loss in cell number after silencing 7SL (Supplementary Figure S4B and C). These data indicated that silencing 7SL caused growth arrest at least in part by inducing p53 levels. Thus, we investigated other p53-regulated cellular processes—cell cycle, senescence and autophagy. By fluorescence-activated cell sorting (FACS) analysis, silencing 7SL led to an increase in the G1 and G2 compartments and a decrease in S-phase cells (Figure 6A). 7SL silencing also lowered [3H]-thymidine incorporation (Figure 6B), further indicating that silencing 7SL inhibited DNA replication, which occurs during S phase; conversely, overexpressing 7SL as explained in Figure 2E increased [3H]-thymidine incorporation (Supplementary Figure S4E).
Figure 6.
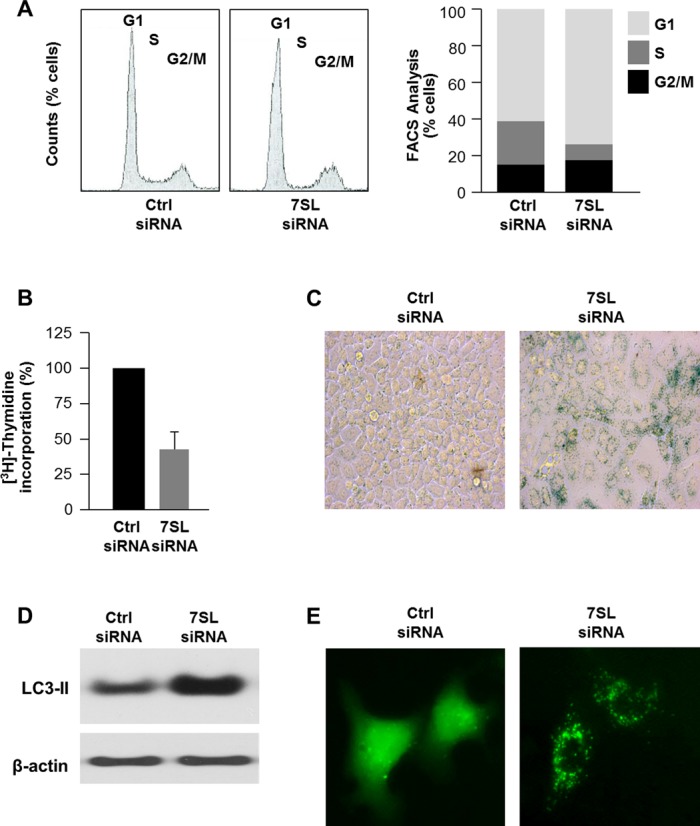
Influence of 7SL silencing on cell phenotype. (A) Forty-eight hours after transfection of HeLa cells with Ctrl siRNA or 7SL siRNA, cells were subjected to FACS analysis (left) and the relative G1, S and G2/M compartments calculated (right). Data are representative of three independent experiments. (B) Measurement of [3H]-thymidine incorporation by 48 h after transfection of HeLa cells with Ctrl siRNA or 7SL siRNA. (C) β-galactosidase activity in HeLa cells 5 days after transfection with either Ctrl siRNA or 7SL siRNA. (D) Western blot analysis of the autophagy marker LC3 in 48 h after transfection of HeLa cells with Ctrl siRNA or 7SL siRNA. (E) HeLa cells were transfected with a plasmid that expresses GFP-LC3 (a fluorescent fusion protein that is recruited to autophagosomes) and with either Ctrl siRNA or 7SL siRNA; 48 h later, GFP-LC3 signals were visualized by fluorescence microscopy.
In keeping with the growth suppression that characterizes senescence, 5 days after silencing 7SL, HeLa cells displayed high activity of the senescence marker SA-β-gal (senescence-associated-β-galactosidase; Figure 6C). Finally, we assessed the effect of 7SL on autophagy, another process influenced by p53 (28). Western blot analysis of the processed form of the microtubule-associated protein 1 light chain 3 (LC3)-II, broadly used to monitor autophagy because it correlates with the number of autophagosomes (29), showed that silencing 7SL selectively elevated LC3-II (Figure 6D). Moreover, silencing 7SL in cells transfected with plasmid pGFP-LC3, which expresses green fluorescent protein (GFP)-tagged LC3, revealed a pattern of distinct GFP-LC3 puncta staining that is characteristic of the presence of autophagosomes (30) (Figure 6E). Together, these data indicate that 7SL silencing reduced cell proliferation and promoted cellular senescence and autophagy, and lend support to the notion that suppression of p53 by 7SL promotes cell growth.
DISCUSSION
Given that the ncRNA 7SL is more abundant in cancer tissues compared to normal tissues and is required for cancer cell growth [(21,22), Figure 1], we have studied the possibility that 7SL promotes cancer cell growth by lowering the expression of the tumor suppressor p53. We identified an RNA-dependent interaction between 7SL and TP53 3′UTR (Figures 2 and 4) that prevents HuR binding to TP53 mRNA and thereby suppresses p53 translation (Figure 5); these effects by 7SL appeared to be independent of its function as a component of the SRP (Figure 3). We postulate that by lowering p53 expression levels, 7SL promotes cell cycle progression and suppresses cellular senescence and autophagy, all of which are key phenotypes of cancer cells (Figure 6).
ncRNAs bind and regulate the fate of target mRNAs by forming complexes that usually contain RBPs. The consequences of these lncRNA–mRNA interactions vary widely. For instance, 1/2-sbsRNAs lncRNAs enhance target mRNA decay (3), while BACE1AS prevents the degradation of its target transcript, BACE1 mRNA (4). The AS Uchl1 lncRNA interacts with Uchl1 mRNA to enhance its translation (6), while lincRNA-p21 represses the translation of JUNB and CTNNB mRNAs (7). Similarly, we found that 7SL interacts with TP53 mRNA and repressed its translation. Our findings support growing evidence that lncRNA–mRNA interactions are a common mechanism to regulate mRNA fate and that RBPs are functionally involved. For instance, the repressive influence of lincRNA-p21 upon JUNB and CTNNB mRNAs required the actions of Rck and Fmrp (7). In cells and in vitro, HuR binding to TP53 mRNA was restricted by the abundance of 7SL, indicating a competitive interaction between HuR and 7SL (Figure 4). A similar competition between HuR and miR-494 was previously reported to regulate NCL mRNA translation (18). However, the present study constitutes the first example, to our knowledge, of a long ncRNA that competes with an RBP to regulate the post-transcriptional outcome (translation in this case) of a shared target mRNA.
The expression of p53 is also regulated by other lncRNAs, including H19, maternally expressed gene 3 (MEG3) and lincRNA-RoR (31). H19, essential for human tumor growth, was found to interact with p53 and suppressed p53 function post-translationally (32,33). In contrast, MEG3, a tumor suppressor lncRNA, regulates the transcriptional activation of p53 by reducing the levels of MDM2 (mouse double minute 2 homolog) (34), which mediates p53 degradation. However, tumor suppression by MEG3 was found to be both p53-dependent and p53-independent (35). In this study, we found that the effect of 7SL silencing on HeLa cell growth is largely p53-dependent, since p53-deficient HCT116 cells were refractory to 7SL silencing (Supplementary Figure S4B and C). Recently, lincRNA-RoR, which is transcriptionally induced by p53, was found to repress p53 translation indirectly by interacting with hnRNP I (36). These findings and our study indicate the existence of more complex regulatory levels of TP53 mRNA and p53 protein by lncRNAs. Additionally, p53 translation is positively regulated via the formation of a partial hybrid between the TP53 5′UTR and 3′UTR (37). Whether this intramolecular interaction is functionally linked to the effects of 7SL or HuR is the subject of ongoing studies.
The discovery that lowering 7SL RNA results in growth arrest raises the possibility that 7SL could enhance the growth of malignant cells. These actions could be mediated by p53 in large part, as p53 potently regulates several cellular processes involved in tumorigenesis, such as cell survival, proliferation, senescence and autophagy (38). Silencing 7SL recapitulated the inhibition of cancer cell growth in models of therapy-induced senescence in which p53 expression is elevated (39). Similarly, the discovery that 7SL is more abundant in cancer tissues suggests that 7SL is required for cancer cell growth and might have oncogenic effects [Figure 1 (21,22)]. Whether 7SL also influences the expression of other proteins involved in proliferation of cancer cells remains to be investigated. An interesting finding of our study is that 7SL silencing-induced cell cycle arrest and senescence occur despite sustained levels of HuR (Figure 6). This finding indicates that high p53 levels force cell cycle arrest and senescence, while HuR can help maintain cell survival, as more cell death is observed in HuR-silenced cells together with 7SL RNA silencing (Supplementary Figure S4A). Although HuR is more abundant in cancer cells (40), p53 levels are usually low in cancer. In light of the high levels of 7SL, HuR may not be able to enhance TP53 mRNA translation in cancer cells if 7SL is elevated and outcompetes HuR. However, the stoichiometry of this competitive interaction awaits further study.
It is worth noting that in 1991, Sakamoto et al. (41) cloned and studied a 510-bp genomic fragment that contained 180 bp upstream and 30 bp downstream from the 7SL sequence. The authors observed that transfection of HeLa cells with this vector suppressed cell proliferation and elevated 7SL levels transiently (by 6 h after transfection). They proposed that the elevated presence of the 510-bp sequence, which contained binding sites for RNA Polymerase III, might ‘sponge’ RNA Pol III and alter its basal activity. In agreement with this possibility, the effect of overexpressing the 510-bp genomic 7SL fragment was abolished by mutation of the Pol III binding site (41).
In light of the influence of 7SL on p53 abundance, future work is warranted to test if 7SL is broadly elevated in cancer cells and if the concentration of 7SL is inversely correlated with that of p53. To test if 7SL is oncogenic, studies are underway to examine if 7SL promotes tumorigenesis in mice and whether such influence is linked to HuR levels. In sum, we have found that by influencing p53 levels, 7SL and HuR affect gene expression programs modulated by p53. In the immediate future, studies are warranted to investigate if 7SL is a valuable target of therapeutic intervention.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank R.P. Wersto (NIA, NIH) for assistance with FACS analysis.
FUNDING
National Institute on Aging-Intramural Research Program [NIA-IRP], National Institutes of Health [NIH].
Conflict of interest statement. None declared.
REFERENCES
- 1.Ulitsky I., Bartel D.P. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoon J.H., Abdelmohsen K., Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013;425:3723–3730. doi: 10.1016/j.jmb.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gong C., Maquat L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470:284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faghihi M.A., Modarresi F., Khalil A.M., Wood D.E., Sahagan B.G., Morgan T.E., Finch C.E., St Laurent G., 3rd, Kenny P.J., Wahlestedt C. Expression of a noncoding RNA is elevated in Alzheimer's disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008;14:723–730. doi: 10.1038/nm1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faghihi M.A., Zhang M., Huang J., Modarresi F., Van der Brug M.P., Nalls M.A., Cookson M.R., St-Laurent G, 3rd, Wahlestedt C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010;11:R56. doi: 10.1186/gb-2010-11-5-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrieri C., Cimatti L., Biagioli M., Beugnet A., Zucchelli S., Fedele S., Pesce E., Ferrer I., Collavin L., Santoro C., et al. (2012) Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 491:454–457. doi: 10.1038/nature11508. [DOI] [PubMed] [Google Scholar]
- 7.Yoon J.H., Abdelmohsen K., Srikantan S., Yang X., Martindale J.L., De S., Huarte M., Zhan M., Becker K.G., Gorospe M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell. 2012;47:648–655. doi: 10.1016/j.molcel.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilkie G.S., Dickson K.S., Gray N.K. Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem. Sci. 2003;28:182–188. doi: 10.1016/S0968-0004(03)00051-3. [DOI] [PubMed] [Google Scholar]
- 9.Wilusz C.J., Wilusz J. Bringing the role of mRNA decay in the control of gene expression into focus. Trends Genet. 2004;20:491–497. doi: 10.1016/j.tig.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Glisovic T., Bachorik J.L., Yong J., Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukherjee N., Corcoran D.L., Nusbaum J.D., Reid D.W., Georgiev S., Hafner M., Ascano M., Jr, Tuschl T., Ohler U., Keene J.D. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples Pre-mRNA processing and mRNA stability. Mol. Cell. 2011;43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srikantan S., Gorospe M. HuR function in disease. Front. Biosci. 2012;17:189–205. doi: 10.2741/3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdelmohsen K., Gorospe M. Posttranscriptional regulation of cancer traits by HuR. WIRES RNA. 2010;1:214–229. doi: 10.1002/wrna.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eberhardt W., Doller A., Pfeilschifter J. Regulation of the mRNA-binding protein HuR by posttranslational modification: spotlight on phosphorylation. Curr. Protein Pept. Sci. 2012;13:380–390. doi: 10.2174/138920312801619439. [DOI] [PubMed] [Google Scholar]
- 15.Yoon J.H., Abdelmohsen K., Srikantan S., Yang X., Martindale J.L., Gorospe M. Tyrosine phosphorylation of HuR by JAK3 triggers dissociation and degradation of HuR target mRNAs. Nucleic Acids Res. 2014;42:1196–1208. doi: 10.1093/nar/gkt903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharyya S.N., Habermacher R., Martine U., Closs E.I., Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 17.Lal A., Mazan-Mamczarz K., Kawai T., Yang X., Martindale J.L., Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23:3092–3102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tominaga K., Srikantan S., Lee E.K., Subaran S.S., Martindale J.L., Abdelmohsen K., Gorospe M. Competitive regulation of Nucleolin expression by HuR and miR-494. Mol. Cell. Biol. 2011;31:4219–4231. doi: 10.1128/MCB.05955-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernstein H.D., Zopf D., Freymann D.M., Walter P. Functional substitution of the signal recognition particle 54-Kda subunit by its Escherichia-coli homolog. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5229–5233. doi: 10.1073/pnas.90.11.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Labuda D., Zietkiewicz E. Evolution of secondary structure in the family of 7sl-like RNAs. J. Mol. Evol. 1994;39:506–518. doi: 10.1007/BF00173420. [DOI] [PubMed] [Google Scholar]
- 21.Chen W., Bocker W., Brosius J., Tiedge H. Expression of neural BC200 RNA in human tumours. J. Pathol. 1997;183:345–351. doi: 10.1002/(SICI)1096-9896(199711)183:3<345::AID-PATH930>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 22.White R.J. RNA polymerase III transcription and cancer. Oncogene. 2004;23:3208–3216. doi: 10.1038/sj.onc.1207547. [DOI] [PubMed] [Google Scholar]
- 23.Mazan-Mamczarz K., Galbán S., López de Silanes I., Martindale J.L., Atasoy U., Keene J.D., Gorospe M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. U.S.A. 2003;100:8354–8359. doi: 10.1073/pnas.1432104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdelmohsen K., Srikantan S., Kuwano Y., Gorospe M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc. Natl. Acad. Sci. U.S.A. 2008;105:20297–20302. doi: 10.1073/pnas.0809376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdelmohsen K., Pullmann R., Jr, Lal A., Kim H.H., Galban S., Yang X., Blethrow J.D., Walker M., Shubert J., Gillespie D.A., et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell. 2007;25:543–557. doi: 10.1016/j.molcel.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Deiry W.S., Tokino T., Velculescu V.E., Levy D.B., Parsons R., Trent J.M., Lin D., Mercer W.E., Kinzler K.W., Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 27.Abdelmohsen K., Lal A., Kim H.H., Gorospe M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle. 2007;6:1288–1292. doi: 10.4161/cc.6.11.4299. [DOI] [PubMed] [Google Scholar]
- 28.Maiuri M.C., Galluzzi L., Morselli E., Kepp O., Malik S.A., Kroemer G. Autophagy regulation by p53. Curr. Opin. Cell. Biol. 2010;22:181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Mann S.S., Hammarback J.A. Molecular characterization of light chain-3—a microtubule-binding subunit of Map1a and Map1b. J. Biol. Chem. 1994;269:11492–11497. [PubMed] [Google Scholar]
- 30.Kabeya Y., Mizushima N., Yamamoto A., Oshitani-Okamoto S., Ohsumi Y., Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell. Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 31.Baldassarre A., Masotti A. Long Non-Coding RNAs and p53 Regulation. Int. J. Mol. Sci. 2012;13:16708–16717. doi: 10.3390/ijms131216708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barsyte-Lovejoy D., Lau S.K., Boutros P.C., Khosravi F., Jurisica I., Andrulis I.L., Tsao M.S., Penn L.Z. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006;66:5330–5337. doi: 10.1158/0008-5472.CAN-06-0037. [DOI] [PubMed] [Google Scholar]
- 33.Yang F., Bi J., Xue X., Zheng L., Zhi K., Hua J., Fang G. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 2012;279:3159–3165. doi: 10.1111/j.1742-4658.2012.08694.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X., Zhou Y., Mehta K.R., Danila D.C., Scolavino S., Johnson S.R., Klibanski A. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocr. Metab. 2003;88:5119–5126. doi: 10.1210/jc.2003-030222. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Y., Zhong Y., Wang Y., Zhang X., Batista D.L., Gejman R., Ansell P.J., Zhao J., Weng C., Klibanski A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007;282:24731–24742. doi: 10.1074/jbc.M702029200. [DOI] [PubMed] [Google Scholar]
- 36.Zhang A., Zhou N., Huang J., Liu Q., Fukuda K., Ma D., Lu Z., Bai C., Watabe K., Mo Y.Y. The human long non-coding RNA-RoR is a p53 repressor in response to DNA damage. Cell Res. 2013;23:340–350. doi: 10.1038/cr.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J., Kastan M.B. 5′-3′-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev. 2010;24:2146–2156. doi: 10.1101/gad.1968910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bieging K.T., Attardi L.D. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012;22:97–106. doi: 10.1016/j.tcb.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berns A. Senescence: a companion in chemotherapy. Cancer Cell. 2002;1:309–311. doi: 10.1016/s1535-6108(02)00063-6. [DOI] [PubMed] [Google Scholar]
- 40.Abdelmohsen K., Kim M.M., Srikantan S., Mercken E.M., Brennan S.E., Wilson G.M., de Cabo R., Gorospe M. miR-519 suppresses tumor growth by reducing HuR levels. Cell Cycle. 2010;9:1354–1359. doi: 10.4161/cc.9.7.11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakamoto K., Fordis C.M., Corsico C.D., Howard T.H., Howard B.H. Modulation of HeLa cell growth by transfected 7SL RNA and Alu gene sequences. J. Biol. Chem. 1991;266:3031–3038. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.