


Abstract
ATP-dependent chromatin remodeling complexes regulate nucleosome organizations. In Drosophila, gene Brm encodes the core Brahma complex, the ATPase subunit of SWI/SNF class of chromatin remodelers. Its role in modulating the nucleosome landscape in vivo is unclear. In this study, we knocked down Brm in Drosophila third instar larvae to explore the changes in nucleosome profiles and global gene transcription. The results show that Brm knockdown leads to nucleosome occupancy changes throughout the entire genome with a bias in occupancy decrease. In contrast, the knockdown has limited impacts on nucleosome position shift. The knockdown also alters another important physical property of nucleosome positioning, fuzziness. Nucleosome position shift, gain or loss and fuzziness changes are all enriched in promoter regions. Nucleosome arrays around the 5′ ends of genes are reorganized in five patterns as a result of Brm knockdown. Intriguingly, the concomitant changes in the genes adjacent to the Brahma-dependent remodeling regions have important roles in development and morphogenesis. Further analyses reveal abundance of AT-rich motifs for transcription factors in the remodeling regions.
INTRODUCTION
Eukaryotic genomic DNA is densely packaged in nucleus through high-order chromatin architecture. Nucleosomes are the fundamental structural unit of chromatin, composed of a histone octamer wrapped by ∼147 bp DNA (1). Nucleosome positioning plays a critical role in numerous biological processes mainly through regulating the accessibility of DNA. There are various factors affecting nucleosome positioning such as DNA sequence, histone variants, histone tail modifications, ATP-dependent chromatin remodeling enzymes and histone chaperones. It has been reported that the AA/TT dinucleotides occurred in a biased and/or periodic arrangement across nucleosomal DNA around the 5′ end of genes in yeast (2). In contrast, the nucleosome positioning sequence pattern in Drosophila is CC/GG (3). Both histone variants and histone modifications often exist in the nucleosomes around the 5′ end of genes that could facilitate nucleosome eviction (4).
The SWItch/Sucrose NonFermentable (SWI/SNF) family, one of the ATP-dependent chromatin remodeling complex families, has diverse roles. Conditional knockout study in mice showed that Brg1, a member of the SWI/SNF family, played an important role in neural development through regulating sonic hedgehog signaling (5). The ablation of Brg1 in neural stem cells in mice failed to maintain neural stem cells in a gliogenic state and switched from neurogenesis to gliogenesis (6). Other studies implicated that SWI/SNF complexes also functioned in self-renewal and pluripotency of mouse embryonic stem cells, heart and thymocyte development (7–9). Inactivation of the key subunits of the mouse SWI/SNF complex (Snf5 or Brg1) resulted in a loss of nucleosome occupancy at target promoters (10). In Drosophila, there are two distinct SWI/SNF complexes, Brahma-associated proteins (BAP) and polybromo-containing BAP (PBAP), that are characterized by specific accessory subunits. Studies have found that the Drosophila Brahma complex played a role in wing development (11), cell cycle control (12), self-renewal of neural stem cells (13) and intestinal stem cell proliferation (14). The signature subunit Osa of BAP complex was required to repress the expression of Wingless target genes (15). The subunit Osa was also needed for the Drosophila wing development by regulating the epidermal growth factor receptor (EGFR) pathway (16) and mediating the expression of Apterous target genes (17). The signature subunit Bap170 of PBAP complex was required in normal eggshell development (18). Snr1, a highly conserved subunit of Brahma complex also functioned in the development of wing vein (19).
In spite of recent substantial effort, the role of Brahma complex, the common ATP-dependent catalytic subunit of Drosophila SWI/SNF class of chromatin remodelers, in affecting nucleosome landscape in vivo is unclear. To gain insights into this question, we employed the UAS-Gal4 system driven by the promoter of heatshock protein in Drosophila. We optimized a heating program to precisely control the timing of Brm knockdown in Drosophila third instar larvae. Then we generated and compared the high-resolution maps of nucleosome positions in the genome before and after Brm knockdown. Our results showed that knockdown of Brm led to extensive changes in nucleosome landscape. The resultant remodeled regions were enriched in the promoter regions. The differentially expressed genes adjacent to the remodeled regions had functions for disc or tissue morphogenesis.
MATERIALS AND METHODS
Fly stocks and crossing
Stocks used in this paper are UAS-mCD8GFP, UAS-Brm-IR (#31712) and Hs-Gal4 from Bloomington Stock Center (http://flystocks.bio.indiana.edu). All transgenes are on the third chromosome. We crossed UAS-Brm-IR males with Hs-Gal4 females to generate the transgenic strain containing UAS-Brm-IR and Hs-Gal4 for Brm knockdown (experimental group, short for BrmIR). Similarly, the transgenic strain containing UAS-mCD8GFP and Hs-Gal4 (control group, short for GFP) was generated by crossing UAS-mCD8GFP males with Hs-Gal4 females and expected to remove the influence of UAS-Gal4 system.
RNAi by heatshock at the third larvae stage of D. melanogaster
We collected the fly fertilized eggs of the control and experimental group, respectively, and cultured them at normal temperature (25°C) for 83.5 h (i.e. the third larvae stage). Next, we cultured the larvae at 37°C for 0.5 h. During the heatshock, Brm was knocked down by the expression of Brm inverted repeat (IR) transgene in the experimental group (denoted as BrmIR37). In contrast, Brm was not knocked down in the control group (denoted as GFP37) because of no expression of Brm IR transgene. Then we recovered the larvae at 25°C for 4.5 h. Repeat the heatshock-and-recovery treatment twice, but with recovery time of 3.5 and 2 h, respectively. After the heatshock treatment, the larvae were collected and used in this study. It is noteworthy that we tried other heatshock programs, e.g. heatshock in an earlier stage or longer heatshock time, leading to embryonic development arrest at early stages. The current heatshock program significantly reduced the Brm transcription and the embryos could develop until the late pupae. Therefore, this heatshock program was used in our study.
Total RNA extraction and qRT-PCR
Total RNA was extracted from the larvae using TRIzol (Invitrogen) that underwent the above heatshock treatment. Reverse transcription was performed on 500 ng of total RNA in 10 μl reaction system for each sample using PrimeScript RT Master Mix Perfect Real Time Kit (TAKARA, Code: DRR036A). The real-time polymerase chain reaction (PCR) was carried out in triplicate using F-416L DyNAmoTM ColorFlash SYBR® Green qPCR Kit (Thermo). Relative quantitative analysis of Brm expression levels was calculated using on 2−ΔΔCt formula described in a previous study (20) with gene tub as the reference gene. Primers for Brm are sense 5′-CCAATGCCGAGCGTGAAC-3′ and antisense 5′-ACTCATCTGTCTGCGACAGTAGGA-3′. Primers for tub are sense 5′-GCTGTTCCACCCCGAGCAGCTGATC-3′ and antisense 5′-GGCGAACTCCAGCTTGGACTTCTTGC-3′.
RNA-seq data analysis
The RNA sequencing libraries were constructed from the extracted RNA using standard Illumina libraries prep protocols. RNA-seq was performed on Illumina HiSeq2000 platform. Sequencing reads were aligned to annotated Drosophila transcripts (FlyBase r5.43) using TopHat with zero mismatch. The statistics of raw reads, mapped reads were summarized in Supplementary Table S1. The differentially expressed genes were identified by the tool Cuffdiff. For each transcript, reads per kilobase per million (RPKM) mapped reads were calculated to evaluate the expression level.
Western blotting
Total protein was extracted from 30 third instar larvae by homogenization in 600 RIPA lysis buffer 600 μl (Beyotime Biotechnology, China). Polyacrylamide gel electrophoresis and western blotting were performed as described previously (14). Antibodies used were as follows: rabbit anti-α tublin (1:1000, ab52866, Abcam), rabbit anti-Brm (1:500, a gift from Dr Lei Zhang, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China), secondary goat anti-rabbit IgG H&L (HRP) (1:1000, ab6721, Abcam).
Nuclei preparation and MNase-seq
We collected 18 larvae for each sample to prepare nuclei for MNase digestion, similarly to what has been described previously (21). Briefly, larvae were crosslinked in a 1.5-ml tube containing 0.5 ml A1 buffer (60 mM KCl, 15 mM NaCl, 4 mM MgCl2, 15 mM 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (pH 7.6), 0.5% Triton X-100, 0.5 mM DTT, 1×EDTA (Ethylene Diamine Tetraacetic Acid)-free protease inhibitor cocktail (Roche 04693132001)) + 1.8% formaldehyde. The tube was gently shaken for 15 min at room temperature. The crosslinked larvae were homogenized using a PRO200 homogenizer 220 V (PROScientific Inc., Oxford, CT, USA). Crosslinking was stopped by adding glycine (final concentration was 0.125 M) at room temperature for 5 min. The mixture was centrifuged at 2000g for 5 min at 4°C, and the supernatant was discarded. The pellet was washed as follows: once with 500 μl A1 buffer, and once with 500 μl A2 buffer (140 mM NaCl, 15 mM HEPES (pH 7.6), 1 mM EDTA, 0.5 mM ethylene glycol tetraacetic acid (EGTA), 1% Triton X-100, 0.1% sodium deoxycholate, 0.5 mM DTT, 1×EDTA-free protease inhibitor cocktail (Roche 04693132001)). For each wash, the tube was shaken for 1 min and centrifuged as before, and the supernatant was discarded. The pellet was resuspended in 500 μl A2 buffer + 0.1% sodium dodecyl sulfate (SDS) and incubated on a rotating wheel at 4°C for 10 min. Then the mixture was centrifuged at 16 000g for 5 min at 4°C and the supernatant was discarded.
The pellet was washed with 500 μl MNase digestion buffer (10 mM Tris–HCl (pH 7.5), 15 mM NaCl, 60 mM KCl, 1 mM CaCl2, 0.15 mM spermine, 0.5 mM spermidine, 1×EDTA-free protease inhibitor cocktail) and centrifuged at 16 000g for 10 min at 4°C. The nuclei were resuspended in 500 μl MNase digestion buffer plus 15 U MNase (Worthington, LS004797) at 37°C for 30 min. The MNase digestion was terminated on ice by adding EDTA to a final concentration of 10 mM for 10 min. The mixture was centrifuged at 16 000g for 10 min at 4°C. The supernatant was discarded. The pellet was resuspended in 500 μl A3 buffer (140 mM NaCl, 15 mM HEPES (pH 7.6), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 1×EDTA-free protease inhibitor cocktail). RNA was removed by digestion with 5 μl 10 mg/ml RNaseA at 37°C for 1 h in waterbath and with 3 μl 20 mg/ml Proteinase K at 65°C overnight in waterbath, respectively. Nucleosomal DNA was extracted by phenol–chloroform and dissolved in ddH2O. Mononucleosomal DNA fragments were extracted on a 2% agarose gel (Supplementary Figure S1) and purified by the MinElute Gel Extraction Kit (QIAGEN, 28604). The purified mononucleosomal DNA was subjected to massively parallel DNA sequencing on Illumina HiSeq2000 using single end protocol.
Nucleosome prediction and analysis of positioning dynamics
Sequencing reads were aligned to Drosophila melanogaster reference genome (dm3) using Bowtie with up to two mismatches. The statistics of raw reads, mapped reads were summarized in Supplementary Table S1. Only the uniquely mapped reads were retained in this study. The resulting sequence read distribution was used to identify nucleosomes using the peak-calling tool GeneTrack (22,23). Each nucleosome was assigned to either of promoter, genic or intergenic regions depending on in which region the midpoint of the nucleosome located. The nucleosome coordinates were given in Supplementary data file 1 (GFP37) and file 2 (BrmIR37).
We located the closest nucleosome in GFP37 sample for each nucleosome in BrmIR37 sample. The two nucleosomes were defined the same nucleosome if the distance between their midpoint was <80 bp. Otherwise, nucleosome gain and loss occurred. For each pair of nucleosomes, position shift was calculated by subtracting the midpoint coordinate of the nucleosome in GFP37 from that in BrmIR37. Nucleosome fuzziness was calculated as the standard deviation of the midpoint of the set of reads defining the same nucleosome as described previously (3). It measures how spread out a nucleosome position is. Nucleosome fuzziness difference was calculated by subtracting the nucleosome fuzziness in GFP37 from that in BrmIR37.
Nucleosome occupancy change analysis
We scanned the Drosophila genome with a 200-bp window, and calculated nucleosomal sequencing read count (RPKM) in each window as its nucleosome occupancy. The ratio of nucleosome occupancy after to before Brm knockdown in each window represents the nucleosome occupancy change. We retained the windows with at least 2-fold nucleosome occupancy change, i.e. ratio ≥2 (increase) or ratio ≤0.5 (decrease), for the next analysis. Each window was assigned to either of promoter, genic or intergenic region depending on which genomic region overlaps with ≥50% of the window length. Then, the enrichment of nucleosome occupancy increase in each genomic region is the total number of nucleosome occupancy increase windows normalized by the total length of corresponding genomic regions. The enrichment of nucleosome occupancy decrease was analyzed in the same way.
Nucleosome organization change around TSS
The annotation of all Drosophila genomic features were downloaded from FlyBase release 5.43 (ftp://ftp.flybase.net/genomes/dmel/dmel_r5.43_FB2012_01/fasta/). The nucleosome array within ±1 kb of the Transcription Start Site (TSS) was plotted as the previous study (24). Briefly, nucleosomes within ±1 kb of the TSS formed the nucleosome array of a gene. The nucleosome position was defined by its dyad. The length was equal to the fuzziness value that measured the spread-out of a nucleosome. We compared the nucleosome array of each gene before and after Brm knockdown. As a result, each site had either of the two possible nucleosomal states: occupied by or depleted of a nucleosome. There was no difference in nucleosome organization at a site that had the same nucleosomal state before and after Brm knockdown. Otherwise, nucleosomal state at a site was changed after Brm knockdown. Then nucleosome organization difference was clustered by K-means (K = 5). Note that we tried several K values and K = 5 gave distinct patterns among clusters and least variation within each cluster. The clustering results were plotted in heatmap.
Composite distribution of nucleosome relative to TSS
The composite nucleosome occupancy landscapes were calculated by aggregating read count at each distance relative to the TSS as described previously (3). Briefly, given a subset of TSSs, GFP37 and BrmIR37 nucleosome reads within ±1 kb of each subset of TSSs were collected, respectively. Each read represented a nucleosome. Thus, we extended each read toward 3′ end to a length of 147 bp. The midpoint of extended read defined the nucleosome position. Nucleosome distances to the TSS were binned in 10-bp intervals. Nucleosome occupancy was the total read count in the corresponding bin. Finally, nucleosome occupancy in each bin was further normalized as RPKM, and smoothed with five bins.
Brahma-dependent chromatin remodeling regions
Global Brahma-dependent chromatin remodeling regions were detected by the similar method used in the previous study (25). In brief, the normalized nucleosome signal of each genomic site, Ni, is the ChIP tag count at coordinate i normalized by total tag count in the sample. Then the nucleosome signal change of site i, Si, was measured by the log2 transformed ratio of Ni(BrmIR37) to Ni(GFP37). The average length of linkers in Drosophila is 18 bp (3). Thus, the midpoint distance of two adjacent nucleosomes is 165 bp on average. To reduce the false positive rate, we required a chromatin remodeling region that span at least three nucleosomes, and defined remodeling score at coordinate i as Zi = Si(Si−165 +Si+165). The sites whose absolute value of the Z score is ≥1 (FDR = 0.05) were defined as remodeling blocks. Blocks <165 bp apart were merged to one remodeling region. This analysis identified 3758 distinct regions, typically of 500–700 bp in length.
Accession numbers
The RNA-seq and MNase-seq data sets have been deposited in ArrayExpress database under accession numbers E-MTAB-1966 and E-MTAB-1967, respectively.
RESULTS
Brm knockdown leads to perturbation on gene transcription with diverse functions
We generated the control fly strain (UAS-mCD8GFP/Hs-Gal4) and the Brm RNAi fly strain (UAS-Brm-IR/Hs-Gal4). The Brm was knocked down by the expression of Brm IR transgene through heatshock (see ‘Materials and Methods’ section for the details). The qRT-PCR results showed that the expression level of Brm was significantly decreased by knockdown (Figure 1A). The western blotting result further confirmed the largely reduced level of Brahma complex in the knockdown sample (Figure 1B). The normalized read count on Brm gene body confirmed the high expression level of Brm IR transgene and low expression level of Brm in the Brm RNAi fly strain when compared to the control fly strain (Figure 1C). This suggested that the UAS-Gal4 system worked well to knockdown Brm in vivo in our study. To investigate the effect of Brm knockdown on global gene expression, we identified 872 significantly differentially expressed genes whose expression change was >2-fold after Brm knockdown (Figure 1D). The differentially expressed genes included the hormone-responsive Ecdysone-induced genes (Eig) and homeotic genes that were reported to be the targets of Brahma in the previous studies (26–28). Gene Ontology (GO) analysis found that the differentially expressed genes had diverse functions including molting cycles, de novo protein folding, immune response, sensory perception and more (Supplementary Figure S2). A previous study showed that the Brm mRNA level was significantly higher in 0–12 h Drosophila embryos than at later developmental stages (27). Consistent with it, knockdown of Brm in early embryos arrested the development of Drosophila embryos in our study. Moreover, Brm depletion from the zygote was lethal at late stages of embryonic development (29). Therefore, Brm has vital functions for the development of Drosophila embryos.
Figure 1.
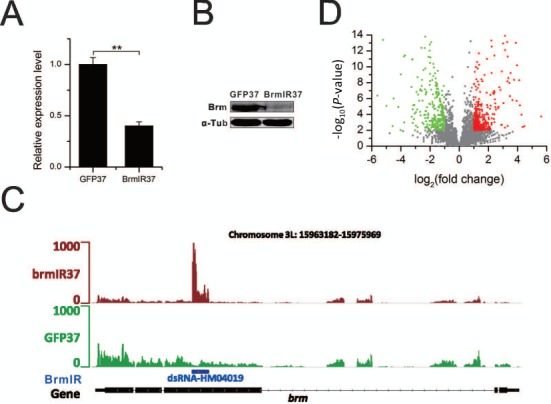
Impact of Brm knockdown on gene expression. (A) qPCR results confirm the significantly reduced expression level of Brm through its knockdown (**P-value < 0.01, two-tailed t-test). (B) Western blotting analysis quantifies relative abundance of Brahma without (GFP37) and with (BrmIR37) Brm knockdown. (C) The screenshot of RNA-seq read density shows the high expression level of Brm inverted repeat (IR) transgene by heatshock induction. The blue bar indicates the position of Brm IR in Brm gene. (D) The volcano plot presents the significantly differentially expressed genes (red and green dots) with two or more folds change and P-value < 0.01.
Brahma alters global nucleosome organization and the physical properties associated with nucleosome positions
We aligned the nucleosomal reads from MNase-seq against Drosophila genome. The read count in a given region indicates its nucleosome occupancy. We compared the genome-wide nucleosome occupancy before and after Brm knockdown. The results showed that nucleosome occupancy changes spread out the entire genome after Brm knockdown (Figure 2A and Supplementary Figure S3). To examine whether there is a bias in the distribution of nucleosome occupancy change in different genomic regions, we calculated the enrichment of nucleosome occupancy change in promoter, genic and intergenic regions. Our results showed a similar level of nucleosome occupancy increase in the all three genomic regions. In contrast, promoter and intergenic regions had higher enrichment of nucleosome occupancy decrease than genic regions. Moreover, nucleosome occupancy decrease was sufficiently higher than occupancy increase in either of the genomic regions (Figure 2B). This was partially attributed to the fact that Brahma-containing remodeling complexes (P)BAP promoted formation of nucleosome at their target sequences (30).
Figure 2.
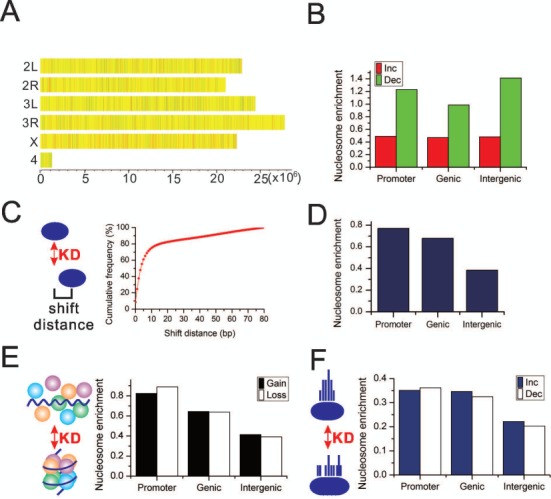
Brm knockdown alters nucleosome organizations. (A) Nucleosome occupancy changes throughout each chromosome. The genome is scanned with a 200-bp window. The ratio of normalized nucleosome occupancy after over before Brm knockdown within each window is presented by a color. Red indicates the regions where occupancy increases by two or more folds after knockdown. Green indicates the regions where occupancy decreases by two or more folds after knockdown. Yellow indicates the regions with unchanged occupancy, i.e. the ratio is <2-fold. (B) The enrichment of nucleosome occupancy change in different genomic regions. The enrichment of nucleosome occupancy increase/decrease in promoters equals to the number of red/green windows in (A) locating in promoters normalized by the length of promoters. The enrichment of nucleosome occupancy change in genic and intergenic regions are calculated in the same way. (C) Left schematic diagram illustrates nucleosome position shift. The curve plot shows the cumulative frequency of position shift after knockdown. (D) The enrichment of the shifted nucleosomes in the different genomic regions. (E) Left schematic diagram illustrates nucleosome gain and loss. The bar plot shows the enrichment of the nucleosome gain and loss in the different genomic regions. (F) Left schematic diagram illustrates nucleosome fuzziness change. The bar plot shows the enrichment of the nucleosomes whose fuzziness is changed in the different genomic regions. The nucleosome enrichment in (D, E, F) equals to the number of each category of nucleosomes in a given genomic region normalized by the length of corresponding genomic regions (see ‘Materials and Methods’ section for details). KD, knockdown.
To further examine the impact of Brm knockdown on the physical properties associated with nucleosome positions, we employed GeneTrack (23) to generate genome-wide maps of mononucleosome positions before and after Brm knockdown for comparison, respectively. We first investigated how Brm knockdown affected nucleosome position shift. 89.6% of total nucleosomes (406 286) shifted after Brm knockdown. More specifically, 66.4% of nucleosomes shifted <10 bp. There was no shift in only 8.5% of nucleosomes (Figure 2C). Thus, Brm knockdown led to a limited change in nucleosome positions. Unlike occupancy change, these shifted nucleosomes were enriched in promoter regions and gene body (Figure 2D). In addition to nucleosome shift, Brm knockdown also caused nucleosome disassembly (loss) and assembly (gain). Intriguingly, there was also an enrichment of nucleosome gain or loss in promoter regions and gene body (Figure 2E). There was similar number of nucleosome gain and loss within each genomic regions. Although nucleosome gain or loss occurred in 7381 genes, only 379 (5%) genes were significantly differentially transcribed. These findings suggested that nucleosome organization changes was not tightly correlated with gene transcription changes. The similar findings were also reported in yeast (31). GO term analysis found that these genes had a key role in positive regulation of kinase activity (Supplementary Figure S4).
Fuzziness is another important physical property of nucleosome positions. It measures the delocalization of a nucleosome position. The larger the fuzziness, the more delocalized a nucleosome positioning is. Conversely, a highly phased nucleosome has a very small fuzziness value and its position is fixed irrespective of cell state or developmental stage. Interestingly, Brm knockdown significantly changed the fuzziness of 19 081 nucleosomes (Student's t-test, P-value < 0.01). Consistent with nucleosome shift and gain/loss, nucleosomes with fuzziness change were enriched in promoter and gene body (Figure 2F). The fuzziness change was bidirectional. Some nucleosome positioning became delocalized after Brm knockdown and some was opposite. Unlike nucleosome occupancy change, there was no bias in fuzziness increase or decrease.
Nucleosome landscape changes around TSS regions arise from knockdown of Brm
The −1, NFR (nucleosome free region), +1, +2, +3, etc. canonical nucleosome arrangement around TSS regions plays an important role in gene transcription regulation and is linked to many biological processes (3,4,22,32,33). To gain insights on how Brm knockdown alters nucleosome profiles around TSS regions, we explored nucleosome organization difference in the regions surrounding TSS. The unsupervised clustering of nucleosome organization changes around TSS regions revealed five distinct patterns (Figure 3). The original composite distribution of nucleosomes around TSS of approximately two-thirds of genes (Cluster I) showed a very similar pattern before and after Brm knockdown. There was only slight shift in +3, +4, +5, etc. downstream nucleosomes and the nucleosomes >500 bp upstream of TSS (Cluster I). In contrast, Brm knockdown resulted in pronounced shift to 3′ end in +3, +4, +5, etc. downstream nucleosomes. The positioning phase of highly phased −1, +1 and +2 nucleosomes remained unchanged (Cluster III). Another pattern of nucleosome profiles showed position shift of nucleosomes nearby 1-kb upstream of TSS (Cluster VI). There was also marked shift of the entire canonical nucleosomes around TSS (−1, +1, +2, +3, +4, +5, etc. nucleosomes). The shift direction was towards 5′ end in a group of genes (Cluster II) and toward 3′ end in the other group of genes (Cluster V). The functional analysis showed that the different classes of genes had the enrichment of distinct GO terms (Supplementary Figure S5). For example, respiratory system development, cell motion, cuticle development, etc. were enriched in Cluster II genes. Oxidation reduction, mesoderm development, gastrulation, etc. were enriched in Cluster V genes.
Figure 3.
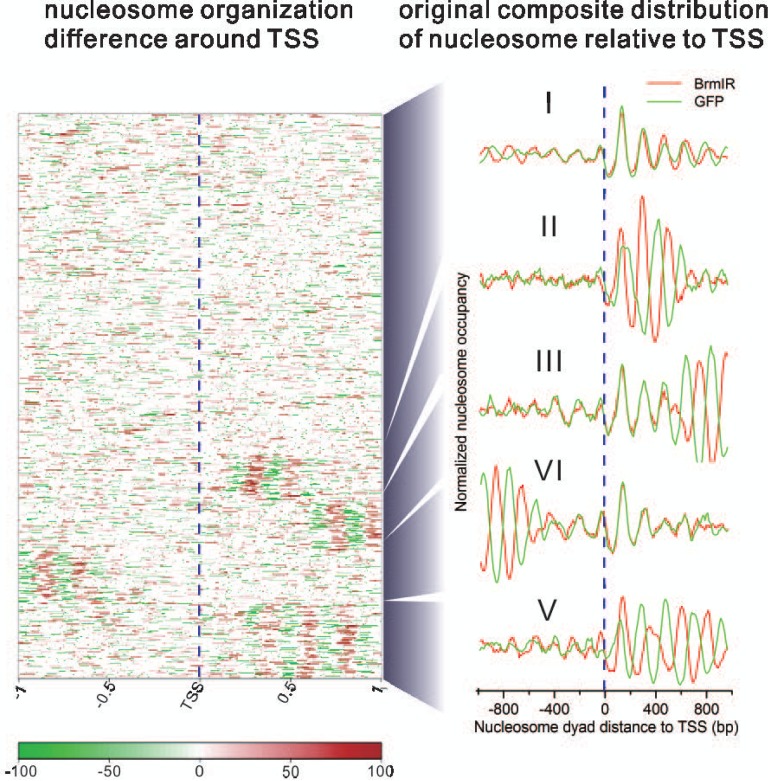
Nucleosome landscape changes around TSS by Brm knockdown. Cluster view (left panel) shows changes in nucleosome organizations around the 5′ end of genes. Red indicates nucleosome occupancy increase after knockdown. Green indicates nucleosome occupancy decrease after knockdown. White implies no change in nucleosome occupancy after knockdown. Curve plots (right panel) show the original composite distribution of nucleosome relative to TSS before and after knockdown, respectively. On the basis of nucleosome change, there are five distinct patterns of altered nucleosomal phasing arising from Brm knockdown. The vertical dotted line indicates TSS.
AT-rich motifs for transcription factors are abundant in Brahma-dependent chromatin remodeling regions
To systematically identify Brahma-dependent chromatin remodeling regions, we developed a comparative approach to detect difference in nucleosome occupancy before and after Brm knockdown. The analysis identified 3758 remodeling regions that were enriched in the promoter regions. Particularly, there was abundance of the remodeling regions adjacently upstream of TSS (Figure 4A and B). We further collected 1301 genes whose TSS located within 1 kb of these remodeling regions. Only 14.3% of these genes were differentially expressed. Seventy genes were up-regulated and 116 genes were down-regulated after Brm knockdown (Figure 4C). Intriguingly, changes of these gene expression affected embryonic development and morphogenesis (Figure 4D).
Figure 4.
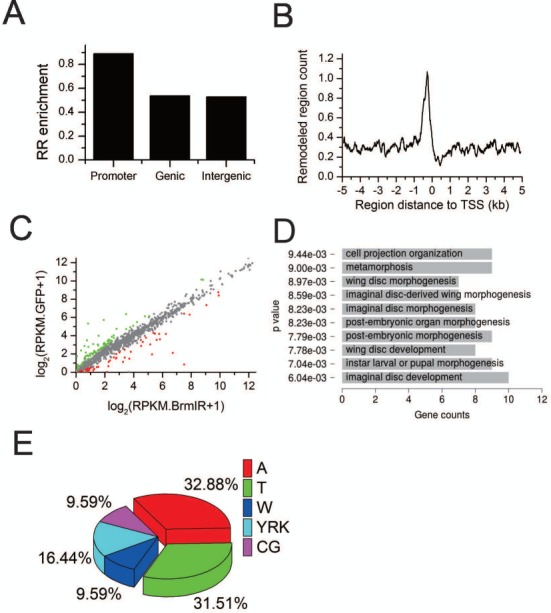
Distribution of Brahma-dependent remodeling regions (RRs) regulating gene transcription. (A) RRs are enriched in promoter regions. (B) Distribution of RRs around TSS. (C) Scatter plot compares the transcription level of the genes whose TSS located within 1 kb of these RRs before and after Brm knockdown, and highlights the differentially expressed ones in red and green (2-fold, P < 0.01, t-test). (D) GO term analysis for the differentially expressed genes in (C). (E) Sequence composition of the top 10 motifs identified in RRs. W = AT, Y = CT, R = AG, K = GT.
The key function of nucleosome positioning is to occlude many regulatory DNA elements. Therefore, we scanned the remodeling regions to identify motifs as the potential binding sites for transcription factors (TFs) using Cistrome (34). The motif discovery results found a bunch of motifs for TFs (Supplementary Table S2). This suggested that Brahma complex regulate the accessibility of these motifs through chromatin remodeling. Interestingly, sequence composition analysis revealed that the sequences of these motifs contained an extremely high level of A and T nucleotides (Figure 4E). The previous work have reported that the rigid poly (dA:dT) tracts disfavored nucleosome formation (35,36). Coincidently, the target sites of Drosophila SWI/SNF complexes disfavored nucleosome formation (30). This partially explained Brm knockdown resulted in more nucleosome occupancy decrease than nucleosome occupancy increase (Figure 2A and B). Notably, presence of TF motifs in the remodeling regions provided a mechanism by which TFs helped recruit chromatin remodelers to their target sites. For example, yeast ISW2 chromatin remodeling complex repressed early meiotic genes by establishing an inaccessible chromatin structure upon recruitment by Ume6p (37).
DISCUSSION
Nucleosomes are the primary repeating units of eukaryotic chromatin structure, controlling access of DNA. Thereby, nucleosome organization plays important roles in many biological processes. ATP-dependent chromatin remodeling enzymes are one of the key factors modulating nucleosome positioning. Drosophila Brm gene encodes the ATPase subunit of the two forms of SWI/SNF chromatin remodeling complexes (BAP and PBAP). Our data demonstrated that Brm knockdown resulted in extensive changes in nucleosome occupancy throughout the genome. Nucleosome position shift, fuzziness change, assembly and disassembly were enriched in the promoter regions. Motif sequences in the remodeling regions possessed the characteristics of disfavoring nucleosome formation. The accompanied gene expression changes had a variety of functions that may cause the arrest of Drosophila embryonic development after Brm knockdown.
Chromatin remodelers counteract DNA sequence-driven nucleosome positioning through binding to their target sites. Interestingly, ChIP-chip analysis found the binding overlapped between chromatin remodeling complexes (P)BAP, NURD and INO80, but not ISWI (30). We also compared our Brahma-dependent chromatin remodeling regions with the reported binding sites of (P)BAP, NURD, INO80 and ISWI (30). Intriguingly, the remodeling regions were enriched on the binding sites of (P)BAP, NURD and INO80, but depleted on the binding sites of ISWI (Supplementary Figure S6). This results could be partially attributed to the fact that the target sits of (P)BAP, NURD and INO80 disfavored nucleosome position whereas the remodeling sites of ISWI promoted nucleosome placement (30). The enrichment of Brahma-dependent chromatin remodeling regions on the binding sites of (P)BAP, NURD and INO80 may also implicate their potential involvement in Brahma-dependent chromatin remodeling. However, more validation assays are required to resolve whether there is an interplay between the different families of chromatin remodelers during Brahma-dependent chromatin remodeling. For example, the identification of the binding sites of Brahma by ChIP-seq in larvae and overlapping analysis of their binding sites.
Chromatin remodeling changes nucleosome positioning and consequently gene expression. However, the extent of impact on gene expression of nucleosome positioning depends on the genomic site where nucleosome remodeling occurs. It was reported that the canonical arrangement of −1, NFR, +1, +2, +3, etc. nucleosomes around TSS played important roles in gene expression (3,4). The active genes possessed this canonical nucleosome organization around their TSSs whereas the TSSs of silent genes were occupied by a nucleosome blocking their access (33,38). In our study, 14.3% of the genes whose TSS located within 1 kb of the remodeling regions expressed differentially after brm knockdown whereas only 5% genes with nucleosome gain or loss on the gene body expressed differentially. There was significantly over proportion of the genes nearby the remodeling regions that expressed differentially after brm knockdown (χ2 test, P-value = 2.45E−29). This further confirmed that nucleosome occupancy change in TSS had a stronger correlation with gene expression. Although nucleosome occupancy usually occludes access to DNA sequences, regulatory DNA elements on nucleosome surface could also be bound by TFs through nucleosomal rotational setting (22). Thus, the occlusion effect of nucleosome occupancy on DNA access was weakened. Of note, in addition to nucleosome occupancy, other factors including histone modifications and DNA methylation also regulate gene expression. As a matter of fact, these factors cooperated to regulate gene expression through complex cross talks (39). Therefore, there was not always a strong association between nucleosome remodeling alone and gene expression change, and vice versa. For example, inactivation of Isw1 and Chd1 in yeast obtained the similar findings that the resulting perturbation in nucleosome organizations was not correlated with changes in gene transcription (31). In summary, Brahma-dependent chromatin remodeling alters nucleosome landscape and regulates gene transcription in an intricate way.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to thank the anonymous reviewers for their valuable comments and suggestions. We thank Dr Zhang for providing us with rabbit anti-Brm antibody.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Ministry of Science and Technology of China [2010CB944901, 2011CB965104, 2012AA020405]; National Natural Science Foundation of China [91019017, 31271373, 31200952]; Aurora Talent Project of Shanghai [10SG24]; the Program for Eastern Scholar of Shanghai, the Fundamental Research Funds for the Central Universities [20113048, 20113109]. Funding for open access charge: Ministry of Science and Technology of China.
Conflict of interest statement. None declared.
REFERENCES
- 1.Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Ioshikhes I.P., Albert I., Zanton S.J., Pugh B.F. Nucleosome positions predicted through comparative genomics. Nat. Genet. 2006;38:1210–1215. doi: 10.1038/ng1878. [DOI] [PubMed] [Google Scholar]
- 3.Mavrich T.N., Jiang C., Ioshikhes I.P., Li X., Venters B.J., Zanton S.J., Tomsho L.P., Qi J., Glaser R.L., Schuster S.C., et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453:358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang C., Pugh B.F. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhan X., Shi X., Zhang Z., Chen Y., Wu J.I. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc. Natl. Acad. Sci. U.S.A. 2011;108:12758–12763. doi: 10.1073/pnas.1018510108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsumoto S., Banine F., Struve J., Xing R., Adams C., Liu Y., Metzger D., Chambon P., Rao M.S., Sherman L.S. Brg1 is required for murine neural stem cell maintenance and gliogenesis. Dev. Biol. 2006;289:372–383. doi: 10.1016/j.ydbio.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 7.Ho L., Ronan J.L., Wu J., Staahl B.T., Chen L., Kuo A., Lessard J., Nesvizhskii A.I., Ranish J., Crabtree G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. U.S.A. 2009;106:5181–5186. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chi T.H., Wan M., Lee P.P., Akashi K., Metzger D., Chambon P., Wilson C.B., Crabtree G.R. Sequential roles of Brg, the ATPase subunit of BAF chromatin remodeling complexes, in thymocyte development. Immunity. 2003;19:169–182. doi: 10.1016/s1074-7613(03)00199-7. [DOI] [PubMed] [Google Scholar]
- 9.Lickert H., Takeuchi J.K., Von Both I., Walls J.R., McAuliffe F., Adamson S.L., Henkelman R.M., Wrana J.L., Rossant J., Bruneau B.G. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- 10.Tolstorukov M.Y., Sansam C.G., Lu P., Koellhoffer E.C., Helming K.C., Alver B.H., Tillman E.J., Evans J.A., Wilson B.G., Park P.J., et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc. Natl. Acad. Sci. U.S.A. 2013;110:10165–10170. doi: 10.1073/pnas.1302209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herr A., McKenzie L., Suryadinata R., Sadowski M., Parsons L.M., Sarcevic B., Richardson H.E. Geminin and Brahma act antagonistically to regulate EGFR-Ras-MAPK signaling in Drosophila. Dev. Biol. 2010;344:36–51. doi: 10.1016/j.ydbio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 12.Moshkin Y.M., Mohrmann L., van Ijcken W.F., Verrijzer C.P. Functional differentiation of SWI/SNF remodelers in transcription and cell cycle control. Mol. Cell. Biol. 2007;27:651–661. doi: 10.1128/MCB.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumuller R.A., Richter C., Fischer A., Novatchkova M., Neumuller K.G., Knoblich J.A. Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell. 2011;8:580–593. doi: 10.1016/j.stem.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin Y., Xu J., Yin M.X., Lu Y., Hu L., Li P., Zhang P., Yuan Z., Ho M.S., Ji H., et al. Brahma is essential for Drosophila intestinal stem cell proliferation and regulated by Hippo signaling. eLife. 2013;2:e00999. doi: 10.7554/eLife.00999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins R.T., Treisman J.E. Osa-containing Brahma chromatin remodeling complexes are required for the repression of wingless target genes. Genes Dev. 2000;14:3140–3152. doi: 10.1101/gad.854300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terriente-Felix A., de Celis J.F. Osa, a subunit of the BAP chromatin-remodelling complex, participates in the regulation of gene expression in response to EGFR signalling in the Drosophila wing. Dev. Biol. 2009;329:350–361. doi: 10.1016/j.ydbio.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Milan M., Pham T.T., Cohen S.M. Osa modulates the expression of Apterous target genes in the Drosophila wing. Mech. Dev. 2004;121:491–497. doi: 10.1016/j.mod.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Carrera I., Zavadil J., Treisman J.E. Two subunits specific to the PBAP chromatin remodeling complex have distinct and redundant functions during drosophila development. Mol. Cell. Biol. 2008;28:5238–5250. doi: 10.1128/MCB.00747-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marenda D.R., Zraly C.B., Dingwall A.K. The Drosophila Brahma (SWI/SNF) chromatin remodeling complex exhibits cell-type specific activation and repression functions. Dev. Biol. 2004;267:279–293. doi: 10.1016/j.ydbio.2003.10.040. [DOI] [PubMed] [Google Scholar]
- 20.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Li L.M., Arnosti D.N. Fine mapping of chromatin structure in Drosophila melanogaster embryos using micrococcal nuclease. Fly. 2010;4:213–215. doi: 10.4161/fly.4.3.12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albert I., Mavrich T.N., Tomsho L.P., Qi J., Zanton S.J., Schuster S.C., Pugh B.F. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 23.Albert I., Wachi S., Jiang C., Pugh B.F. GeneTrack—a genomic data processing and visualization framework. Bioinformatics. 2008;24:1305–1306. doi: 10.1093/bioinformatics/btn119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z., Wippo C.J., Wal M., Ward E., Korber P., Pugh B.F. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science. 2011;332:977–980. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitehouse I., Rando O.J., Delrow J., Tsukiyama T. Chromatin remodelling at promoters suppresses antisense transcription. Nature. 2007;450:1031–1035. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- 26.Brizuela B.J., Elfring L., Ballard J., Tamkun J.W., Kennison J.A. Genetic analysis of the brahma gene of Drosophila melanogaster and polytene chromosome subdivisions 72AB. Genetics. 1994;137:803–813. doi: 10.1093/genetics/137.3.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamkun J.W., Deuring R., Scott M.P., Kissinger M., Pattatucci A.M., Kaufman T.C., Kennison J.A. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell. 1992;68:561–572. doi: 10.1016/0092-8674(92)90191-e. [DOI] [PubMed] [Google Scholar]
- 28.Zraly C.B., Middleton F.A., Dingwall A.K. Hormone-response genes are direct in vivo regulatory targets of Brahma (SWI/SNF) complex function. J. Biol. Chem. 2006;281:35305–35315. doi: 10.1074/jbc.M607806200. [DOI] [PubMed] [Google Scholar]
- 29.Brown E., Malakar S., Krebs J.E. How many remodelers does it take to make a brain? Diverse and cooperative roles of ATP-dependent chromatin-remodeling complexes in development. Biochem. Cell Biol. 2007;85:444–462. doi: 10.1139/O07-059. [DOI] [PubMed] [Google Scholar]
- 30.Moshkin Y.M., Chalkley G.E., Kan T.W., Reddy B.A., Ozgur Z., van Ijcken W.F., Dekkers D.H., Demmers J.A., Travers A.A., Verrijzer C.P. Remodelers organize cellular chromatin by counteracting intrinsic histone-DNA sequence preferences in a class-specific manner. Mol. Cell. Biol. 2012;32:675–688. doi: 10.1128/MCB.06365-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gkikopoulos T., Schofield P., Singh V., Pinskaya M., Mellor J., Smolle M., Workman J.L., Barton G.J., Owen-Hughes T. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science. 2011;333:1758–1760. doi: 10.1126/science.1206097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mavrich T.N., Ioshikhes I.P., Venters B.J., Jiang C., Tomsho L.P., Qi J., Schuster S.C., Albert I., Pugh B.F. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18:1073–1083. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schones D.E., Cui K., Cuddapah S., Roh T.Y., Barski A., Wang Z., Wei G., Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu T., Ortiz J.A., Taing L., Meyer C.A., Lee B., Zhang Y., Shin H., Wong S.S., Ma J., Lei Y., et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson J.D., Widom J. Poly(dA-dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites. Mol. Cell. Biol. 2001;21:3830–3839. doi: 10.1128/MCB.21.11.3830-3839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan G.C., Liu Y.J., Dion M.F., Slack M.D., Wu L.F., Altschuler S.J., Rando O.J. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- 37.Goldmark J.P., Fazzio T.G., Estep P.W., Church G.M., Tsukiyama T. The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell. 2000;103:423–433. doi: 10.1016/s0092-8674(00)00134-3. [DOI] [PubMed] [Google Scholar]
- 38.Teif V.B., Vainshtein Y., Caudron-Herger M., Mallm J.P., Marth C., Hofer T., Rippe K. Genome-wide nucleosome positioning during embryonic stem cell development. Nat. Struct. Mol. Biol. 2012;19:1185–1192. doi: 10.1038/nsmb.2419. [DOI] [PubMed] [Google Scholar]
- 39.Jones P.A., Baylin S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.