

Abstract
We describe a proof-of-principal, immune sandwich assay in which immune complexes link micron-size beads via DNA tethers to a sensor surface. The number of tethered beads, counted using low-magnification microscopy, provides a measure of the concentration of analyte. The prototype assay was sensitive to pM concentration of analyte. In theory, the assay could be sensitive to sub-fM analyte because beads attached via single-immune complexes and DNA strands form tethers, and tether formation in the absence of analyte is extremely rare. The limiting step at present is binding of streptavidin at the end of DNA to biotin on capture beads. Potential advantages of this type of sensor are discussed.
Keywords: thered-bead immunoassay, DNA tethers, flow-mediated DNA extension, optomicrofluidics, PSA immunoassay, single-molecule immunoassay
1. Introduction
Over the past few decades several methods have been developed that can detect very low concentrations of analyte molecules. For immunoassays, these methods often involve immune capture of analyte on a solid phase followed by binding of a labeled, second antibody (“immune sandwich”) and sensitive detection of the label. Transduction methods for label detection include total internal reflection-fluorescence microscopy (Yanagida et al., 2000; Jain et al., 2011), mechanical oscillators sensitive to the mass of single gold nanoparticle labels (Burg et al., 2007), field effect transistors (Liu and Guo, 2012), whispering gallery mode optical sensors (Vollmer and Arnold, 2008), magnetic sensors sensitive to micro- or nanometer sized magnetic particles (Osterfeld et al., 2008; Mulvaney et al., 2007), and enzyme-based assays with enhanced sensitivity resulting from confinement of enzyme products to micron-sized chambers (Rissin et al., 2010). The enhanced sensitivity of these methods often requires expensive, cumbersome detection equipment. This paper describes a simple transduction method potentially sensitive to single molecules, based on optical detection of the motion of micron-sized beads attached via immune sandwiches to the ends of single molecules of DNA (“tethered bead” technology).Tethered beads can be detected with a low-magnification imaging system and inexpensive camera.
Tethered bead technology was initially developed to study biophysical properties of DNA molecules and enzymes that act on DNA, at the single molecule level (Schafer et al., 1991; Cluzel et al., 1996; Larsen et al., 1997; Bustamante et al., 2003; Abbondanzieri et al., 2005; Kim et al., 2007; Neuman and Nagy, 2008). A typical experimental set-up is shown in Figure 1A. DNA molecules of known length are attached via one end to a surface in a flow cell. The other end of the DNA is attached to a micron-sized bead that can be pulled to stretch the DNA. Pulling can be effected via liquid flow (drag force on the bead), laser trapping, or magnetic force if the bead is paramagnetic. Tethered beads are easily identified in light microscopy by their characteristic motion – they exhibit damped and confined Brownian motion in the absence of external force, and translation by a predictable distance in the presence of an external force. In single-molecule studies of DNA, the tether length is usually measured very precisely to provide quantitative information about DNA conformation and elasticity. In the method described here, tether length, measured with relatively low precision, provides a means of identifying beads that have captured analyte molecules.
Figure 1.

A, typical configuration of tethered bead in single-molecule pulling experiment. B, attachment configuration in immune-sandwich tethered bead assay. C, flow cell assembly.
To use tethered bead technology in a biosensor, we attached DNA tethers to beads via immune sandwiches. One way of doing this is illustrated in Figure 1B. One end of a linear DNA is attached to a flow cell surface, and the other end of the DNA is labeled with biotin and then streptavidin. Beads coated with antibody against an analyte are used as a solid phase to capture analyte molecules from a sample fluid. The beads are then washed and exposed to a different, biotin-labeled, “detection” antibody that can bind to captured analyte molecules, forming an immune sandwich. These beads are introduced into the flow cell and the number of beads that become tethered provides a measure of the number of captured analyte molecules.
We describe a “proof-of-principle” for this type of assay using prostate specific antigen (PSA) as the analyte. Although the clinical utility of measuring PSA levels for screening purposes is controversial (US Preventive Services Task Force, 2012), PSA tests are used to monitor response of prostate cancer to treatment and PSA is commonly used to evaluate new assay methods (Rissin et al., 2010). Serial dilution of PSA showed that the lowest detectable concentration in our prototype assay was ~1pM PSA (30pg/ml) in a 50μl sample, or ~300 analyte molecules/bead. This is lower than needed in most clinical assays, but since bead motion strongly suggested that most tethered beads were attached via single analyte molecules and single-molecule detection would predict a much smaller, lowest detectable concentration, we investigated the efficiency of various steps in the assay. We describe technical challenges that must be overcome to decrease the lower limit of detection to ~1 molecule of analyte per bead. Very sensitive detection methods have applications in the diagnosis and treatment of infectious diseases and cancer.
2. Experimental
2.1 Flow cells
Flow cells were made by attaching coverslips to microscope slides via ~100μm thick gaskets made of double-sided sticky tape (3M Company, 300SLE adhesive) (Figure 1C). The coverslips had two ~1mm holes drilled by bombardment with 50μm AlO2particles using a Comco Inc. MicroBlaster. Nylon adaptors (McMaster Carr #91145A133) were attached over the holes with epoxy glue or melted parafilm to provide inflow and outflow ports. Slides and coverslips were cleaned by sonicating in 15% KOH in ethanol and deionized water before assembling into flow cells. Alternatively, flow cells were made by bonding air-plasma treated polydimethylsiloxane (PDMS) slabs to microscope slides; the PDMS slabs were cast over scotch tape strips to form shallow channels ~40mm×2mm×0.1mm; inflow and outflow ports were formed by puncturing the PDMS with syringe needles. Flow cell volumes were ~30μl.
2.2 DNA
DNA for tethers was prepared from a 17kb plasmid derived from pBluescript (Stratagene); the sequence is available on request. The linearized plasmid was labeled with digoxigenin at one end (for attachment to flow cell surfaces via anti-digoxigenin antibody), and biotin at the other end (for attachment to capture beads via streptavidin), as follows: The plasmid was linearized with restriction enzyme BsaI, which cuts the plasmid once leaving overhangs 5'GTTT--and 5'AAAC--. The GTTT overhang was filled in with biotin-dATP using Klenow DNA polymerase lacking 3'-exonuclease activity (New England Biolabs) followed by column purification (Qiagen) to remove unincorporated biotin-dATP. This should attach a maximum of 3 biotins at the end of each DNA. Since a streptavidin molecule binds 4 biotins, each streptavidin bound to biotin at the end of a DNA would have at least one unoccupied site capable of binding an additional biotin. The AAAC overhang at the other end of the DNA was then filled in with dGTP and digoxigenin-dUTP (Roche Applied Science) and unincorporated nucleotides removed as above. Flow cells were filled with 0.1μM polyclonal sheep antibody to digoxigenin (Roche, #11333089001) in phosphate buffered saline (PBS) and incubated for ~1h to non-specifically adsorb anti-digoxigenin antibody to the flow cell surfaces. Flow cell surfaces were then passivated by washing with wash buffer (WB): 0.3% bovine serum albumin, 0.5mM EDTA, 0.01% Tween-20, and 0.02% sodium azide in PBS. 108–109 molecules of biotin/digoxigenin-end labeled DNA were introduced into the flow cell and incubated for 2–4h to attach to the anti-digoxigenin antibodies. Unattached DNA was removed by washing with WB. Streptavidin (20nM in WB) was added to bind biotin at the end of the DNAs, and unattached streptavidin was removed by washing with ~20 flow cell volumes of WB under gravity flow.
2.3 Capture beads
Capture beads were made by covalently attaching anti-PSA monoclonal antibody to 2.8μm- paramagnetic beads using hydrazine-aldehyde chemistry (Solulink). Anti-PSA monoclonal antibody (Biospacific #8311) was modified by covalent attachment of hydrazine-nicotinamide to primary amino groups. The hydrazine-derivatized antibody was incubated with 4-fluorobenzene-derivatized paramagnetic beads (Magnalink beads, Solulink) according to the manufacturer's instructions. Efficiency of antibody attachment to beads was estimated spectroscopically by measuring the antibody (protein) concentration before and after incubation: about 90% of antibody attached to beads, equivalent to ~106 antibody molecules/bead.
2.4 Biotinylation
Detection antibody (Biospacific, monoclonal 8301) was biotinylated using NHS-PEG4 biotin (Thermo Scientific #21329). For analytic purposes (see below), a batch of PSA analyte was biotinylated in the same way.
2.5 Enzyme-linked immunoassay (ELISA)
The efficiency with which capture beads bound low concentrations of PSA, detection antibody, and streptavidin was estimated by ELISA as described below, using streptavidin-peroxidase and Sure Blue enzyme substrate (KPL Inc). Bead-bound streptavidin-peroxidase was determined from light-absorption of enzyme-modified substrate using a Nanodrop spectrophotometer.
2.6 Optical detection of tethered beads
Flow cells were imaged with 5x or 10x objectives using transmitted light or darkfield illumination. Movies of bead motion were taken at 7.5 frames per second with a Thorlabs CCD camera (model DC310-C) or an Imaging source CMOS camera (model DMK 72AUC02) while fluid was pushed alternately to the left and right for ~1–2 seconds in each direction using a syringe and tubing connected to one of the ports of a flow cell. Flow rate was adjusted manually while watching the motion of free beads. Beads were deemed tethered if they moved more than 1 bead diameter and less than 5 bead diameters in each direction under flow that was maintained in a given direction for a few seconds. Free beads moved >>5 bead diameters in each directions under these conditions.
3. Results and Discussion
3.1 Tethered bead immune sandwich assay
105 anti-PSA capture beads were incubated with 1nM PSA in 50μl WB for 1 hour on a rotary mixer. At 106 antibody molecules/bead (see methods), the capture antibody concentration is nominally 3nM. Since this is greater than the PSA concentration-and 10 times the reported dissociation constant of the antibody for PSA (KD= 0.1nM, Biospacific), most PSA molecules in the incubation mixture should be captured on beads at equilibrium. More specifically, the fraction, f, of PSA molecules that should be captured at equilibrium can be estimated from the well-known relationship between free ligand and fractional receptor occupancy, f (e.g., see Squires et al, 2008): f=1/(1+KD/[Ab]) =.9, where [Ab] is the concentration of “free” antibody molecules. A characteristic time to reach equilibrium for many antibody-antigen reactions at nM concentration is ~20–60 minutes (Sadana, 1995). The beads were pulled to the side of the tube using a magnet, washed, and incubated with 50μl of 1nM biotinylated detection antibody for 1 hour. The capture and detection antibodies bind to different epitopes on PSA and do not interfere with each other's binding (Ferguson et al., 1996). Since the detection antibody was in excess over the potentially captured analyte, and its concentration was ~100x the KD of its binding to PSA (0.01nM), almost all of the captured PSA molecules should bind detection antibody at equilibrium. After washing, the beads were introduced into flow cells coated with DNA strands that had streptavidin at their distal ends (see methods). Beads were introduced by pipeting ~105 beads suspended in ~1 flow cell volume of WB into the inflow port of a flow cell from which excess WB had been drained, inverting the flow cell several times to mix the added bead suspension with WB retained in the flow cell. Flow cells were not further washed, so unattached beads remained in the flow cell. ~50μl of WB was added to entry and exit port reservoirs and a syringe and tubing filled with air was attached to one of the ports. Gentle pushing and pulling on the syringe plunger caused fluid in the reservoirs to flow back and forth. Movies S1 and S2 show examples of bead motion observed using an inverted microscope with a 10X objective and darkfield illumination. Movie S1 shows beads that had been exposed to 1nM PSA. Most of the beads appeared tethered, moving ~12μm (judged from an ocular calibration ruler) as fluid flowed back and forth. By contrast, unattached beads, which appear as streaks in some frames of the movie due to rapid movement, moved ~120μm before reversing direction with the flow. Movie S2 shows beads from a control experiment in which PSA was omitted. Almost all of the beads in this movie are unattached, as expected.
The expected travel distance for tethered beads was calculated using a model (appendix) that takes into account DNA length in base pairs, DNA extension as a function of drag force based on the worm-like chain model (Bustamante et al., 1994), and drag force as a function of fluid velocity (Goldman et al., 1967). The calculation determines the position of DNA attachment on the bead surface that balances drag force and torque when a tether is extended by flow. The model predicts a center-to-center bead travel distance of 12μm for fluid flow of 120μm/s around a tethered bead. The fluid flow speed was estimated in movies like S1 from the speed of unattached beads in the same focal plane (height above the flow cell surface) as tethered beads, and was ~100–120μm/s. The agreement between observed and predicted tether length and flow speed provides strong support that the tether structures are as depicted in figure 1.
Both movies show a few beads that move <1 bead diameter during flow. These probably represent beads that have attached to the surface non-specifically due to incomplete passivation of the bead and flow cell surfaces. One bead in the upper right in movie S1B shows characteristic tethered motion even though beads in this movie were not exposed to PSA; such tethers could result from detection antibody sticking non-specifically to a capture bead, or a capture bead sticking non-specifically to DNA. In movie S1A there are also a few beads that move > 1 bead diameter but < 12μm. These shorter-than-expected tether lengths could result from beads being attached via more than one DNA, or DNA sticking to glass or bead surfaces at positions internally along the DNA. The movies also show some bead aggregates, which result from non-specific interactions since they are seen in beads not exposed to PSA (Movie S2).
The paramagnetic capture beads we used contain iron, are denser that water, and settle to the lower surface of flow cells by gravity within minutes. Tethers also formed within minutes, with no noticeable increase in the number of tethers after ~5 minutes.
3.2 Limits of detection
When this experiment was performed with 10-fold serial dilutions of PSA, the number of tethered beads was maximal at ~10pM and decreased as PSA concentration decreased, with a ~1pM lower limit of detection (table 1). At higher concentrations of analyte the number of tethers might be limited by the density of capture DNAs on the sensor surface. At 1pM PSA the 50μl sample volume contained ~300 PSA molecules/bead. Since only ~3% of beads formed tethers at this concentration despite potentially hundreds of PSA molecules per bead, we investigated the efficiency of various steps in the assay.
Table 1.
Average # (± sd) of tethered, stuck, and total beads per 5x field of view*
| PSA concentration | tethered beads | stuck beads | total beads |
|---|---|---|---|
| 100pM | 96 ±21 | 9±4 | 260±21 |
| 10pM | 82 ±21 | 3±1 | 314±37 |
| 1pM | 8±3 | 9±2 | 257±38 |
| 0 | 0.2 ±0.4 | 12±3 | 262±24 |
5 fields of view averaged for 100pM sample, 20 fields of view for other samples
3.3 Efficiency of bead capture of PSA, detection antibody, and streptavidin
We used an enzyme-linked immune sandwich assay (ELISA) with anti-PSA beads as the solid phase to estimate the efficiency of capturing PSA and detection antibody, as shown schematically in Figure 2. In the first experiment, we used PSA that had been labeled with biotin so that a single binding step would capture biotin on beads. We estimated how efficiently anti-PSA beads bound PSA by measuring how completely anti-PSA beads removed biotinylated PSA from an incubation mixture, as illustrated in the first two rows in Figure 2. When we incubated 105 capture beads with 108 biotinylated PSA molecules in 50μl (3pM) for 1.5 hours (sample 1 most of the biotin was captured as determined by incubating the supernatant of these beads with fresh capture beads (sample 2). In the representative experiment reported in Table 2, the OD452 was 0.170 for beads incubated with biotin-PSA (sample 1) versus 0.044 for beads incubated with the supernatant (sample 2), and 0.052 for control beads incubated with WB without PSA in the first step (sample 3). Thus, we estimate the first step captured ~75% or more of the target PSA molecules.
Figure 2.

Schematic of ELISA assay to determine efficiency of capturing PSA, detection antibody, and streptavidin. Row 1, ELISA assay of sample with known concentration of biotin-labeled PSA. Row 2, ELISA assay of supernatant from row 1 assay. Reduction in ELISA signal indicates how much biotin-labeled PSA is captured by anti-PSA beads. Row 4, ELISA assay of unlabeled PSA at same concentration as PSA in row 1, followed by biotin-labeled detection antibody. Comparing ELISA signal in row 4 vs. row 1 indicates how efficiently biotin-detection antibody is captured by bound PSA.
Table 2.
Enzyme-linked immunoassay to monitor efficiency of capture of analyte, detection antibody and streptavidin-peroxidase (SA-perox.) per Figure 2. Assay volumes were 44 μl.
| sample | # anti-PSA beads | target molec. | # biot.-anti-PSA molec. added | # SA-perox. molec. added | OD452 |
|---|---|---|---|---|---|
| 1 | 2*105 | 2*108 bio-PSA | none | 4*109 | 0.170 |
| 2 | 2*105 | supernatant of #1 after 1.5h | none | 4*109 | 0.044 |
| 3 | 2*105 | none | none | 4*109 | 0.052 |
| 4 | 2*105 | 2*108 PSA | 2*1010 | 4*109 | 0.181 |
| OD452 as function of number of SA-peroxidase molecules in solution | |||||
| 5 | 2*108 | 0.764 | |||
| 6 | 2*107 | 0.189 | |||
| 7 | 2*106 | 0.036 | |||
To estimate the efficiency of binding of detection antibody, we compared the amount of biotin-labeled detection antibody captured by beads that had been incubated with 3pM unlabeled PSA to the amount of biotin-labeled PSA captured by beads incubated with 3pM biotin-labeled PSA (sample 4 in Figure 2). The ELISA signals were similar (OD452 0.170 vs. 0.181, sample 1 vs. sample 4, Table 2), indicating that most of the unlabeled PSA molecules captured on beads bound detection antibody. To be more quantitative, we estimated the number of biotins per labeled antibody (~8) and per labeled PSA molecule (~3) using a HABA-avidin reagent (Sigma #H2153). Based on these numbers, we estimate that 37% of captured PSA molecules bound detection antibody under our conditions. This estimate is conservative in the sense that if steric interference prevented SA-peroxidase from binding all the biotins on the detection antibody, we would estimate that more than 37% of PSA molecules bound detection antibody. This implies that a 1pM PSA test sample would lead to at least ~.37*.75*300 = 80 biotinylated detection antibody molecules/bead.
To estimate the efficiency with which biotin on capture beads bound streptavidin, we compared the ELISA signal from beads that captured biotin-PSA to the signal from serially diluted streptavidin-peroxidase in solution. In the above experiment, 2×107 streptavidin-peroxidase molecules in solution resulted in an OD452 of 0.189 (Table 2, sample 6). Since this is comparable to the ELISA signal generated when ~7.5×107–108 molecules of biotinylated-PSA were captured on beads (Table 2, sample 1), we estimate that 20% of biotin-labeled PSA molecules captured on beads bound streptavidin-peroxidase. The streptavidin-peroxidase concentration in these experiments was 30pM.
The above experiments show that PSA analyte and detection antibody are captured efficiently, as are free streptavidin-peroxidase molecules. However, only a few percent of beads estimated to have bound at least 80 molecules of biotinylated detection antibody (those exposed to 1pM PSA) formed tethers (table 1), suggesting that these beads bind relatively inefficiently to streptavidin at the ends of tethered DNA.
3.4 Discussion
The correspondence between observed and predicted travel distance for many tethered beads in the system described here provides strong evidence that binding via single analyte molecules to single DNA molecules is sufficient to form tethers. This conclusion is consistent with a large body of literature in single-molecule biophysics using DNA-tethered beads.
If the affinity of streptavidin on DNA for biotin on beads was the same as its affinity in solution, one would expect almost all beads with at least one biotinylated detection antibody to form tethers at equilibrium. This follows from the fact that the concentration of streptavidin on DNA in the vicinity of beads should be much greater than the solution KD. The concentration of DNA-bound streptavidin in the vicinity of beads in flow cells is not known precisely but can be estimated as follows. The beads settle by gravity to the surface. For beads that settle “on top of” a DNA, the local concentration of streptavidin is one molecule per volume of a hemisphere with radius equal to a DNA length= 1 molecule/250μm3 =7pM. The local streptavidin concentration can also be estimated from the number of DNAs per unit area of surface divided by the maximal length (height) of a DNA. The number of DNAs on the surface must be ≥ the number of tethers that formed when streptavidin-coated beads were introduced into flow cells containing biotin-dig-labeled DNAs. This number was ~106 tethers/flow cell. 106 DNAs in a volume = flow cell area (3cm2) × 1 DNA length (~5μm) corresponds to a concentration ~1pM. Both of these concentration estimates are well over the solution KD of biotin-streptavidin (~1fM) and roughly equal to the concentration of streptavidin-peroxidase that bound efficiently to beads in the ELISA experiments. This suggests that streptavidin at the end of DNA is less accessible to beads than free streptavidin.
Streptavidin at the ends of long DNA molecules may be less accessible due to “masking” from negative charge or steric effects of adjacent DNA. Raising the salt concentration to 1M NaCl to shield charge did not lead to significantly more tethers. Steric interference from adjacent DNA likely increases with DNA length. For example, if the DNA is modeled as a “blob” made up of n, statistically independent segments, capture by biotin on a bead might require the streptavidin-labeled DNA segment to be closer to the bead that any other DNA segment, in which case binding efficiency would decrease proportional to 1/n, i.e. inversely with DNA length. This suggests efficiency might be improved by shortening the DNA. However, shortening the DNA makes it more difficult to distinguish tethered beads from non-specifically stuck beads.
Another possible contributor to inefficient tether formation is reduced capture rate as the number of biotins/bead decreases. For molecules in solution, reducing the area of absorption from the entire surface of a sphere to a single, small absorbing patch reduces the absorption rate by the ratio of the radius of the patch to that of the sphere (Berg, 1993). For a 5nm antibody and 2.8μm bead, this would be about 500-fold. In our system, 2.8μm beads completely coated with streptavidin (~106molecules/bead, Dynal, M280 beads) formed tethers with DNAs on flow cell surfaces without noticeable delay upon settling to the surface (i.e. ≤ minutes), which sets an upper limit to delay from this phenomenon of several hours. However, we did not notice an increase in number of tethers after overnight incubation. If time to bind DNA is limiting, it should decrease proportionally to bead radius, but smaller beads are more difficult to detect in low magnification, large field-of-view microscopy. Other approaches to increasing efficiency of tether formation include increasing the concentration of biotin or streptavidin (e.g. putting multiple biotins on each DNA, using aggregated forms of streptavidin rather than molecular streptavidin, using higher concentrations of DNA, or incubating DNA with beads in suspension before introduction into flow cells). Each of these approaches entails other challenges, such as avoiding cross linking or formation of multiple tethers/bead. Preliminary experiments with these approaches have not, so far, significantly reduced the lowest detectable concentration of analyte.
If the last step in tether formation can be made more efficient, the tethered bead transduction method has several potential advantages compared to previously reported, highly sensitive methods. First, it is inherently a single molecule counting method since beads can be tethered via single analyte molecules. Second, the method allows one to disregard signal from beads or molecules sticking non-specifically to flow cell surfaces as these do not lead to tethers of the expected length. Non-specific sticking of reporter molecules to the sensor surface limits the sensitivity of fluorescence-based sensors since there is no way to distinguish fluorescent reporter molecules stuck non-specifically to sensor surfaces from those stuck to analyte molecules captured on the sensor surface. Non-specific sticking to DNA does pose a problem for the tethered bead method but the surface area of the DNA is much less than the surface area of the flow sensor, so non-specific sticking to DNA may be quantitatively less of a problem. In our experiments, non-specific sticking in the absence of analyte led to ~30 tethers per 105 beads. This imposes a floor on the lowest detectable concentration of ~100 molecules/50μl sample, or ~3aM. A third potential advantage of the tethered bead method is that detection of tethered beads does not require advanced technology – at present only ~5–10× magnification, simple illumination, and a low cost video camera. The rapid development of inexpensive consumer CMOS cameras with >10M pixels of ~1micron pixel size promises to make detection of micron-size beads easier, potentially achievable with a cell-phone camera.
4. Conclusions
We provide a “proof-of-principal” for a single-molecule immune sandwich assay in which read-out is based on counting micron-size beads tethered by molecules of DNA. The motion of tethered beads in flow is recognizable with a low-power optical microscope and distinguishable from that of beads that stick non-specifically to the flow cell surface. Analyte molecules are captured efficiently by capture beads and labeled efficiently by biotinylated detection antibody. Tether formation is limited at present by low efficiency of binding of streptavidin at the ends of DNA to biotin-labeled detection antibody on beads. If the efficiency of this step can be increased, this type of assay could be sensitive to very low concentration of analyte because beads displaying tethered motion are very rare in the absence of analyte and tethers are linked to beads via single immune complexes.
Supplementary Material
We demonstrate a “single-molecule” immunoassay in which analyte molecules link micron-size beads to DNA strands on a sensor surface
beads tethered by single DNA molecules are recognized by their characteristic motion under alternating fluid flow using low magnification microscopy
The number of tethered beads provides a measure of analyte concentration
Assay sensitivity is limited by the efficiency with which immune complexes on beads bind to capture molecules at the ends of DNA strands
Acknowledgments
We thank Liusongsen Yang for help constructing flow cells and image analysis.
Appendix. Supporting Information
Quantitative model of tether attachment geometry and tether extension as a function of flow rate. Liquid flow around a tethered bead exerts a drag force on the bead in the direction of flow and also a torque because the liquid flows faster over the portion of the bead farther from the glass surface (Poiseuille flow). Fluid dynamics theory predicts how the force and torque near a surface are modified compared to their bulk values. For example, (Goldman et al., 1967) predicts that stationary spherical beads touching a surface experience a drag force =α*(6πηrv) and torque =β*(4πηr2v) where is η the fluid viscosity, r the radius of the bead, v the fluid velocity at a height r above the surface, α = 1.7, and β = 0.94. Thus, the ratio, R, of the torque to r*drag force = (2/3)(β/α) = 0.37. The DNA tether must exert a force and a torque equal and opposite to that of the shear flow at the trajectory end points where tethered beads are stationary. The position of the attachment point on a bead (see figure A.1) determines the component of tether force in the direction of flow and the torque exerted by the DNA tether:
| (A.1) |
| (A.2) |
where FT is the tension in the DNA and ϕ and κ are angles defined in Figure A.1. Geometry determines a relationship between ϕ and κ:
| (A.3) |
(1), (2) and (3) can be combined to yield:
| (A.4) |
The center-to-center travel distance, Dcc, for a tethered bead can be expressed as:
| (A.5) |
The well-validated worm-like chain model of DNA predicts a relationship between tension and relative extension of a piece of DNA. To good approximation (Bustamante et al., 1994):
| (A.6) |
where p is the persistence length of DNA (~50nm), kB is Boltzmann's constant, T is temperature in degrees Kelvin, and L is the contour length of the DNA, ~0.3nm/base pair.
In our experiment we observed Dcc = 12μm. We take r =1.4μm, L = 5.1 μm, p = 50nm, and kBT = 4*10−21J. If we consider R =0.37 as given from fluid dynamics, the four equations (A3), (A4), (A5) and (A6) have four remaining variables ϕ, κ, FT, and x, which are therefore all determined. We find ϕ = 16°, 2κ = 95°, FT = 5.8N, and x = 4.8μm. From (A1), we then calculate Fdrag = 5.6pN. If we take α=1.7, then v is predicted from (A1): for η = 10−3Ns/m2, we find v = 125μm/s. But v is also estimated from the observed motion of free beads, vobs = 100 – 120μm/s. The agreement between the predicted and observed v provides an independent test of the model.
Figure A.1.
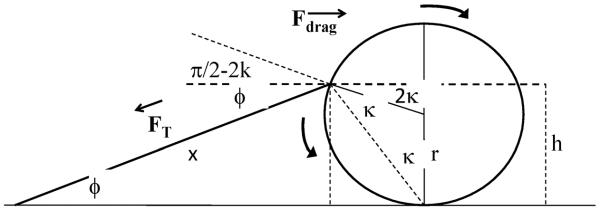
Diagram of tethered bead with tether stretched by flow to the right, for calculation of tether length × as a function of drag force Fdrag. Bead of radius r is attached to flow cell surface via a single DNA tether. Tension in DNA FT increases until it balances the drag force on bead. When the bead is stationary in flow, the height h of the tether attachment point is such that torque from shear flow (clockwise arrow) balances that from off-center tether attachment (counter-clockwise arrow) and FT cos(ϕ) = Fdrag. Angles ϕ and κ are defined in the figure. A relation between force and extension from the worm-like chain model of DNA and fluid dynamics predictions of force and torque on a sphere as a function of flow rate for a sphere resting on a surface allow all variables to be determined as a function of lateral flow rate (see appendix).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, Landick R, Block SM. Nature. 2005;438:460–465. doi: 10.1038/nature04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg HC. Random Walks in Biology p.30. Princeton University Press; 1993. [Google Scholar]
- Burg TP, Godin M, Knudsen SM, Shen W, Carlson G, Foster JS, Babcock K, Manalis SR. Nature. 2007;446:1066–1069. doi: 10.1038/nature05741. [DOI] [PubMed] [Google Scholar]
- Bustamante C, Marko JF, Siggia ED, Smith S. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- Bustamante C, Bryant Z, Smith SB. Nature. 2003;421:423–427. doi: 10.1038/nature01405. [DOI] [PubMed] [Google Scholar]
- Cluzel P, Lebrun A, Heller C, Lavery R, Viovy J-L, Chatenay D, Caron F. Science. 1996;271:792–794. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- Ferguson RA, Yu H, Kalyvas M, Zammit S, Diamandis EP. Clin Chem. 1996;42:675–684. [PubMed] [Google Scholar]
- Goldman AJ, Cox RG, Brenner H. Chemical Engineering Science. 1967;22:653–660. [Google Scholar]
- Jain A, Liu R, Ramani B, Arauz E, Ishitsuka Y, Ragunathan K, Park J, Chen J, Xiang YK, Ha T. Nature. 2011;473:484–488. doi: 10.1038/nature10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Blainey PC, Schroeder CM, Xie XS. Nat. Methods. 2007;4:397–399. doi: 10.1038/nmeth1037. [DOI] [PubMed] [Google Scholar]
- Larson RG, Perkins TT, Smith DE, Chu S. Physical Review E. 1997;55:1794–1797. [Google Scholar]
- Liu S, Guo X. NPG Asia Materials. 2012;4:e23. [Google Scholar]
- Mulvaney SP, Cole CL, Kniller MD, Malito M, Tamanaha CR, Rife JC, Stanton MW, Whitman LJ. Biosensors and Bioelectronics. 2007;23:191–200. doi: 10.1016/j.bios.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Neuman KC, Nagy A. Nature Methods. 2008;5:491–505. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterfeld SJ, Yu H, Gaster RS, Caramuta S, Xu L, Han S-J, Hall DA, Wilson RJ, Sun S, White RL, Davis RW, Pourmand N, Wang SX. PNAS. 2008;105:20637–20640. doi: 10.1073/pnas.0810822105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, Piech T, Patel PP, Chang L, Rivnak AJ, Ferrell E, Randall JD, Provuncher GK, Walt DR, Duffy DC. Nature Biotechnology. 2010;28:595–599. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadana A. Biotechnol. Prog. 1995;11:50–57. doi: 10.1021/bp00031a007. [DOI] [PubMed] [Google Scholar]
- Schafer DA, Gelles J, Sheetz MP, Landick R. Nature. 1991;352:444–448. doi: 10.1038/352444a0. [DOI] [PubMed] [Google Scholar]
- Squires TM, Messinger RJ, Manalis SR. Nature Biotechnology. 2008;26:417–426. doi: 10.1038/nbt1388. [DOI] [PubMed] [Google Scholar]
- [accessed 3/31/2014];U.S. Preventive Services Task Force recommendation on screening tests for prostate cancer. 2012 May; http://www.uspreventiveservicestaskforce.org/prostatecancerscreening.htm.
- Vollmer F, Arnold S. Nature Methods. 2008;5:591–596. doi: 10.1038/nmeth.1221. [DOI] [PubMed] [Google Scholar]
- Yanagida T, Sako Y, Minoghchi S. Nature Cell Biology. 2000;2:168–172. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.