

Abstract
Ribosomes control the missense error rate of ~10−4 during translation though quantitative contributions of individual mechanistic steps of the conformational changes yet to be fully determined. Biochemical and biophysical studies led to a qualitative tRNA selection model in which ribosomal A-site residues A1492 and A1493 (A1492/3) flip out in response to cognate tRNA binding, promoting the subsequent reactions, but not in the case of near cognate or non-cognate tRNA. However, this model was recently questioned by X-ray structures revealing conformations of extrahelical A1492/3 and domain closure of the decoding center in both cognate and near-cognate tRNA bound ribosome complexes, suggesting that the non-specific flipping of A1492/3 has no active role in tRNA selection. We explore this question by carrying out molecular dynamics (MD) simulations, aided with fluorescence and NMR experiments, to probe the free energy cost of extrahelical flipping of 1492/3 and the strain energy associated with domain conformational change. Our rigorous calculations demonstrate that the A1492/3 flipping is indeed a specific response to the binding of cognate tRNA, contributing 3 kcal/mol to the specificity of tRNA selection. Furthermore, the different A-minor interactions in cognate and near-cognate complexes propagate into the conformational strain and contribute another 4 kcal/mol in domain closure. The recent structure of ribosome with features of extrahelical A1492/3 and closed domain in near-cognate complex is reconciled by possible tautomerization of the wobble base pair in mRNA-tRNA. These results quantitatively rationalize other independent experimental observations and explain the ribosomal discrimination mechanism of selecting cognate versus near-cognate tRNA.
Keywords: ribosome, A-site, decoding, tRNA selection
Introduction
Ribosomes translate mRNAs and synthesize proteins with high fidelity (error rate of ~10−4)[1, 2] and high efficiency (~22 codons per second)[3] in order to sustain cellular functions. Ribosomes achieve 10−3−10−2 missense frequencies in discrimination of near-cognate versus cognate tRNA[4-6] during the decoding process, in which quantitative contributions of individual mechanistic steps remain to be fully determined. Biochemical studies together with NMR and X-ray structures of the ribosomal A-site reveal that the extrahelical flipping of the universally conserved adenine nucleobases at the ribosomal A-site, A1492 and A1493 (A1492/3), is a fundamental aspect of codon recognition.[7-18] The prevailing model posits that A1492/3 actively monitor the conformation of the minihelix formed between the mRNA codon and the tRNA anti-codon.[19-22] Formation of canonical base pairs between cognate mRNA and tRNA induces extrahelical flipping of A1492/3, which form A-minor interactions with the minor groove of the mRNA-tRNA minihelix, thereby stabilizing the tRNA and promoting GTP hydrolysis through mechanisms that are thought to involve global conformational changes, including 30S domain closure, tRNA distortion, and EF-Tu rearrangement.[15, 16, 22] By contrast, binding of near-cognate or non-cognate tRNA results in non-Watson-Crick base pairing in the minihelix, which disrupts the A-minor interactions and consequently fails to induce A1492/3 flipping and subsequent GTPase activation.[15] In this manner, near-cognate and non-cognate tRNAs are discriminated against due to a combination of destabilized binding affinity and reduced GTP hydrolysis rate. This model also explains the effects of aminoglycoside antibiotics that bind the ribosomal A-site and induce the extrahelical flipping of A1492/3 and 30S domain closure.[12, 15, 23-25]
However, the above decoding model, and in particular the specificity of A1492/3 flipping, was recently questioned by X-ray structures of the 70S ribosome with cognate and near-cognate tRNAs.[26] For both cognate and near cognate complexes, A1492/3 were observed in extrahelical conformations, forming A-minor interactions with the minihelix. The mRNA-tRNA minihelix adopts a Watson-Crick like geometry even for the near-cognate tRNA, possibly through nucleobase tautomerization that allows a G:U mismatch to adopt a Watson-Crick like base-pair.[26] Moreover, both structures also feature 30S domain closure. These structures were interpreted as evidence that the extrahelical flipping of A1492/3 is a non-specific response to tRNA binding and thus has no active function in tRNA selection.
Using all-atom molecular simulations of both oligonucleotide and ribosomal A-site models, as well as fluorescence and NMR experiments, we have computed the energetics of microscopic events during decoding, including the free energy cost of A1492/3 flipping and the strain in domain closure of the decoding center. We demonstrate that the A1492/3 flipping in the ribosomal A-site is indeed a specific response to the binding of cognate tRNA, contributing 3 kcal/mol to the specificity of tRNA selection. In addition, we show that the different A-minor interactions in cognate and near-cognate complexes propagate into the strain of domain closure in the decoding center and contribute another 4 kcal/mol in discriminatory free energy. Furthermore, our thermodynamic integration calculations characterize the free energies associated with the possible tautomerization of the wobble base pair in mRNA-tRNA minihelix. We find that the Watson-Crick like conformation of the G:U tautomer is only about 3 kcal/mol higher than the normal wobble pair conformation in the closed form of ribosomal decoding center, thus the cognate-like conformation of the near-cognate complex is conjectured to be a transient state captured in the crystallographic experiments. We demonstrate the validity of these computed free energy values by building a complete thermodynamic model for the initial selection stage that quantitatively accommodates other independent experimental observations and explains the ribosomal decoding error rate of 10−3−10−2 in selecting cognate versus near-cognate tRNA.
Results and discussion
Intrinsically endothermic A1492/3 flipping in empty A-site
To explore the role of the A1492/3 flipping in tRNA selection, we first examined the intrinsic energetic cost of the A1492/3 flipping. The dynamics and energetics of the A1492/3 flipping in the empty A-site or oligonucleotide analogues were intensively examined in the previous experimental and theoretical studies,[10-12, 14, 27-34] reaching a common agreement that A1492/3 disfavors the fully extrahelical conformation in the absence of tRNA or ligand. However, it is important to calculate the intrinsic free energy cost of the A1492/3 flipping in the empty A-site (APO state) here because this provides self-consistent reference states for comparison with cognate and near-cognate tRNA bound A-sites. An oligonucleotide A-site model[10-12] was employed to capture this intrinsic energetic cost before we advance to the more realistic A-site model in the context of ribosomal decoding center (Fig. S1).
In our free energy calculations, we adapted a progress variable called the center-of-mass pseudo-dihedral angle (CPD) for each adenine to characterize the extrahelical flipping (Fig. S2).[35] The CPD was found to be an efficient progress variable to characterize similar motions studied in other nucleic acid systems.[36] Two-dimensional (2D) umbrella sampling was performed to explore the two CPDs of A1492 and A1493 simultaneously and generate 2D free energy landscapes for A1492/3 flipping in various A-site models. For empty A-site, our simulations show that the most favorable conformations with free energies of 0–2 kcal/mol (dark blue region in Fig. 1a) correspond to both A1492 and A1493 being intrahelical, in agreement with a previous free energy simulation of the A-site analogue.[30] This feature is consistent with prior X-ray and NMR structures of empty A-sites, which are marked on the free energy landscape using red cross symbols. Other favorable regions with free energies of 2–4 kcal/mol represent conformations in which A1493 adopts a wide range of CPDs undergoing excursions between intra- and extrahelical positions. The lower barrier for A1493 flipping compared to that for A1492 is consistent with NMR measurements of order parameters reported here (Table S3), which show greater mobility for A1493 compared to A1492. The coexistence of intra- and extrahelical conformations of A1493 has also been reported in recent experimental and theoretical studies.[12, 14, 27-34]
Figure 1.
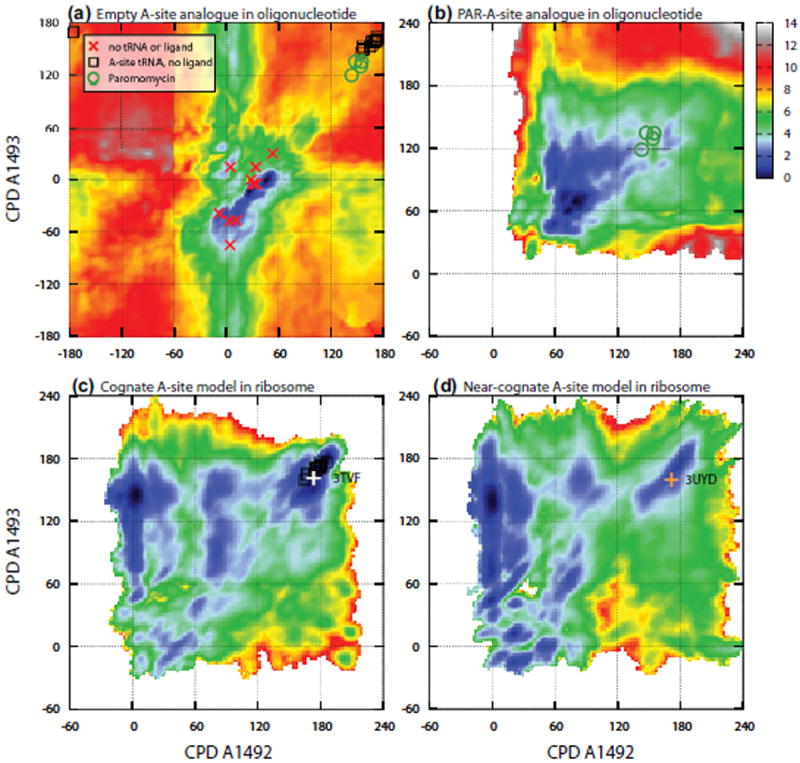
2D free energy landscape along the CPDs of A1492/3 of the A-site with color coded free energy values. Free energy landscapes of (a) ligand-free (APO) and (b) paromomycin bound oligonucleotide A-site analogues, respectively. The CPDs of A1492/3 from the crystal structures are marked on the free energy landscapes. Red crosses are structures of 30S without tRNA or ligand bound to the A-site; black squares represent tRNA bound (but no aminoglycosides) A-sites; green circles are paromomycin bound A-sites; and orange plus symbols represent 30S with both paromomycin and tRNA bound. (c) and (d), free energy landscapes of cognate and near-cognate tRNA bound A-site models in the context of 70S subunits, respectively. The free energy costs of A1492/3 flipping were determined to be: (a) 7 ± 0.3 kcal/mol, (b) 3 ± 0.2 kcal/mol, (c) −2 ± 0.3 kcal/mol, and (d) 1 ± 0.4 kcal/mol, respectively. See Materials and Methods section for details of the determination of free energies and simulation precisions.
The agreement with prior X-ray/NMR structures and other biophysical measurements strongly suggests that the computed free energy landscape captures the thermodynamics of A1492/3 flipping in the empty (APO) A-site analogue in oligonucleotide. The region with fully extrahelical A1492/3 (CPDs > 120°) resembles the conformations of cognate tRNA bound A-site, as indicated by the black squares (Fig. 1a). The free energy difference between this region and the most favorable region is 7±0.3 kcal/mol[37] and represents the intrinsic free energy cost of the endothermic A1492/3 flipping. This provides a reference value to interpret the thermodynamics of A1492/3 flipping in cognate and near-cognate complexes within the ribosome.
Active role of A1492/3 in Codon Recognition
Although A1492/3 flipping is endothermic in empty A-site, the free energy landscape is expected to be altered by the binding of tRNA due to the establishment of A-minor interactions. In an early theoretical study of the structural dynamics of A1492/3 in cognate and near-cognate complexes, it was noticed that the A1492 tends to adapt the flipped-out conformation.[38] In order to capture the energetics in the ribosomal environment, we performed umbrella sampling simulations of the decoding center of the 70S subunit with cognate and near-cognate tRNA bound to the A-site,[39] applying similar restraints on the CPDs of A1492/3. To achieve satisfactory convergence of the 2D free energy landscapes, simulations of μs timescale are needed as previously shown in the oligonucleotide A-site analogue system.[30] The need for quantitative free energy calculations makes it is feasible to simulate only the decoding center of the 70S ribosome, where all the codon specific responses initiate. As shown in Fig. S1, segments of 30S, 50S, mRNA-tRNA and S12 within a radius of 25 Å from A1492/3, solvated in spherical boundary potentials,[40, 41] were used to model the decoding center. The chemical configurations of all structures were assumed to be in the most stable tautomers, i.e., the keto forms for guanine and uracil. For the near-cognate complex containing a G:U wobble pair at the first codon-anticodon position, additional simulations were performed assuming uracil is in the uncommon enol form.
Fig. 1c and 1d show the free energy landscapes of the A1492/3 flipping transitions in cognate and near-cognate tRNA bound A-site complexes, respectively. The most striking feature in the free energy landscape is that the A1492/3 flipping, from the intrahelical (CPDs < 60°) to the extrahelical region (120° < CPDs < 240°), now becomes an energetically favorable process for the cognate complex. However, the same conformational change remains slightly unfavorable in the near-cognate complex. Thus, in contrast to the recent X-ray structures suggesting that A1492/3 undergo non-specific extrahelical flipping,[26, 42] our simulations indeed show significant free energy discrimination between cognate and near-cognate complexes. The free energy changes in the extrahelical flipping of A1492/3 (ΔGflip) are −2±0.3 kcal/mol and +1 ± 0.4 kcal/mol in cognate and near-cognate complexes, respectively.[37]
The free energy landscape obtained in the context of the mRNA-tRNA minihelix and ribosomal environment indicates that the free energy gain from the newly formed A-minor interactions overcomes the intrinsic free energy cost of the flipping motions of A1492/3 in the cognate complex, but is not sufficient to compensate the cost in the near-cognate complex. Given the intrinsic free energy cost of 7 kcal/mol, we estimate that the newly formed A-minor interactions contribute −9 kcal/mol and −6 kcal/mol to the thermodynamic stabilities of cognate and near-cognate complexes, respectively. The 3 kcal/mol free energy difference is certainly a result of the thermodynamic average of various interactions; however, we find that the change in the H-bond network between A1493 and the first codon-anticodon pair is an important factor. As shown in Fig. 2a, A1493 forms four H-bonds with the G:C pair of the cognate mRNA-tRNA minihelix, monitoring the base pairing conformations of mRNA and tRNA simultaneously. In the near-cognate complex (with nucleotides in the regular keto form), two of the four H-bonds are preserved (Fig. 2b), but the other two H-bonds, between the 2′-OH of A1493 and the first codon residue U4 (numbering in 3UYD), cannot be maintained due to the G:U wobble base-pair between the codon and anticodon.
Figure 2.
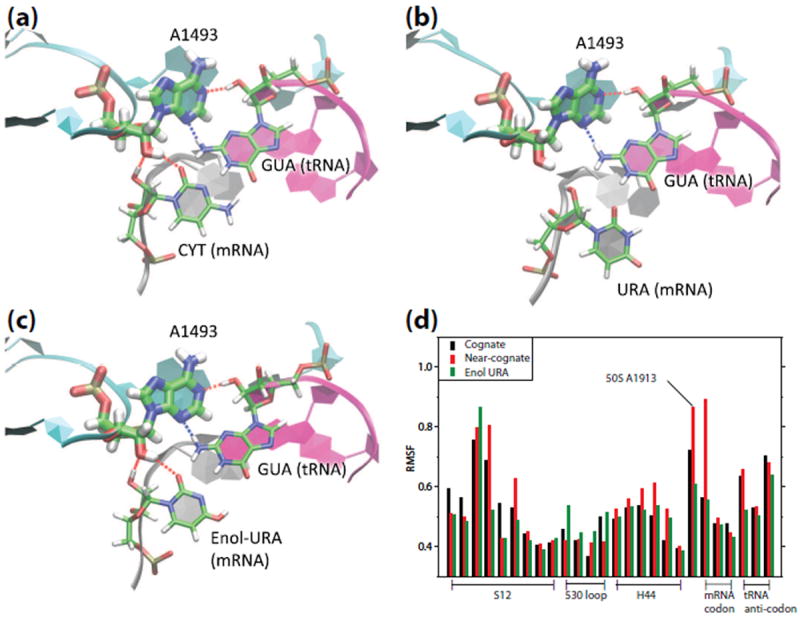
Different interaction networks in cognate and near-cognate tRNA bound A-site. (a) A typical conformation of extrahelical A1493 interacting with the cognate mRNA-tRNA minihelix, forming four H-bonds. (b) A conformation for the near-cognate mRNA-tRNA bound A-site. Two H-bonds are broken due to the flexible wobble G:U pair. (c) In the enol form of near-cognate complex, G:enol-U forms a Watson-Crick-like base pair, four H-bonds with the extrahelical A1493 are maintained. (d) Structural stabilities characterized by large root-of-mean-square fluctuations (RMSF) in the residues around the decoding center in free MD simulations. RMSF are shown for the S12 segment (residues 41 to 49 in cognate complex 3TVF and 44 to 52 in near-cognate complex 3UYD), part of 530 loop (residues 518, 519, 529 and 530 in 30S), H44 segment (residues 1490 to 1495 in 30S), A1913 in 50S, mRNA codon and tRNA anticodon.
The G:U wobble base-pair demonstrates a moderate displacement from the X-ray structure of the near-cognate complex (3UYD),[26] which shows a Watson-Crick like conformation of the G:U pair. Considering the possibility of H-bond rearrangement due to tautomerization, an additional simulation was performed assuming the enol form of U4. The simulation shows that the G:enol-U base pair indeed forms a stable Watson-Crick like conformation and interacts with the extrahelical A1492/3, as shown in Fig. 2c, resembling the structure and the A-minor interactions of the cognate complex. Analysis of the root-of-mean-square fluctuations (RMSF), as shown in Fig. 2d, suggests that the near-cognate tRNA complex in the keto form is less stable (i.e., showing larger RMSF) than the cognate complex, especially in the structures of H44, A1913 (50S) and the C4/U4 on mRNA, due to the weakened A-minor interactions. By contrast, the enol form near-cognate complex shows stable structure, with fluctuations comparable overall to the cognate complex. The extraordinarily large RMSF of A1913 in near-cognate complex suggests that its interaction with H44 is not as stable as that in the cognate complex, suggesting a possible role of A1913 in the tRNA binding.[22, 26, 42, 43]
The stable structure of extrahelical A1492/3 in the enol form near-cognate complex agrees with the observation from previous experiments that A-site monitors the codon-anticodon mismatch through a structure-specific rather than a sequence-specific mechanism.[4, 11] If a Watson-Crick like structure is formed in the G:enol-U pair, it will be treated by the A-site as a cognate complex. In the RMSF analysis, the chemical configurations were either assumed to be keto or enol-form without explicitly including the cost of tautomerization. To examine the free energy cost of the keto-enol tautomerization, alchemical thermodynamic integration (TI) calculations were performed and the results suggest that the free energy cost associated with keto-to-enol tautomerization of U4 is 7 ± 1.0 kcal/mol in the ribosome complex (Fig. S3).[37] The conformation of the near-cognate complex in the X-ray structure is likely less energetically favorable than that of the cognate complex or the near-cognate complex with the wobble geometry. Therefore, it is possible that the X-ray structure captured a high energy conformational state that exists transiently in ribosome translation (e.g., before tRNA rejection) or possibly accounts for some of the errors seen in translation,[44, 45] which is similar to the G:T tautomerization implicated in erroneous DNA replication.[46-48]
Paromomycin binding eliminates discrimination
We also examined whether our model could explain how the aminoglycoside antibiotic paromomycin affects the specificity of A1492/3 flipping. Structural and biochemical studies show that binding of paromomycin to the ribosomal A-site facilitates extrahelical flipping of A1492/3 and domain closure in near-cognate complexes,[12, 15] and also accelerates GTP hydrolysis.[23-25] If the energetics of A-site flipping are truly critical to codon recognition, paromomycin should alter the free energy landscape of A1492/3 flipping. Thus, we performed simulations of the oligonucleotide A-site analogue bound to paromomycin. As shown in Fig. S7, paromomycin binds to the major groove of the RNA helix with its ring I buried in the adenine bulge. In agreement with X-ray structures,[15, 49] ring I of paromomycin displaces the noncanonical AA base pairing and forms stable H-bonds with A1408, firmly occupying the volume that is otherwise taken by the A1492/3 nucleobases.
By restricting either of the A1492/3 bases from adopting intrahelical positions, paromomycin substantially changes the free energy landscape of the A-site. The free energy landscape of the paromomycin bound A-site, as presented in Fig. 1b, shows a vast low free energy (blue) region with high CPDs (60° < CPDs < 180°). The free energy landscape indicates more populated extrahelical conformations than in the empty A-site, in qualitative agreement with previous[50] and current fluorescence experiments (Fig. S8) as well as recent simulations of the gentamicin bound A-site.[30] Based on these simulations, we find that the binding of paromomycin reduces the intrinsic free energy cost of flipping out A1492/3 from 7 ± 0.3 kcal/mol to 3 ± 0.2 kcal/mol.[37] Since paromomycin binds similarly in the oligonucleotide and ribosome, and the binding site is more than 10 Å away from the interface of A-minor interactions, we suggest that the energetic effect of paromomycin should be very similar in the oligonucleotide and ribosome. Borrowing the free energies from the paromomycin bound oligonucleotide, we estimate that the overall free energy change for A1492/3 flipping in the presence of paromomycin becomes about −6 kcal/mol in the cognate complex and −3 kcal/mol in the near-cognate complex. The free energy changes of A1492/3 flipping under different conditions, as shown in Fig. 3, quantifies the effect of paromomycin on the specificity of A1492/3 flipping, and supports the critical role of A1492/3 in the previous structural model.[20] Because the intrinsic energy cost to flip-out A1492/3 is partially compensated by paromomycin, even the suboptimal A-minor interactions between A1492/3 and the near-cognate mRNA-tRNA minihelix become sufficient to maintain the fully extrahelical conformation, in agreement with crystal structures.[14, 51] In the presence of paromomycin, the near-cognate tRNA is stabilized by the A-minor interactions with the extrahelical A1492/3, in agreement with the kinetic observations that paromomycin reduces the dissociation rates of tRNAs.[23] Without the extrahelical A1492/3 extruded by paromomycin, near-cognate tRNAs may rapidly dissociate from the decoding center due to the strain of tRNA distortion in GTPase activation.[16] Our simulations illustrate the thermodynamic basis of paromomycin interference in ribosomal tRNA selection. By turning the extrahelical flipping of A1492/3 into a non-specific and spontaneous process, paromomycin eliminates the thermodynamic discriminatory functionality of A1492/3 against near-cognate tRNA.
Figure 3.
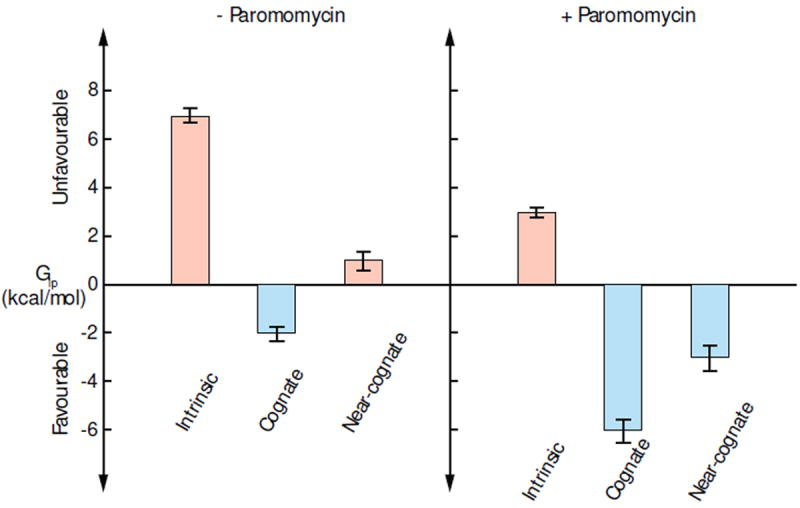
Free energy profiles of the extrahelical flipping of A1492/3 under different conditions with the statistical errors from the simulations. In the absence and presence of paromomycin, the free energy costs of A1492/3 flipping in cognate and near-cognate ribosomal complexes are compared with the intrinsic costs, respectively.
Quantitative model of the tRNA initial selection
Combining our simulation results with the experimentally determined kinetic rate constants and binding affinities,[4, 15] we constructed the mechanistic model shown schematically in Fig. 4, which quantitatively describes the free energy changes in the initial selection of cognate versus near-cognate tRNA. The pre-hydrolysis state (state B) is expanded into three substates (shown in bold lines in Fig. 4a): substate B1 after codon-anticodon matching, substate B2 after A1492/3 flipping, and substate B3 after domain closure in A-site, preceded by GT-Pase activation and GTP hydrolysis.[20]
Figure 4.
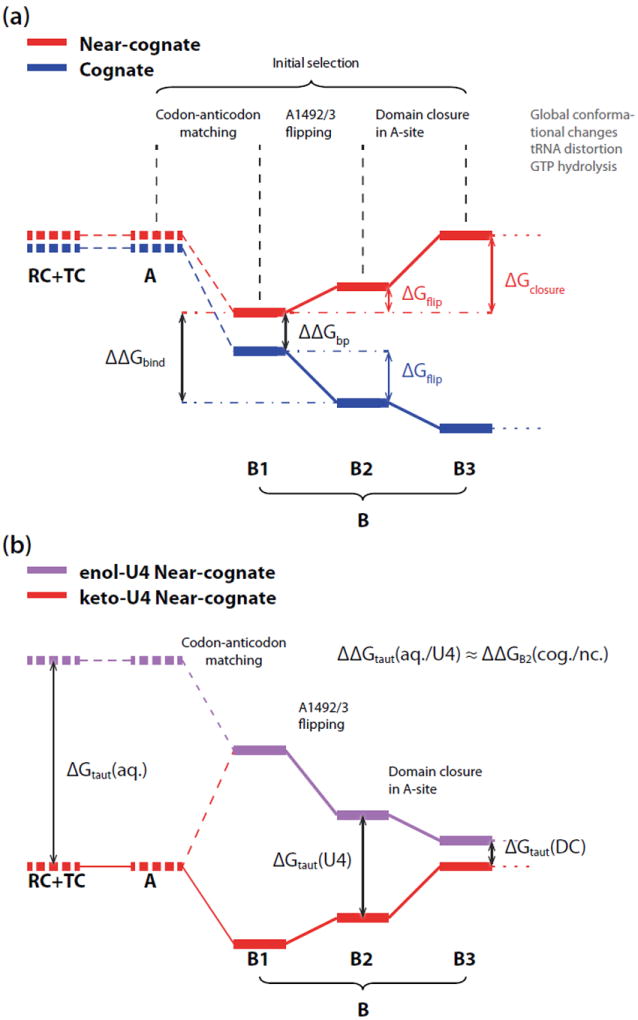
Schematic free energy profile of the initial selection stage. (a) Initial selection of cognate and near-cognate complexes, with state B (pre-hydrolysis state) expanded into three substates shown as solid bold lines: (B1) after codon-anticodon matching, (B2) after A1492/3 flipping and (B3) after domain closure in A-site. The difference between B1 and B2 represents the free energy change of A1492/3 flipping, ΔGflip. For the near-cognate tRNA complex, the most stable pre-hydrolysis substate is B1, which requires energy for further conformational changes to evolve into the active form. Those states for which direct estimates of free energy changes were computed in the calculations presented herein are shown as emboldened solid line. Estimates that are taken from experimental studies[4, 15, 23] are indicated by the emboldened dashed lines. (b) Initial selection models for keto (res) and enol form (purple) of U4 in near-cognate complex. The Watson-Crick like conformation of G:enol-U4 leads to the profile of codon recognition similar to that of cognate complex with an offset of the intrinsic cost of the keto-to-enol tautomerization.
The free energy difference between the near-cognate and cognate complex in substate B1 corresponds to the inherent base-pairing specificity, ΔΔGbp, which is reported to be 1.1 ± 0.3 kcal/mol for wobble mismatches.[52, 53] The free energy changes between substate B1 and B2, ΔGflip, calculated in our simulations, are −2 kcal/mol and +1 kcal/mol for cognate and near-cognate complexes, respectively.
The transition from B2 to B3 represents the conformational reorganization of A-site that determines the strain of 30S domain closure. In order to characterize the strain in the decoding center during 30S domain closure, we performed further umbrella sampling simulations based on an RMSD (root-mean-square deviation) progress variable[54] to calculate the free energy for the conformational strain in the A-site reorganization after the A1492/3 flipping. A crystal structure of the near-cognate complex with features of the inactive open 30S domain (PDB: 1N34) was used as a reference for the open form, while the crystal structures of the complexes with the active closed 30S domain (PDB: 3UYD and 3TVF) were used as a reference for the closed forms. As shown in Fig. S4, the free energy profiles illustrate that the cognate complex favors conformations similar to the closed form while the near-cognate complex more favors the open form. Quantitatively, a free energy cost of about 4±0.3 kcal/mol[37, 55] is required for the near-cognate complex to evolve into the active closed conformation similar to the cognate complex. Despite the artificially flipped-out A1492/3, the near-cognate complex still disfavors closed state due to the wobble base pair mismatching and the suboptimal A-minor interactions. This finding agrees with the crystallographic observations of the active domain closed form in the cognate complexes, while the near-cognate complex adopts the inactive open form.[15, 16] Therefore, we conclude that the domain closure is likely to be a spontaneous process in the cognate complex following codon recognition, and it does not contribute an additional free energy cost to the apparent GT-Pase activation barrier. For the near-cognate complex, however, the free energy of the conformational strain and A1492/3 flipping in the A-site, 5 kcal/mol in total, contributes to the overall barrier prior to GTP hydrolysis.
The quantitative initial selection model shown in Fig. 4 illustrates the origin of the thermodynamic and kinetic specificities between cognate and near-cognate tRNA. The base-pairing specificity (ΔΔGbp = 1 kcal/mol)[52, 53] and the spontaneous A1492/3 flipping in the cognate complex (ΔGflip = −2 kcal/mol) together produce a ~3 kcal/mol difference in the thermodynamic stability of tRNA binding ΔΔGbind. This thermodynamic stabilization is essential to secure the anticodon stem loop of tRNA at the A-site. In the near-cognate complex, the combination of the endothermic A1492/3 flipping and the strain in domain closure (A-site reorganization) result in a 5 kcal/mol free energy component contributing to the overall activation barrier prior to GTP hydrolysis as the kinetic specificity. The quantitative energetics of these two aspects of selectivity show excellent agreement with the experimental measurements of the cognate and near-cognate tRNAs, which report about 3.0 kcal/mol thermodynamic specificity in the binding affinities (with G:U mismatching at the 1st codon)[15] and 2.7-4.5 kcal/mol kinetic specificity in the GTP hydrolysis rate constants (in various mismatch types).[4] Based on the kinetic equations derived from rate constant measurements,[ 4, 56] our simulations show that the ribosomal selectivity of cognate against near-cognate tRNA in the initial selection stage is about 1.8 × 103, in close agreement with the the experimental estimation.[5]
Based on previous experimental measurements and our TI calculations (Fig. S3) on the keto-to-enol tautomerization free energies, we also constructed the mechanistic model of the near-cognate complex with enol-U4 in the mRNA and compared it with the keto form model, as shown in Fig. 4b. Experimental measurements suggest that the keto-to-enol tautomerization of aqueous uracil consumes ~10 kcal/mol in free energy [ΔGtaut(aq.)].[57] Our TI calculations show that the similar keto-to-enol tautomerization of mRNA U4 consumes 3 kcal/mol less than that of aqueous uracil, suggesting ΔGtaut(U4) is about ~7 kcal/mol. The relative tautomerization free energy difference matches very well with the changes in the local ribosomal interactions in the A-site of the cognate and near-cognate complexes. As shown in Fig. 4a, the free energy difference between the cognate and near-cognate complexes in the codon recognition state B2 [ΔΔGB2(cog./nc.)] is about 4 kcal/mol corresponding to the summation of the base-pairing specificity ΔΔGbp (1 kcal/mol[52, 53]) and the difference in the A1492/3 flipping ΔΔGflip (3 kcal/mol), which represents the different local interactions in the ribosomal A-site between Watson-Crick like and wobble base-pair matching geometry. In the near-cognate complex, the keto-to-enol tautomerization of mRNA U4 is lowered by 3 kcal/mol mainly due to the compensation of the H-bonds formed not only between G:U4 but also between A1493 and U4, which are stable only in the Watson-Crick like G:enol-U conformation (Fig. 2b-c). The agreement between ΔΔGtaut(aq./U4) and ΔΔGB2(cog./nc.) again proves that the ribosome senses the codon-anticodon mismatch on the geometric basis instead of the sequential basis. From substate B2 to B3, the relative free energy difference between the cognate and near-cognate complex is 4 kcal/mol for the strain in domain closure. If we assume the ribosome response to the Watson-Crick like G:enol-U pair the same as a normal G:C cognate pair in the subsequent domain closure, the free energy difference between the keto and enol form of near-cognate complex ΔGtaut(DC) will be further reduced from 7 kcal/mol to 3 kcal/mol. Nevertheless, our constructed model still indicates that the transformation to enol-U4 is a less favorable process during the codon recognition stage, which is likely captured as a transient state in crystal structures.
Conclusion
In this work, we simulated the conformations and energetics of A-site models in the presence of cognate tRNA, near-cognate tRNA and paromomycin, with special emphasis on the role of A1492/3 flipping. We find that A1492/3 in the A-site models indeed shows different responses to the binding of cognate and near-cognate tRNAs. The extrahelical flipping of A1492/3 is found to be an intrinsically endothermic process with ΔGflip of 7 kcal/mol. The flipping becomes a spontaneous process in the cognate tRNA bound A-site while it remains endothermic in the near-cognate complex, with ΔGflip of −2 kcal/mol and +1 kcal/mol, respectively, due to the different thermodynamic driving forces from the A-minor interaction networks. This model is further substantiated by the finding that paromomycin reduces the energetic cost of A1492/3 flipping and thus results in similar responses to either cognate or near-cognate tRNA binding. After codon recognition, the codon-anticodon mismatch and the sub-optimal A-minor interactions lead near-cognate complexes to adopt a conformation similar to the inactive open form, which requires about 4 kcal/mol to overcome the strain and evolve to the active closed form. The free energy in A1492/3 flipping and the strain in domain closure of the decoding center well explain the mechanism that the A-site senses the codon-anticodon mismatch through thermodynamic and kinetic specificities. Our TI calculations revealed the profile of possible tautomerization in the near-cognate complex. The tautomerization allows formation of more H-bonds in the near-cognate complex; however, the intrinsic cost still keeps the near-cognate complex less favorable than the cognate complex. Although computational cost limits the current simulations to the ribosomal A-site instead of the entire ribosome, all the specific ribosomal responses in the initial selection stage originate from conformational changes in the A-site, and are captured in our simulations. The findings in our model support the active role of A1492/3 flipping in tRNA selection, providing a comprehensive theoretical perspective in supplement to structural information to help us interpret the function and mechanism of the A-site in ribosomal translation.
Materials and Methods
Preparation and purification of A-site RNA
Uniformly 13C/15N-labeled elongated A-site (E-A-site) sample for NMR relaxation measurement was prepared by standard in vitro transcription using T7 RNA polymerase (Takara Mirus Bio, Madison, WI), uniformly 13C/15N-labeled nucleotide triphosphates (ISOTEC, Miamisburg, OH), unlabeled nucleotide triphosphates (Sigma, St. Louis, MO), and synthetic DNA templates (Integrated DNA Technologies, Coralville, IA) containing the T7 promoter and sequence of interest as described previously.[58] E-A-site sample was purified using 20% (w/v) denaturing polyacrylamide gel electrophoresis with 8 M urea and Tris-Borate-EDTA buffer. The RNA was eluted from the gel in 20 mM Tris pH 8 buffer followed by ethanol precipitation. E-A-site RNA pellet was dissolved in water and annealed by heating to 95 °C for 5 min and rapid cooling on ice for 30 min before being exchanged into NMR buffer (15 mM Phosphate, 25 mM NaCl, 0.1 mM EDTA, pH 6.4 in 10% 2H2O). The final RNA concentration of E-A-site sample was maintained at 0.5 mM using a Centricon Ultracel YM-3 concentrator (Millipore, Bedford, MA). 2-aminopurine (2-AP) labeled 29mer oligonucleotide A-site samples (2-AP1492, 2-AP1493 and A1408G-2AP1492) were purchased from Dharmacon. Lyophilized powder was dissolved in H2O and annealed by heating to 95 °C for 5 min and rapid cooling on ice for 30 min before exchanging into buffer (15 mM Phosphate, 25 mM NaCl, 0.1 mM EDTA, pH 6.4). The final RNA concentration of all three A-site samples was maintained at 20 nM. The concentrations of all the single-stranded oligonucleotides were determined using the calculated extinction coefficients from the absorbance at 260 nm measured with a CARY 300 UV-Visible spectrophotometer controlled by CARY WinUV software package.
NMR spectroscopy
All NMR experiments were conducted at 298K on Avance Bruker (Billerica, MA) 600 MHz NMR spectrometer equipped with a 5 mm triple-resonance cryogenic probe. Spectra were processed using NMRPipe/NMRDraw[59] and analyzed using SPARKY 3.[60] Longitudinal (R1) and transverse (R1ρ) relaxation rates for aromatic 13C8-H8 and 13C2-H2 were measured in E-A-site samples using a TROSY-detected carbon relaxation experiment.[61] All relaxation data was measured in an interleaved manner. Relaxation rates and errors were measured by fitting intensities against the relaxation delays to a monoexponential decay using a Mathematica 6.0 script (Wolfram Research, Inc.).[62] The R1 and R1ρ relaxation delays used are summarized in Table S2. The value of 2R2-R1 was computed using the R2 values measured at a spinlock power of 1000 Hz and an offset of 3500 Hz to minimize contribution from chemical exchange while suppressing Hartman-Hahn type transfers due to scalar coupled carbon spins. 2R2-R1 is a good estimate of order parameters S2 as described previously.[63] Relative S2 ( ) value is then calculated for each residue to that of the largest value obtained in the well-structured A-form helices.
Fluorescence intensity measurements
Fluorescence emission spectra were recorded with Fluoro Max-2 controlled by ISA Datamax (v 2.10) software. The excitation wavelength was set at 310 nm in all the experiments, with the excitation and emission slit widths set at 5 nm. A quartz cell with a 1-cm path length in both the excitation and emission directions was used for all the measurements. Fluorescence signal was measured from emission wavelengths 325 nm to 450 nm. Fluorescence maxima were detected at 370 nm and then time-based fluorescence was recorded for 15 s at an emission wavelength of 370 nm at steps of 0.1 s and signal intensity was averaged over 15 s. Samples (2.8 ml) of 2-AP labeled A-site (20 nM) were prepared in above described buffer and spectra were recorded at 298K. Spectra of Paromomycin bound (3 μM) and Neomycin bound (3 μM) 2-AP labeled A-site (20 nM) were recorded after incubation of RNA and aminoglycoside for 1 min.
Molecular simulations of A-site models
The molecular systems of the A-site models used in our simulations are shown in Fig. S1. The sequence of the oligonucleotide A-site model used in the fluorescence and NMR experiments was an elongated helical loop based on E. Coli A-site. (See supplementary information for detailed sequence.) MD simulations on the oligonucleotide A-site model were also based on the same sequence used in experiments. MD simulations on ribosomal A-site models were based on the T. Thermophilus ribosome sequence. A temperature of 310K was maintained in the simulations using the Nose-Hoover thermostat.[64, 65] The latest CHARMM force field for nucleotides was used in the MD simulations.[66] The RNA and protein were solvated using explicit solvent using the TIP3P[67] model. The oligonucleotides were built from the crystal structures 1J5E and 1J7T for the empty and paromomycin bound A-site, respectively.[49, 68] Each oligonucleotide model was solvated in a cubic water box of 67 Å in edge length with ~9600 water molecules. Periodic boundary conditions with the particle-mesh Ewald method (64 grid points on each side) were used in the simulations. A cutoff of 12 Å was used to build the non-bond list and 9 Å was used for non-bond interaction calculations in the real space. To mimic the 25 mM salt concentration in experiment, 4 pairs of Na+ and Cl− were added into the solution. In order to balance the negative charge on the oligonucleotides, 28 additional Na+ were added. (For paromomycin bound molecules only 23 additional Na+ were added because paromomycin was assumed to be fully protonated with a 5+ net charge.) Divalent cations were not included due to the known artifacts in simulations.[69] The A-site models in the context of 70S ribosome were built based on crystal structures 3TVF(30S)/3TVE(50S) (cognate) and 3UYD(30S)/3UYE(50S) (near-cognate), respectively.[26] The ribosomal residues within a radius of 25 Å from the center of A1492/3 nucleobases were included to model the ribosomal A-site. The ribosomal A-site model was solvated in spherical boundary potentials[40, 41] with a radius of 36 Å. Harmonic restraints of 0.05 and 0.01 kcal/mol·Å−1·amu−1 were applied to the backbone and side-chain of the ribosomal atoms, selected by residue, beyond 18 Å from the center of the solvation sphere. In the simulations with spherical boundary potentials, a cutoff of 16 Å for the generation of non-bond list, a cutoff of 12 Å for the non-bond interactions, and the FSHIFT and VSHIFT functions were used on atoms beyond 10 Å.[70] The SHAKE algorithm[71] was used to constrain the bonds involving hydrogen atoms in all simulations.
In the umbrella sampling, a force constant of 100 kcal/mol/rad2 was used to restrain the CPDs of A1492/3, with a 10° increment between the windows, covering the range −180° ~ +180° (1296 windows) for empty A-site, 0° ~ 240° (625 windows) for paromomycin bound A-site model, and 0° ~ 210° (484 windows) for cognate and near-cognate A-site complexes. 2 ns NVT MD was carried out for each window with a timestep of 2 fs, generating trajectories of the time scale of about 2.5 μs for empty A-site model, 1.2 μs for paromomycin bound A-site model, and 1.0 μs for both cognate and near-cognate A-site complexes, respectively. The weighted histogram analysis method (WHAM) was used to construct the 2D free energy landscapes from the umbrella sampling data.[72-74] The sampling convergence and precision were examined by dividing the trajectory in each umbrella sampling window into 4 sequential blocks, constructing the free energy landscapes from each set of data, and calculating the average of the root of mean square error (RMSE) of the free energy landscapes obtained from the 4 data sets. The average RMSE is 0.2-0.4 kcal/mol which satisfies the precision of our analysis. Based on the simulations with different conformations of A1493, the overall free energy landscape of each oligonucleotide reported in Fig. 1 was obtained by combining the two free energy landscapes of A1493 in both anti and syn conformations with their corresponding statistical weight.
Free energy changes of A1492/3 flipping in different systems were calculated using the differences between the free energies of the fully intrahelical (−60° < CPDs < +60°) and the fully extrahelical regions (120° < CPDs < 240°). In the paromomycin bound A-site analogue, the difference between the partially extrahelical (60° < CPDs < 120°) and the fully extrahelical regions was used.
The free energy of keto-to-enol tautomerization was calculated using alchemical thermodynamic integration (TI) switching the U4 base between the keto and enol form. The tautomerization of a uridine in aqueous solution serves as the reference to define the tautomerization free energy of the U4 in the near-cognate ribosome complex. A potential scaling parameter λ is used in TI as the transformation coordinate connecting the end-states. Eleven TI simulations with the timescale of 250 ps and 900 ps for each window were carried out for aqueous uridine and mRNA U4, respectively, corresponding to λ changes from 0 to 1 with an increment of 0.1. Out TI calculations suggest that the relative free energy difference between the tautomerization in aqueous uridine and mRNA U4 is about −3 ± 1.0 kcal/mol, as discussed in detail in SI. Previous experimental measurements report that the tautomerization of aqueous uracil is ~10 kcal/mol.[57] Therefore, we estimate the keto-to-enol tautomerization free energy is ~7 kcal/mol for the mRNA U4.
In the RMSD-based umbrella sampling simulations,[54] the coordinates of the backbone atoms of the near-cognate complex from PDB: 1N34 were used to define the open form Xopen. The closed form was defined by the coordinates of the backbone atoms from PDB: 3UYD and 3TVF for near-cognate and cognate complexes, respectively. Umbrella sampling was performed to construct the free energy profiles of domain closure transitions, and a progress variable of ΔRMSD = RMSD(X:Xopen) − RMSD(X:Xclosed) was used in the umbrella sampling. A harmonic potential of kf ΔRMSD2 with the force constant kf of 100 kcal/mol/Å2 was used. The free energy profile of each complex was constructed using WHAM from 10 umbrella sampling windows with a separation of 0.2 Å in ΔRMSD for a total of 4 ns.
Estimation of ribosomal selectivity at the initial selection stage
The kinetic scheme of the initial selection has been determined in previous biochemical experiments as,[4, 75]
| (1) |
where A is the tRNA ternary complex and B is the ribosome complex. C, D, and E represent the ribosome complex after the initial binding, codon recognition and GTP hydrolysis, respectively. The kinetic rate constants for the cognate complex were adapted from Ref.[4], and the values for the near-cognate complex were assumed to be the same except for k−2 and k3, which were constructed using the free energy differences calculated in this work, ΔΔGbind and . The ribosomal selectivity at the initial selection, I, is calculated according to the following definition,[ 4, 56]
| (2) |
| (3) |
Supplementary Material
Highlights.
computational modeling and simulation combined with NMR and fluorescence experiments inform and resolve a controversy on tRNA selectivity in the ribosome
free energy calculations of adenine flipping for A-site adenines 1492/1493 indicate 3-4 kcal/mol selectivity arises from coupling adenines with the mRNA-tRNA mini-helix
the strain associated with moving from the more open form and the closed form accounts for nearly 4 kcal/mol in selectivity
the free energy cost of keto to enol tautomerization is about 7 kcal/mol in the context of the ribosome with bound tRNA and mRNA, thus suggesting it is unlikely
Acknowledgments
The financial support the NIH through RR012255 and GM037554 (CLB) is greatly appreciated. We thank Dr. Sarah Woodson, Dr. Douglas Jacobsen, Anthony Mustoe and Joseph Yesselman for discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosenberger RF, Foskett G. An estimate of the frequency of in vivo transcriptional errors at a nonsense codon in escherichia coli. Mol Gen Genet. 1981;183:561–563. doi: 10.1007/BF00268784. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberger RF, Hilton J. The frequency of transcriptional and translational errors at nonsense codons in the lacz gene of escherichia coli. Mol Gen Genet. 1983;191:207–212. doi: 10.1007/BF00334815. [DOI] [PubMed] [Google Scholar]
- 3.Johansson M, Bouakaz E, Lovmar M, Ehrenberg M. The kinetics of ribosomal peptidyl transfer revisited. Mol Cell. 2008;30(5):589–598. doi: 10.1016/j.molcel.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Gromadski KB, Daviter T, Rodnina MV. A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol Cell. 2006;21(3):369–377. doi: 10.1016/j.molcel.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 5.Mittelstaet J, Konevega AL, Rodnina MV. Distortion of tRNA upon near-cognate codon recognition on the ribosome. J Biol Chem. 2011;286(10):8158–8164. doi: 10.1074/jbc.M110.210021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodnina MV. Quality control of mRNA decoding on the bacterial ribosome. In: Marintchev A, editor. Fidelity and Quality Control in Gene Expression; of Adv Prot Chem Struct Bio. Vol. 86. Academic Press; 2012. pp. 95–128. [DOI] [PubMed] [Google Scholar]
- 7.Moazed D, Noller HF. Binding of tRNA to the ribosomal A and P sites protects two distinct sets of nucleotides in 16 S rRNA. J Mol Bio. 1990;211(1):135–145. doi: 10.1016/0022-2836(90)90016-F. [DOI] [PubMed] [Google Scholar]
- 8.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JHD, et al. Crystal structure of the ribosome at 5.5 Å resolution. Science. 2001;292(5518):883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 9.Potapov AP, Triana-Alonso FJ, Nierhaus KH. Ribosomal decoding processes at codons in the A or P sites depend differently on 2-OH groups. J Biol Chem. 1995;270(30):17680–17684. doi: 10.1074/jbc.270.30.17680. [DOI] [PubMed] [Google Scholar]
- 10.Recht MI, Fourmy D, Blanchard SC, Dahlquist KD, Puglisi JD. Rna sequence determinants for aminoglycoside binding to an a-site rrna model oligonucleotide. J Mol Bio. 1996;262(4):421–436. doi: 10.1006/jmbi.1996.0526. [DOI] [PubMed] [Google Scholar]
- 11.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Structure of the a site of escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science. 1996;274(5291):1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 12.Fourmy D, Yoshizawa S, Puglisi JD. Paromomycin binding induces a local conformational change in the A-site of 16 s rRNA. J Mol Bio. 1998;277(2):333–345. doi: 10.1006/jmbi.1997.1551. [DOI] [PubMed] [Google Scholar]
- 13.Wimberly BT, Brodersen DE, Clemons WM, Morgan-Warren RJ, Carter AP, Vonrhein C, et al. Structure of the 30S ribosomal subunit. Nature. 2000;407(6802):327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 14.Ogle JM, Brodersen DE, Clemons WM, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292(5518):897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 15.Ogle JM, IV FVM, Tarry MJ, Ramakrishnan V. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111(5):721–732. doi: 10.1016/s0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- 16.Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy FV, Weir JR, et al. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326(5953):688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voorhees RM, Schmeing TM, Kelley AC, Ramakrishnan V. The mechanism for activation of GTP hydrolysis on the ribosome. Science. 2010;330(6005):835–838. doi: 10.1126/science.1194460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenner L, Demeshkina N, Yusupova G, Yusupov M. Structural rearrangements of the ribosome at the tRNA proofreading step. Nat Struct Mol Biol. 2010;17(99):1072–1078. doi: 10.1038/nsmb.1880. [DOI] [PubMed] [Google Scholar]
- 19.Rodnina MV, Wintermeyer W. Fidelity of aminoacyl-tRNA selection on the ribosome: Kinetic and structural mechanisms. Annu Rev Biochem. 2001;70(1):415–435. doi: 10.1146/annurev.biochem.70.1.415. [DOI] [PubMed] [Google Scholar]
- 20.Ogle JM, Carter AP, Ramakrishnan V. Insights into the decoding mechanism from recent ribosome structures. Trends Biochem Sci. 2003;28(5):259–266. doi: 10.1016/S0968-0004(03)00066-5. [DOI] [PubMed] [Google Scholar]
- 21.Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu Rev Biochem. 2005;74(1):129–177. doi: 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- 22.Voorhees RM, Ramakrishnan V. Structural basis of the translational elongation cycle. Annu Rev Biochem. 2013;82(1):203–236. doi: 10.1146/annurev-biochem-113009-092313. [DOI] [PubMed] [Google Scholar]
- 23.Pape T, Wintermeyer W, Rodnina MV. Conformational switch in the decoding region of 16S rRNA during aminoacyl-tRNA selection on the ribosome. Nat Struct Biol. 2000;7(2):104–107. doi: 10.1038/72364. [DOI] [PubMed] [Google Scholar]
- 24.Gromadski KB, Rodnina MV. Streptomycin interferes with conformational coupling between codon recognition and GTPase activation on the ribosome. Nat Struct Mol Biol. 2004;11(4):316–322. doi: 10.1038/nsmb742. [DOI] [PubMed] [Google Scholar]
- 25.Matt T, Akbergenov R, Shcherbakov D, Böttger EC. The ribosomal A-site: Decoding, drug target, and disease. Isr J Chem. 2010;50(1):60–70. [Google Scholar]
- 26.Demeshkina N, Jenner L, Westhof E, Yusupov M, Yusupova G. A new understanding of the decoding principle on the ribosome. Nature. 2012;484(7393):256–259. doi: 10.1038/nature10913. [DOI] [PubMed] [Google Scholar]
- 27.Shandrick S, Zhao Q, Han Q, Ayida BK, Takahashi M, Winters GC, et al. Monitoring molecular recognition of the ribosomal decoding site. Angew Chem Int Ed. 2004;43(24):3177–3182. doi: 10.1002/anie.200454217. [DOI] [PubMed] [Google Scholar]
- 28.Kaul M, Barbieri CM, Pilch DS. Aminoglycoside-induced reduction in nucleotide mobility at the ribosomal RNA A-site as a potentially key determinant of antibacterial activity. J Am Chem Soc. 2006;128(4):1261–1271. doi: 10.1021/ja056159z. [DOI] [PubMed] [Google Scholar]
- 29.Sanbonmatsu KY. Energy landscape of the ribosomal decoding center. Biochimie. 2006;88(8):1053–1059. doi: 10.1016/j.biochi.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 30.Vaiana AC, Sanbonmatsu KY. Stochastic gating and drug-ribosome interactions. J Mol Bio. 2009;386(3):648–661. doi: 10.1016/j.jmb.2008.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Réblová K, Lankaš F, Rázga F, Krasovska MV, Kocča J, Sponer J. Structure, dynamics, and elasticity of free 16s rRNA helix 44 studied by molecular dynamics simulations. Biopolymers. 2006;82(5):504–520. doi: 10.1002/bip.20503. [DOI] [PubMed] [Google Scholar]
- 32.Meroueh SO, Mobashery S. Conformational transition in the aminoacyl t-RNA site of the bacterial ribosome both in the presence and absence of an aminoglycoside antibiotic. Chem Biol Drug Des. 2007;69(5):291–297. doi: 10.1111/j.1747-0285.2007.00505.x. [DOI] [PubMed] [Google Scholar]
- 33.Romanowska J, Setny P, Trylska J. Molecular dynamics study of the ribosomal A-site. J Phys Chem B. 2008;112(47):15227–15243. doi: 10.1021/jp806814s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romanowska J, McCammon JA, Trylska J. Understanding the origins of bacterial resistance to aminoglycosides through molecular dynamics mutational study of the ribosomal A-site. PLoS Comput Biol. 2011;7(7):e1002099. doi: 10.1371/journal.pcbi.1002099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song K, Campbell AJ, Bergonzo C, de los Santos C, Grollman AP, Simmerling C. An improved reaction coordinate for nucleic acid base flipping studies. J Chem Theory Comput. 2009;5(11):3105–3113. doi: 10.1021/ct9001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikolova EN, Kim E, Wise AA, O’Brien PJ, Andricioaei I, Al-Hashimi HM. Transient hoogsteen base pairs in canonical duplex DNA. Nature. 2011;470(7335):498–502. doi: 10.1038/nature09775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.See Methods section for details on the determination of free energies and simulation precisions.
- 38.Sanbonmatsu K, Joseph S. Understanding discrimination by the ribosome: Stability testing and groove measurement of codonanticodon pairs. J Mol Bio. 2003;328(1):33–47. doi: 10.1016/s0022-2836(03)00236-5. [DOI] [PubMed] [Google Scholar]
- 39.The cognate complex was built based on the structure of PDB entry 3TVE and 3TVF, with codon-anticodon as mRNA/Phe 50-CUC′30 and tRNALUE 30-GAG-50. The near-cognate com-2 plex was built based on PDB entry 3UYD and 3UYE with mRNA/Phe 50-UUU′30 and tRNALUE 30-GAG-50
- 40.Brooks CL, III, Karplus M. Deformable stochastic boundaries in molecular dynamics. J Chem Phys. 1983;79(12):6312–6325. [Google Scholar]
- 41.Brünger A, Brooks CL, III, Karplus M. Stochastic boundary conditions for molecular dynamics simulations of ST2 water. Chem Phys Lett. 1984;105(5):495–500. [Google Scholar]
- 42.Demeshkina N, Jenner L, Westhof E, Yusupov M, Yusupova G. FEBS Letters. 13. Vol. 587. St. Petersburg: 2013. New structural insights into the decoding mechanism: Translation infidelity via a g:u pair with watson-crick geometry; pp. 1848–1857. [DOI] [PubMed] [Google Scholar]
- 43.Selmer M, Dunham CM, Murphy FV, Weixlbaumer A, Petry S, Kelley AC, et al. Structure of the 70s ribosome complexed with mRNA and tRNA. Science. 2006;313(5795):1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 44.Topal MD, Fresco JR. Base pairing and fidelity in codonanticodon interaction. Nature. 1976;263:289–293. doi: 10.1038/263289a0. [DOI] [PubMed] [Google Scholar]
- 45.Goodman MF. Mutations caught in the act. Nature. 1995;378:237–238. doi: 10.1038/378237a0. [DOI] [PubMed] [Google Scholar]
- 46.Watson J, Crick F. Genetical implications of the structure of deoxyribonucleic acid. Nature. 1953;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- 47.Echols H, Goodman MF. Fidelity mechanisms in dna replication. Annu Rev Biochem. 1991;60(1):477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 48.Bebenek K, Pedersen LC, Kunkel TA. Replication infidelity via a mismatch with watson-crick geometry. Proc Natl Acad Sci USA. 2011;108(5):1862–1867. doi: 10.1073/pnas.1012825108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vicens Q, Westhof E. Crystal structure of paromomycin docked into the eubacterial ribosomal decoding a site. Structure. 2001;9(8):647–658. doi: 10.1016/s0969-2126(01)00629-3. [DOI] [PubMed] [Google Scholar]
- 50.Dibrov SM, Parsons J, Hermann T. A model for the study of ligand binding to the ribosomal RNA helix H44. Nucleic Acids Res. 2010;38(13):4458–4465. doi: 10.1093/nar/gkq159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407(6802):340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 52.Xia T, SantaLucia J, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, et al. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 1998;37(42):14719–14735. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]
- 53.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Bio. 1999;288(5):911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 54.Woo HJ, Roux B. Calculation of absolute protein-ligand binding free energy from computer simulations. Proc Natl Acad Sci USA. 2005;102(19):6825–6830. doi: 10.1073/pnas.0409005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Please find more details in the supplemental information.
- 56.Pape T, Wintermeyer W, Rodnina M. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J. 1999;18(13):3800–3807. doi: 10.1093/emboj/18.13.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsuchiya Y, Tamura T, Fujii M, Ito M. Keto-enol tautomer of uracil and thymine. J Phys Chem. 1988;92(7):1760–1765. [Google Scholar]
- 58.Zhang Q, Sun X, Watt ED, Al-Hashimi HM. Resolving the motional modes that code for RNA adaptation. Science. 2006;311(5761):653–656. doi: 10.1126/science.1119488. [DOI] [PubMed] [Google Scholar]
- 59.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 60.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 61.Hansen AL, Al-Hashimi HM. Dynamics of large elongated RNA by NMR carbon relaxation. J Am Chem Soc. 2007;129(51):16072–16082. doi: 10.1021/ja0757982. [DOI] [PubMed] [Google Scholar]
- 62.Spyracopoulos L. A suite of mathematica notebooks for the analysis of protein main chain 15n nmr relaxation data. J Biomol NMR. 2006;36:215–224. doi: 10.1007/s10858-006-9083-0. [DOI] [PubMed] [Google Scholar]
- 63.Dethoff EA, Hansen AL, Musselman C, Watt ED, Andricioaei I, Al-Hashimi HM. Characterizing complex dynamics in the transactivation response element apical loop and motional correlations with the bulge by NMR, molecular dynamics, and mutagenesis. Biophys J. 2008;95(8):3906–3915. doi: 10.1529/biophysj.108.140285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nose S. A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys. 1984;81(1):511–519. [Google Scholar]
- 65.Hoover WG. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 66.Denning EJ, Priyakumar UD, Nilsson L, Mackerell AD. Impact of 2’-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J Comp Chem. 2011;32(9):1929–1943. doi: 10.1002/jcc.21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 68.Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, et al. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat Struct Mol Biol. 2007;14(8):727–732. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]
- 69.Auffinger P. Ions in molecular dynamics simulations of rna systems. In: Leontis N, Westhof E, editors. RNA 3D Structure Analysis and Prediction; of Nucleic Acids and Molecular Biology Springer Berlin Heidelberg. Vol. 27. 2012. pp. 299–318. [Google Scholar]
- 70.Steinbach PJ, Brooks BR. New spherical-cutoff methods for long-range forces in macromolecular simulation. J Comp Chem. 1994;15(7):667–683. [Google Scholar]
- 71.Ryckaert JP, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Chem Phys. 1977;23(3):327–341. [Google Scholar]
- 72.Ferrenberg AM, Swendsen RH. Optimized monte carlo data analysis. Phys Rev Lett. 1989;63(12):1195–1198. doi: 10.1103/PhysRevLett.63.1195. [DOI] [PubMed] [Google Scholar]
- 73.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. The weighted histogram analysis method for free-energy calculations on biomolecules. I. the method. J Comp Chem. 1992;13:1011–1021. [Google Scholar]
- 74.Grossfield A. “WHAM: the weighted histogram analysis method”, version 2.0.6. http://membrane.urmc.rochester.edu/content/wham.
- 75.Kothe U, Rodnina MV. Delayed release of inorganic phosphate from elongation factor tu following gtp hydrolysis on the ribosome. Biochemistry. 2006;45(42):12767–12774. doi: 10.1021/bi061192z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.