

Abstract
Our aim is to characterize the poorly understood mechanisms that influence episomal transgene expression within the nucleus. We found that plasmid DNA microinjected directly into a nucleus moves into a speckled pattern and occupies less nuclear volume than BSA or other inert molecules after 4 hours. In addition, plasmids that contain eukaryotic regulatory sequences and actively transcribe transgenes condense into a few select areas of the nucleoplasm and occupy less nuclear volume than bacterial vectors. This suggests that episomal DNA moves in a sequence and transcription dependent manner. We have also found that plasmids traffic to specific subnuclear domains depending on their sequence. Our experiments show that plasmids with polymerase II regulatory elements will target to nuclear spliceosome regions, while plasmids with the polymerase I promoter often traffic into nucleoli. Further elucidation of intranuclear plasmid trafficking behavior may lead to a better understanding of gene expression that could improve basic laboratory techniques and clinical gene therapies.
Keywords: Nucleus, microinjection, intranuclear trafficking, gene expression, transcription, plasmid, nuclear organization, transfection
INTRODUCTION
Although it is well accepted that DNA must enter the nucleus in order for gene expression to occur during gene transfer, what happens to the DNA once inside the nucleus has not been extensively investigated. Numerous strategies to increase efficiency of gene transfer and transcription have been developed, but almost all have assumed that the end goal of DNA trafficking is to reach the nucleus and that transcription is independent of any other nuclear function. An increasing body of evidence suggests that the eukaryotic genome is regulated by the spatial organization of genes within the nucleus (reviewed in [1, 2]), however, whether the same is true for plasmids has not been investigated. Several studies have suggested that matrix attachment regions, also called scaffold attachment regions (MARs or SARs) can increase both the abundance and duration of gene expression, presumably by controlling the maintenance of chromatin structure on the plasmid and its ability to “stick” to the nuclear matrix, thereby ensuring its retention during cell division and perhaps also aiding directly in transcription [3–6]. However, no direct evidence for intranuclear spatial regulation mediated by these sequences has been presented.
Despite the lack of internal membranes, the nucleus is a highly compartmentalized organelle whose internal structures help to functionally separate DNA into distinct compartments [3, 7]. Interphase chromosomes have been shown to consistently reside within designated territories [8, 9]. Although experiments in mammalian cells show that chromatin is relatively immobile over intranuclear distances longer than 0.5 µm [10, 11], a closer look reveals that chromatin can move within a designated area at rates faster than can be accounted for by diffusion [10]. This dynamic movement of extended lengths of chromatin are believed to influence gene expression, but the mechanisms that govern the relationship between intranuclear chromatin structures and their function remains vague. Several groups have observed changes in expression and large scale chromatin movement within interphase mammalian cells and yeast [10, 12–14]. Most studies have focused on gene silencing and shown that gene poor and silenced heterochromatin is localized to the nuclear periphery while gene rich euchromatin is more centralized [12, 15]. In contrast to the more extensive studies on silencing, only a few groups have begun to look at the potential role of euchromatin movement in the nucleus in relation to gene activation and have shown relationships between chromatin movement and transcriptional upregulation [16–20]. Such chromatin movement could either be targeted towards a sub-nuclear region in order to initiate transcription, or the already transcriptionally active locus could be the first step in assembly of a sub-nuclear domain. Further, it has been demonstrated that inhibition of transcription can disrupt nucleolar structure and can alter the localization of gene rich chromatin “loops” at the ends of designated chromosome territories [19, 21]. These data support transcriptional function as a driving force behind the assembly of sub-nuclear structures.
The majority of these experiments have relied on tagging a chromatin locus and following its movement with fluorescence recovery after photobleaching (FRAP) experiments to determine how cellular chromosomes organize and function within the nucleus [14, 15, 22, 23]. By contrast, relatively little information has been obtained on the nuclear organization and dynamics of episomal DNA following gene delivery. Both virally-derived and nonviral episomes have been shown to associate with histones and other proteins to form chromatin-like structures, thus biochemically mimicking endogenous chromosomes [24–26]. In support of integrated chromatin models, an episomal model of DNA movement has previously shown that plasmids do not exhibit much intranuclear movement [27]. However, this particular study focused on the stabilizing effect scaffold attachment regions (SARs) had on intranuclear plasmid localization and did not follow labeled plasmids that were known to actively express a transgene [27]. By contrast, a number of other studies using transfected and microinjected cells show that expressing plasmids do indeed display discreet staining patterns, suggesting movement and/or localization of the DNA once inside the nucleus [28–32]. This further suggests that gene expression and subnuclear localization of transfected plasmids may be linked. In this study, we have found that the transcriptional capability and transcribed sequence of a plasmid alters its intranuclear trafficking patterns. Specifically, plasmids expressing either RNA polymerase II transcripts (mRNA), RNA polymerase I transcripts (rRNA), or no transcript, all display unique subnuclear organization patters that are dependent on transcription, or vice versa.
RESULTS
A plasmid capable of expression displays intranuclear movement
To determine if plasmids traffic to different subnuclear domains over time, we microinjected a GFP expressing plasmid, pEGFP-N1 (Fig. 1), directly into the nuclei of TC7 cells and fixed the cells at various times post-injection for fluorescence in situ hybridization. Five minutes after injection, the plasmids were diffuse and spread throughout the entire nuclear volume (Fig. 2A). However, over the next 30 to 240 minutes, plasmids showed signs of intranuclear movement (Fig 2). At four hours post injection, the intranuclear pEGFP-N1 traffics into sub-nuclear speckles while occupying less nuclear volume. Even at 30 minutes post-injection, plasmid movement was evident as there were discrete black areas within the injected nuclei where no plasmid DNA was detected. The plasmid signal also showed moderate to near-complete colocalization with the spliceosome component, SC35, at 4 hours post-injection as represented in the two, 240 minute individual cells in Figure 2. There was also a lower level of colocalization with SC35 at the earlier time points. Similar redistribution of pEGFP-N1 was detected in NIH3T3, HeLa, A549, and primary smooth muscle cells (data not shown). Since visually detectable levels of GFP are first detected 30 minutes post nuclear injection [33], these results suggest that plasmid movement may play a role in transgene expression.
Figure 1. Plasmid maps.
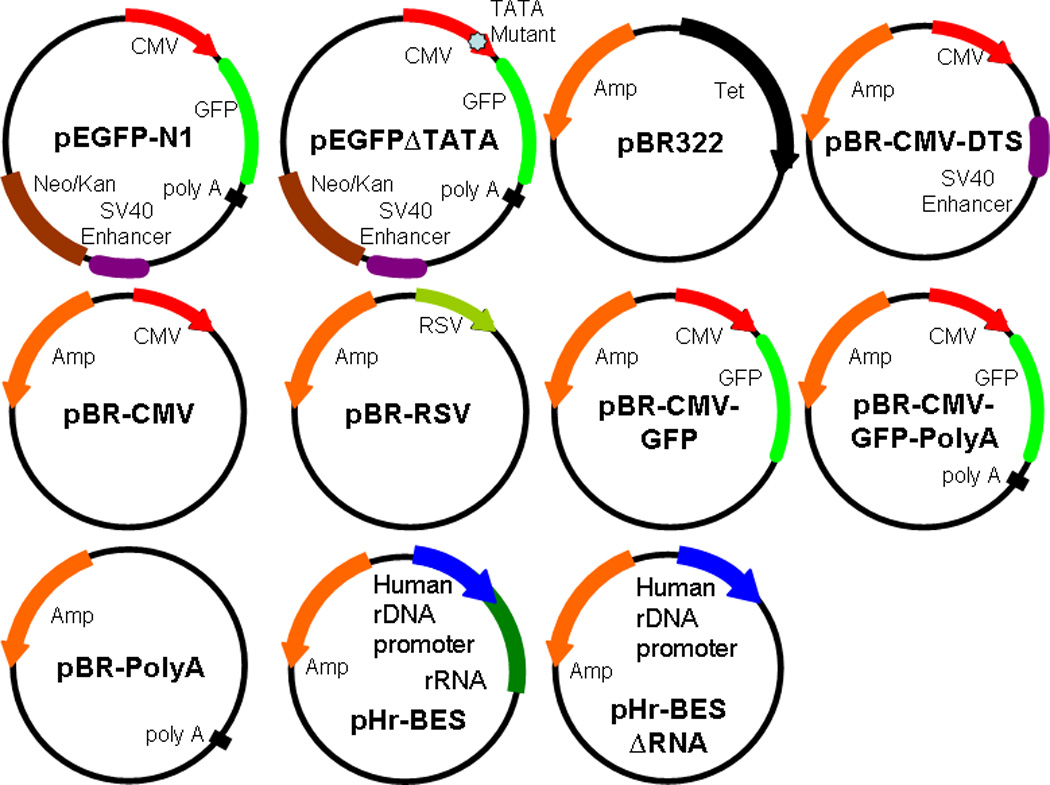
All of the plasmids used have the pEGFP-N1 (Clontech) or pBR322 backbone. pEGFP∆TATA contains a mutant TATA box. All of the pBR plasmids have self-explained eukaryotic regulatory elements cloned into the pBR322 backbone. pHr-BES has a pBR322 backbone and contains the human ribosomal DNA promoter and encodes the first 700 base pairs of the 45S pre-ribosomal RNA. pHr-BES∆RNA is the same as pHr-BES but has the encoded pre-ribosomal RNA sequence deleted.
Figure 2. Time course of microinjected plasmids.

(A) The pEGFP-N1 plasmid was microinjected directly into TC7 nuclei and detected by fluorescence in situ hybridization and confocal microscopy between 5 and 240 minutes after microinjection (green). Colocalization with the spliceosome factor, SC35, was also determined by immunofluorescence (red). Nuclear DNA was visualized with DAPI (blue). Two representative cells are shown for the 240 minute time point to illustrate co-localization with SC35 speckles. (B) Nuclei were microinjected with pBR322 and plasmids were detected four hours later via in situ hybridization as in A. After four hours, the pBR322 plasmid shows little, if any intranuclear plasmid movement. All panels are representative images of over 150 injected and imaged cells for each time point and plasmid. Bar = 10 µm.
A plasmid without eukaryotic sequences shows little intranuclear movement
Based on the experiments in Figure 2A, a logical hypothesis was that transcription factors bind to the eukaryotic regulatory sequences on the plasmid and mediate intranuclear movement. To test this, we microinjected a plasmid without any eukaryotic promoters or enhancers (pBR322) directly into TC7 nuclei and followed the plasmid redistribution over time. Expectantly, pBR322 showed very little, if any, changes in intranuclear distribution over the course of 4 hours (Fig. 2B). The pBR322 plasmid was diffuse and spread throughout the nucleus four hours post injection, much like the pEGFP-N1 appeared 5 minutes after injection.
Eukaryotic regulatory sequences alone are not sufficient to drive intranuclear plasmid redistribution
If eukaryotic regulatory sequences can mediate plasmid redistribution for pEGFP-N1, and pBR322 does not have any of these sequences, cloning such sequences from pEGFP-N1 into the pBR322 backbone should reconstitute plasmid trafficking. However, all of the plasmids created with eukaryotic sequences on the pBR322 backbone visually showed little intranuclear movement by four hours, much like the parental pBR322 plasmid. The differences in intranuclear plasmid localization were quantified by capturing Z-stacks of nuclei and deconvolving the images. Classifiers that consistently select three-dimensional pixels, or voxels, were employed to measure the intensity-independent volume of both fluorescent in situ plasmid signal and the DAPI stained nucleus. The plasmid volume was divided by the DAPI volume to produce the percentage of nuclear volume occupied by the exogenous DNA. With this technique, we were able to measure the differences between pEGFP-N1 at 5 minutes versus four hours post-injection (Fig. 3). Whereas pEGFP-N1 was diffuse and occupied over 50% of the DAPI-based nuclear volume 5 minutes after injection, it occupied about 25% of the nuclear volume four hours later. By contrast, nuclear injected BSA, pBR322 and pBR-CMV-DTS all continued to occupy over 50% of the nuclear volume at four hours (Fig. 3). The same was true for pBR322 based plasmids that contained the CMV immediate early promoter, RSV early promoter, or CMV promoter and SV40 enhancer (Fig. 3). These data suggest that eukaryotic regulatory elements alone are not sufficient to mediate plasmid movement within the nucleus.
Figure 3. Percentage of nuclear volume occupied by microinjected plasmids.

Different plasmids were individually injected into TC7 nuclei and detected by in situ hybridization. Z-stacks of injected nuclei were deconvolved and the voxels of plasmid and DAPI signal were measured. The graph displays the averages of several microinjection experiments for each condition. BSA does not seem to traffic to any specific spot within the nucleus and becomes very diffuse. Compared to pEGFP-N1 5 minutes post-injection, pBR322 and pBR-CMV-DTS all occupy similar nuclear volumes. However, pEGFP-N1 plasmids 4 hours post-injection move into more condensed subnuclear regions and occupy significantly less nuclear volume than all other conditions (p<0.01). Each bar represents the average ± SEM of all in situ positive nuclei from several experiments, and 50–150 nuclei were injected for each condition per experiment.
Transcription is required for intranuclear plasmid localization
While measuring the nuclear volume that pEGFP-N1 occupied at 4 hours post-injection, we noticed that some of the cells had a fluorescent signal that was very diffuse and spread throughout the entire nucleus, much like pBR322, whereas other nuclei showed fluorescent signal limited to small areas of the nuclei. We formed the hypothesis that nuclei showing a diffuse intranuclear plasmid pattern may not be actively expressing the transgene. To address this, we followed injected TC7 nuclei on Eppendorf CELLocate coverslips and found that even though some nuclei were injected with plasmid DNA, they did not produce detectable levels of GFP. The injected nuclei that did not express GFP showed a diffuse intranuclear plasmid staining (Fig. 4A). To further investigate the relationship between DNA trafficking and transcription, we mutated the TATA box in pEGFP-N1 and found that it was also spread throughout the nucleus four hours post-injection (Fig. 4A). Lipofectin-mediated transfections showed that the relative number of pEGFP∆TATA GFP positive cells was only 12% of wild-type pEGFP-N1 transfected cells (data not shown). The intensity of pEGFP∆TATA based expression was also weaker (data not shown). Likewise, the transcription inhibitors actinomycin D and α-amanitin prevented GFP expression in all injected cells and abolished the intranuclear movement of pEGFP-N1 four hours post injection (Fig. 4A). The quantified results are summarized in Figure 4B. The volumes of the nuclei alone between the EGFP expressing and non-expressing cells were statistically identical (780 ± 31 µm3 vs 772 ± 45 µm3, p=0.94). As a result, the nuclear volume occupied by the plasmids was due plasmid movement, and not changes in nuclear size.
Figure 4. Transcription capability influences intranuclear plasmid movement.

(A) Extended focus deconvolved images show that cells expressing GFP have a nuclear pEGFP-N1 pattern that is condensed while cells that do not express GFP have plasmid patterns that are more spread out throughout the entire nucleus. (B) The average plasmid occupied nuclear volume from several experiments for each condition is represented in the graph. Cells that actively express GFP have episomal plasmids that occupy an average of 16% nuclear volume, and is significantly different (p<0.0001) from non-expressing pEGFP-N1, pEGFP∆TATA, or pEGFP-N1 nuclei treated with either actinomycin D or α-amanitin. Each bar represents the average ± SEM of all in situ positive nuclei from several experiments, and 50–150 nuclei were injected for each condition per experiment (p<0.0001).
An active poly-adenylation signal plays a role in episome movement
The previous data suggest that transcription and mRNA production are required for intranuclear plasmid redistribution, but the mRNA signal that controls this redistribution is unknown. Considering that the poly-adenylation (poly(A)) signal is one of the distinguishing features of mRNA, we set out to test its importance in mediating plasmid movement within the nucleus. Three plasmids with the pBR322 backbone, pBR-PolyA, pBR-CMV-GFP, and pBR-CMV-GFP-PolyA, were individually injected into TC7 nuclei and once again fixed 4 hours post-injection. pBR-PolyA was diffuse throughout the nucleus and behaved similar to pBR322 (Fig. 5). pBR-CMV-GFP had detectable GFP levels in less than 1% of lipofectin transfected cells (data not shown), and occupied a high nuclear volume in injected nuclei (Fig. 5). Cells injected with pBR-CMV-GFP-PolyA segregated into expressing and non-expressing categories, similar to pEGFP-N1. The GFP-positive cells had nuclear volumes as low as 3% with an average of 16%, significantly lower than the GFP-negative cells (Fig. 5). Therefore, the data suggest that a poly(A) tail can provide stability for the newly transcribed RNA, which, in turn, may allow access for processing factors to bind and traffic the DNA-RNA complex to a sub-nuclear domain.
Figure 5. The poly-adenylation signal is required for transcription mediated intranuclear plasmid movement.

At 4 hours post-injection, cells that actively express GFP from pBR-CMV-GFP-PolyA have episomal plasmids that occupy an average of 15.6% nuclear volume, and are significantly different (p<0.0001) from pBR322, pBR-PolyA, pBR-CMV-GFP, or non-expressing pBR-CMV-GFP-PolyA. Each bar represents the average ± SEM of all in situ positive nuclei from several experiments, and 50–150 nuclei were injected for each condition per experiment (p<0.0001).
Sequences can direct plasmids to different subnuclear domains
The data in Figures 2, 4 and 5 suggest that plasmids that produce mRNA with a poly(A) tail from a polymerase II (pol II) promoter will traffic within the nucleus and accumulate around SC35 speckles. Since transcription mediates this plasmid redistribution, we investigated the possibility of targeting a plasmid to a different sub-nuclear destination by altering the encoded RNA sequence. The plasmid pHr-BES (Fig 1), based on a pBR322 backbone, contains the human polymerase I (pol I) promoter and the first 700 base pairs of the transcribed 45S pre-ribosomal RNA (rRNA) sequence, and is transcriptionally active [34, 35]. In uninjected control cells, immunofluorescence images displayed a nucleolar localization of the rDNA transcription factor, upstream binding factor (UBF) (Fig. 6A). pHr-BES was injected into HeLa (human cervical carninoma) nuclei and fixed at 2 and 4 hours post-injection. After 2 hours, pHr-BES is present in the nucleoplasm as several large, or many small, round speckles that strongly colocalize with the pol I transcription factors, UBF (Fig. 6A), RPA39/40, and TAF I (data not shown). By 4 hours post-injection, pHr-BES is still colocalized with UBF, but 41.7% of the cells have over 50% of the plasmid DNA within their nucleoli (Fig. 6A). In contrast, pEGFP-N1 is never found in nucleoli and does not colocalize with UBF four hours post-injection (Fig. 6B). Likewise, the human pHr-BES plasmid does not traffic into the nucleoli of green monkey TC7 cells after 4 hours, suggesting species specific protein-DNA interactions, previously shown to be crucial for RNA polymerase I mediated transcription [34] that may also be important in DNA trafficking (data not shown). Altogether, these results suggest that the sub-nuclear distribution of episomes can be altered based on the type of RNA produced.
Figure 6. A plasmid with ribosomal DNA sequences traffics into nucleoli after 4 hours.

(A) The ribosomal transcription factor, UBF (red), is normally found within the nucleoli of uninjected cells (No DNA). pHr-BES, containing the human pol I promoter and encoded rRNA, was injected into HeLa nuclei and observed 2 and 4 hours later. Two hours post-injection, the pHr-BES in situ signal (green) colocalizes with UBF in the DAPI-stained nucleoplasm (blue). However, 4 hours after injection, the plasmid has moved into nucleoli and still remains colocalized with UBF. (B) pEGFP-N1 is not found in nucleoli and does not colocalize with UBF 4 hours post injection. Bar = 10 µm.
rRNA expression and processing is necessary to traffic plasmids into nucleolar space
Based on the redistribution of pHr-BES from nucleoplasm to nucleoli over time, two possible mechanisms could be responsible: movement of plasmids into the nucleolar space by the ribosomal transcription factors, or by active transcription as seen for the trafficking of pEGFP-N1 into condensed nucleoplasmic speckles. pHr-BES∆RNA has the same composition as pHr-BES, but does not encode pre-rRNA (Fig 1). Reverse transcription PCR from HeLa cells transfected with pHr-BES∆RNA shows that this plasmid produces a bacterially-encoded set of non-rRNA that lack any rRNA splicing and processing signals [36]. Four hours after pHr-BES∆RNA was injected into HeLa nuclei, the plasmid in situ signal was observed only in the nucleoplasm, but does colocalize with UBF (Fig. 7) and RPA39/40 (data not shown). The fact that transcription factors colocalize with pHr-BES∆RNA but do not facilitate plasmid movement into nucleoli further suggest that transcription of specific RNA signal sequences are necessary to traffic plasmids to sub-nuclear domains.
Figure 7. Plasmid trafficking into nucleoli is dependent on rRNA transcription.
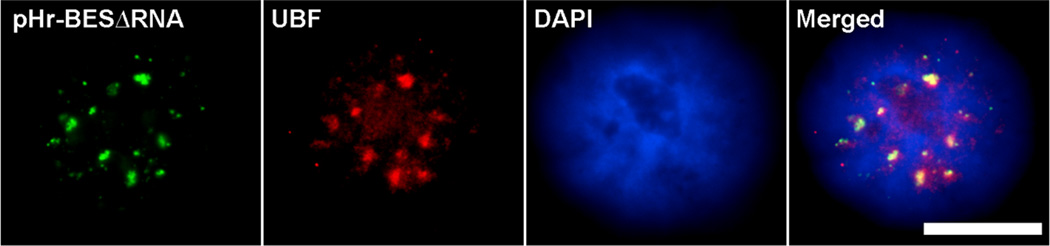
When pHr-BES∆RNA, which does not encode rRNA sequences, is injected into HeLa cell nuclei, it is organized into speckles and colocalizes with transcription factor UBF after 4 hours, but it fails to traffic into nucleolar space. Bar = 10 µm
If transcription is the key element behind episomal transgene movement, RNA processing factors may be the protein links that direct the plasmids to their final destination. The rRNA processing factors Nop58 (Fig. 8A) and fibrillarin (data not shown) are normally found in the nucleoli of uninjected HeLa cells. However, 2 hours post nuclear injection with pHr-BES, UBF and Nop58 (Fig. 8B) and fibrillarin (data not shown) are pulled into the nucleoplasm where they colocalize with round plasmid speckles. When HeLa cells were injected with pHr-BES∆RNA, though, Nop58 (Fig. 8C) and fibrillarin (data not shown) remained in the nucleoli and did not colocalize with the plasmid speckles, despite UBF still being recruited to the pHr-BES∆RNA in the nucleoplasm (Fig. 8C).
Figure 8. rRNA processing factors are only recruited to plasmids with transcribed rRNA sequences.

(A) UBF and the rRNA processing factor Nop58 are found in the nucleoli of uninjected HeLa cells. (B) pHr-BES colocalizes with UBF and Nop58 in the nucleoplasm two hours post nuclear injection. C) pHr-BES∆RNA colocalizes with UBF, but Nop58 remains nucleolar and does not colocalize with the plasmid 2 hours post nuclear injection.
Altogether these data suggest that plasmid redistribution within the nucleus is dependent upon active transcription. Further, these results suggest that plasmid movement can be targeted to discrete subnuclear domains by the active transcription of different encoded RNA signals and the processing factors that recognize them. Thus, subnuclear organization of transfected plasmids and gene expression are linked.
DISCUSSION
Previous studies have shown that DNA can move within the nucleus independent of transcription [12, 37]. Our data support these findings as plasmids 5 minutes after microinjection occupy 60% of nuclear volume while a non-expressing pBR322 plasmid occupies only 55% nuclear volume after four hours (Fig. 3). This relatively low amount of redistribution did not appear to be targeted to spliceosome or nucleolar compartments. Other published studies have suggested that the nucleolus and intranuclear compartments can act as barriers to hold or impede DNA movement [11]. Similar findings were reported by Mearini and colleagues in their studies on the intranuclear dynamics of plasmids [27]. In this study, SAR-containing or lacking plasmids were injected into cell nuclei and observed in fixed cells or by FRAP in living cells. Their data clearly show that the plasmids are relatively immobile and show no redistribution over time. However, it should be stressed that none of these plasmids were transcriptionally active, and thus confirm our findings with non-expressing plasmids. By contrast, our data clearly show that actively transcribing plasmids show much more targeted redistribution, or trafficking (Fig. 4). Based on the current data, and that of previous studies, we propose three possible models for intranuclear plasmid movement. In the first model, plasmids move to a subnuclear domain and are then transcribed. The second model would have the plasmids transcribed first, and the newly transcribed, nascent RNA would recruit transcription and processing domains to the DNA (i.e., the nuclear domains would form de novo around the plasmids). The third, and favored, model is a combination of the previous two, and would be likely to exist in the dynamic environment of the nucleus where macromolecular complexes can organize and self-assemble. In all cases, whether plasmids show directed motor-based movement to specific subnuclear regions or simply diffuse throughout the nucleus and accumulate at specific regions by binding interactions cannot be determined from the present study, but data from others on expression and movement of chromosomal DNA supports both mechanisms [16, 38].
In our favored model, plasmids that are not being transcribed would only show directionless movement within a small contained region of the nucleus. Next, transcription factors that bind to regulatory elements on the plasmids assemble on the promoter along with early processing factors to form a pre-initiation complex (PIC) and begin to inefficiently produce RNA transcripts. The nucleus also contains protein factors that are highly mobile compared to endogenous chromatin. Sequences on the RNA transcripts may then interact with free and highly mobile processing factors [39, 40] which could facilitate trafficking of the entire complex of plasmid, transcription/processing factors, and RNA to the appropriate subnuclear region (e.g., transcriptional center) where additional factors necessary for efficient, continued transcription and processing of the plasmid encoded genes are concentrated. Indeed, transcription and RNA processing are tightly coordinated both spatially and temporally as transcription and processing factors are often found in the same immunoprecipitated complex [41–43]. The end result is greatly increased expression following correct subnuclear localization. A recent study showed that an induced gene can be transcribed in more than one nuclear location, however, the data presented clearly indicated that specific areas exist in the nucleus where more efficient transcription can occur [20].
Although elegant work has shown the recruitment of Pol II transcriptional machinery to defined gene loci in the genome and the sequential recruitment of the processing machinery, none of these studies have addressed whether the genes themselves are moving [44–46]. Rather, most have focused on the movement of the transcription and processing foci as opposed to the DNA [45, 47]. The few other exceptions have been with studies focusing on viral genome transcription and replication where it has been shown that the genomes of herpes simplex virus, human papilloma virus, adenovirus, and SV40 condense and localize into replication centers associated with ND10 domains [31, 32]. Similarly, adenovirus and unintegrated HIV genomes localize with and cause the redistribution of SC35 domains, although the HIV genomes appear to be transcriptionally inactive [48, 49]. However, in most cases, it appears that this localization is dependent on the presence and/or expression of specific viral proteins that cause the redistribution within the nucleus. By contrast, the results presented here demonstrate that even in the absence of expression of viral or exogenous gene products, transcriptionally active plasmids do indeed show specific and regulated patterns of nuclear distribution. As a result, the regulation of DNA trafficking likely resides at the level of protein-nucleic acid interaction and self-assembly.
Transfection experiments are necessary tools for most every molecular biology lab, but the direct effects of having exogenous DNA within the cell are often overlooked. This study shows that even if DNA is introduced into the nucleus, the transgene may not express detectable levels of protein in all cells (Fig. 4). We also show that the first signs of detectable levels of protein also coincide with the first signs of intranuclear episome trafficking. Whether nuclear redistribution of plasmids results in enhanced transcription or transcription and processing drives plasmid movement in the nucleus remains to be seen, although it is likely that both occur simultaneously and continually to lead to enhanced gene expression. Transfections are much more complicated than expected and perhaps the reason why some cell types overexpress a transgene better than others rests with the ability of the cell to target episomal DNA to subnuclear domains. Consequently, we may need to alter our focus to design better viral and non-viral vectors that traffic not just through the plasma and nuclear membranes, but into discrete subnuclear domains to ultimately improve the overall efficacy of gene therapy.
MATERIALS AND METHODS
Plasmids
The pEGFP-N1 plasmid was obtained from Clontech (Mountain View, CA). pEGFP∆TATA was made with primers 5’ -GGTAGGCGTGTACGGTGGGAG GTCgATcgAAGCAGAGCTGGTTTAGTGAACCG- 3’ and 5’ -CGGTTCACTAAA CCAGCTCTGCTTcgATcGACCTCCCACCGTACACGCCTACC- 3’ to mutate the TATA box to a Pvu I site. pBR-CMV, pBR-RSV, and pBR-CMV-SV40 were created as previously described [29]. pBR-CMV-GFP-PolyA was created with primers 5’ -GCACTAGTGGATAACCGTATTACCGCCATGCAT- 3’ and 5’ -CGACTAGTGGACAAACCACAACTAGAATGCAGTG- 3’ and the fragment was digested with Spe I and inserted into the Nhe I site in pBR322. pBR-CMV-GFP was created with primers 5’ -GCACTAGTGGATAACCGTATTACCGCCATGCAT- 3’ and 5’ -CGACTAGTGA GTCGCGGCCGCTTTACTTG- 3’ and the fragment was digested with Spe I and inserted into the Nhe I site in pBR322. pHR-BES∆rRNA was created by PCR amplification of the pol I promoter from pHr-BES [34, 35] using primers 5’-GGAATTCCGAGGCCCTTTCGTCTTCAA-3’ and 5’-CCCAAGCTTGGGCCAGAGGACAGCGTGTCA-3’. The resulting 620 bp fragment which lacks all pre-rRNA coding sequences was cloned into the EcoRI and HindIII restriction sites of pBR322.
Cell Culture
HeLa cells (cervical carcinoma, ATCC CCL-2) were grown in Minimal Essential Medium supplemented with 10% fetal bovine serum, antibiotics and antimycotics. TC7 cells were grown in Dulbecco’s modification of Eagle’s Medium supplemented with 10% fetal bovine serum, antibiotics and antimycotics. Cells were subcultured with the selective mitotic detachment method and seeded into etched coverslips or 175 µm Eppendorf CELLocate etched coverslips (Eppendorf, Hamburg, Germany) in 12-well dishes and allowed to grow in a humidified 37°C incubator with 5% CO2 for 48 hours until they reached 50–60% confluency.
Microinjection
Coverslips of cells were placed in 5ml of fresh media in a 60 mm dish. Labeled or unmodified plasmids were passed through a 0.22 µm filter, diluted in 0.5X phosphate buffered saline (PBS; 68.5 mM NaCl, 1.3 mM Potassium Chloride, 5 mM Phosphate Buffer, pH 7.2) and quantified spectrophotometrically. An inverted Leica microscope fitted with a 37°C acrylic incubation chamber and an Eppendorf Femtojet microinjection system was used to deliver plasmids (0.35mg/ml) into nuclei with an inject pressure of 145 hPa for 0.3 seconds. After microinjection, coverslips were returned to a humidified 37°C incubator and grown with 5% CO2 until the indicated time. For transcription inhibitor experiments, actinomycin D (10 µg/ml) and α-amanitin (5 µg/ml) were added to the media 30 minutes prior to injection and remained in the media until the cells were fixed.
Plasmid in situ hybridation and Immunofluorescence
Cells were rinsed with 1X PBS, permeabilized in 1X PBS plus 0.5% Triton-X100 for 45 seconds, fixed at −20°C in a 1:1 methanol:acetone solution for 5 minutes, and placed in 70% ethanol at 4°C overnight. Plasmid in situ hybridizations were carried out using nick-translated DNA probe from appropriate plasmid backbones with an Alexa 488 fluorophore (Invitrogen, Madison, WI) as previously described [50]. Prior to adding nick-translated fluorescence probe, the cells were heated to 70° C in 70% formamide to separate the injected dsDNA and denature any RNA. After fluorescence in situ hybridization, coverslips were blocked with 1mg/ml BSA in PBS for 1 hour at room temperature. Coverslips were washed for 5 minutes in PBS and then SC35 (1:200), UBF (1:50), and/or Nop58 (1:750) antibodies with 0.5 mg/ml BSA in PBS were added to the coverslips for 2 hours at room temperature. The coverslips were washed with PBS 3 times for 15 minutes. Alexa 555 and/or Alexa 647 secondary antibodies (Invitrogen) were added in PBS with 0.5 mg/ml BSA for 2 hours at room temperature. Slides were washed 3 times for 15 minutes with PBS and mounted on slides with DAPI and the anti-fade reagent, DABCO (Invitrogen).
GFP tracking
TC7 nuclei were seeded onto Eppendorf CELLocate coverslips which have a mapped grid etched onto the surface and nuclearly injected 48 hours later. Four hours post-injection, cells on the mapped surface were imaged and recorded for GFP expression. The cells were then fixed and fluorescence in situ hybridization performed. Since the in situ hybridization process abrogates the native GFP fluorescence, Z-stacks of nuclei with in situ signal were captured, deconvolved, and referred back to the recorded GFP positive cells on the CELLocate grid. Cells that were GFP positive had the plasmid signal scored as expressed transgene, while cells that did not have detectable levels of GFP were scored as non-expressed plasmids.
Microscopy and deconvolution
Confocal images were taken with an inverted Zeiss LSM 510 with UV confocal microscope. Z-stacks (0.3µm increments) were taken with OpenLab software (Improvision, MA) and a Leica (Germany) DMRXA2 epifluorescence microscope outfitted with a Hamamatsu OCRA-ER 12-bit camera (Hamamatsu, Japan) and appropriate DAPI, GFP, Cy3, and Cy5 filter sets. The capture times for each channel were set as the longest exposure that ensured at least one pixel was at maximum brightness (absolute value of 4095 for the 12-bit camera) without allowing any pixels to over saturate the image. Z-stacks were deconvolved with the iterative restoration Velocity module (Improvision, MA) using calculated point spread functions.
Image Analysis and Volume Measurements
Plasmid, DAPI, and BSA classifiers were created with the Velocity Measurements module (Improvision) and applied to each deconvolved Z-stack to consistently and independently measure voxels for each fluorescent channel. The minimum intensity for each classifer was created by subtracting the average intensity value of a 10 × 10 pixel square of background. The maximum intensity value was set to the brightest pixel found in the channel. The Velocity “fill holes” option was selected for the DAPI classifier to obtain a cylindrical volume for the nucleus. The plasmid classifer was used to measure all plasmid voxels above background and was divided by the number of voxels measured by the DAPI classifier to obtain percent nuclear volume occupied by plasmid DNA. The Velocity measurements module was also used to calculate colocalization values and percentage of plasmid DNA within nucleoli.
Statistical Analysis
Paired student’s t-tests were performed to determine statistical significance between individual samples within an experiment.
ACKNOWLEDGEMENTS
Thanks to Sui Huang and the Huang lab for providing us with the UBF and Nop58 antibodies and the pHr-BES∆RNA plasmid. This work was supported in part by NIH grants HL59956 and HL71643. JZG was supported by a predoctoral fellowship 0415541Z from the Midwest Affiliate of the American Heart Association.
REFERENCES
- 1.Verschure PJ. Chromosome organization and gene control: it is difficult to see the picture when you are inside the frame. J Cell Biochem. 2006;99:23–34. doi: 10.1002/jcb.20957. [DOI] [PubMed] [Google Scholar]
- 2.Shav-Tal Y, Darzacq X, Singer RH. Gene expression within a dynamic nuclear landscape. Embo J. 2006;25:3469–3479. doi: 10.1038/sj.emboj.7601226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson DA, Juranek S, Lipps HJ. Designing nonviral vectors for efficient gene transfer and long-term gene expression. Mol Ther. 2006;14:613–626. doi: 10.1016/j.ymthe.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 4.Jenke AC, Stehle IM, Herrmann F, Eisenberger T, Baiker A, Bode J, Fackelmayer FO, Lipps HJ. Nuclear scaffold/matrix attached region modules linked to a transcription unit are sufficient for replication and maintenance of a mammalian episome. Proc Natl Acad Sci U S A. 2004;101:11322–11327. doi: 10.1073/pnas.0401355101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenke BH, Fetzer CP, Stehle IM, Jonsson F, Fackelmayer FO, Conradt H, Bode J, Lipps HJ. An episomally replicating vector binds to the nuclear matrix protein SAF-A in vivo. EMBO Rep. 2002;3:349–354. doi: 10.1093/embo-reports/kvf070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baiker A, Maercker C, Piechaczek C, Schmidt SB, Bode J, Benham C, Lipps HJ. Mitotic stability of an episomal vector containing a human scaffold/matrix-attached region is provided by association with nuclear matrix. Nat Cell Biol. 2000;2:182–184. doi: 10.1038/35004061. [DOI] [PubMed] [Google Scholar]
- 7.Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- 8.Manuelidis L. Individual interphase chromosome domains revealed by in situ hybridization. Hum Genet. 1985;71:288–293. doi: 10.1007/BF00388453. [DOI] [PubMed] [Google Scholar]
- 9.Gasser SM. Positions of potential: nuclear organization and gene expression. Cell. 2001;104:639–642. doi: 10.1016/s0092-8674(01)00259-8. [DOI] [PubMed] [Google Scholar]
- 10.Abney JR, Cutler B, Fillbach ML, Axelrod D, Scalettar BA. Chromatin dynamics in interphase nuclei and its implications for nuclear structure. J Cell Biol. 1997;137:1459–1468. doi: 10.1083/jcb.137.7.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chubb JR, Boyle S, Perry P, Bickmore WA. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr Biol. 2002;12:439–445. doi: 10.1016/s0960-9822(02)00695-4. [DOI] [PubMed] [Google Scholar]
- 12.Andrulis ED, Neiman AM, Zappulla DC, Sternglanz R. Perinuclear localization of chromatin facilitates transcriptional silencing. Nature. 1998;394:592–595. doi: 10.1038/29100. [DOI] [PubMed] [Google Scholar]
- 13.Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, Sedat JW. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol. 1997;7:930–939. doi: 10.1016/s0960-9822(06)00412-x. [DOI] [PubMed] [Google Scholar]
- 14.Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, Belmont AS. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol. 1996;135:1685–1700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilbert N, Boyle S, Fiegler H, Woodfine K, Carter NP, Bickmore WA. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell. 2004;118:555–566. doi: 10.1016/j.cell.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Brown JM, Leach J, Reittie JE, Atzberger A, Lee-Prudhoe J, Wood WG, Higgs DR, Iborra FJ, Buckle VJ. Coregulated human globin genes are frequently in spatial proximity when active. J Cell Biol. 2006;172:177–187. doi: 10.1083/jcb.200507073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chambeyron S, Bickmore WA. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 2004;18:1119–1130. doi: 10.1101/gad.292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, Fraser P. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 19.Mahy NL, Perry PE, Bickmore WA. Gene density and transcription influence the localization of chromatin outside of chromosome territories detectable by FISH. J Cell Biol. 2002;159:753–763. doi: 10.1083/jcb.200207115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taddei A, Van Houwe G, Hediger F, Kalck V, Cubizolles F, Schober H, Gasser SM. Nuclear pore association confers optimal expression levels for an inducible yeast gene. Nature. 2006;441:774–778. doi: 10.1038/nature04845. [DOI] [PubMed] [Google Scholar]
- 21.Haaf T, Ward DC. Inhibition of RNA polymerase II transcription causes chromatin decondensation, loss of nucleolar structure, and dispersion of chromosomal domains. Exp Cell Res. 1996;224:163–173. doi: 10.1006/excr.1996.0124. [DOI] [PubMed] [Google Scholar]
- 22.Bystricky K, Heun P, Gehlen L, Langowski J, Gasser SM. Long-range compaction and flexibility of interphase chromatin in budding yeast analyzed by high-resolution imaging techniques. Proc Natl Acad Sci U S A. 2004;101:16495–16500. doi: 10.1073/pnas.0402766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gasser SM. Visualizing chromatin dynamics in interphase nuclei. Science. 2002;296:1412–1416. doi: 10.1126/science.1067703. [DOI] [PubMed] [Google Scholar]
- 24.Archer TK, Lefebvre P, Wolford RG, Hager GL. Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation. Science. 1992;255:1573–1576. doi: 10.1126/science.1347958. [DOI] [PubMed] [Google Scholar]
- 25.Griffith J, Dieckmann M, Berg P. Electron microscope localization of a protein bound near the origin of simian virus 40 DNA replication. J Virol. 1975;15:167–172. doi: 10.1128/jvi.15.1.167-172.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeong SW, Lauderdale JD, Stein A. Chromatin assembly on plasmid DNA in vitro. Apparent spreading of nucleosome alignment from one region of pBR327 by histone H5. J Mol Biol. 1991;222:1131–1147. doi: 10.1016/0022-2836(91)90597-y. [DOI] [PubMed] [Google Scholar]
- 27.Mearini G, Nielsen PE, Fackelmayer FO. Localization and dynamics of small circular DNA in live mammalian nuclei. Nucleic Acids Res. 2004;32:2642–2651. doi: 10.1093/nar/gkh587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dean DA. Import of plasmid DNA into the nucleus is sequence specific. Exp. Cell Res. 1997;230:293–302. doi: 10.1006/excr.1996.3427. [DOI] [PubMed] [Google Scholar]
- 29.Dean DA, Dean BS, Muller S, Smith LC. Sequence requirements for plasmid nuclear entry. Exp. Cell Res. 1999;253:713–722. doi: 10.1006/excr.1999.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebestyén MG, Ludtke JL, Bassik MC, Zhang G, Budker V, Lukhtanov EA, Hagstrom JE, Wolff JA. DNA vector chemistry: the covalent attachment of signal peptides to plasmid DNA. Nature Biotech. 1998;16:80–85. doi: 10.1038/nbt0198-80. [DOI] [PubMed] [Google Scholar]
- 31.Swindle CS, Zou N, Van Tine BA, Shaw GM, Engler JA, Chow LT. Human papillomavirus DNA replication compartments in a transient DNA replication system. J Virol. 1999;73:1001–1009. doi: 10.1128/jvi.73.2.1001-1009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Q, Bell P, Tegtmeyer P, Maul GG. Replication but not transcription of simian virus 40 DNA is dependent on nuclear domain 10. J Virol. 2000;74:9694–9700. doi: 10.1128/jvi.74.20.9694-9700.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dean DA. Gene Delivery by Direct Injection and Facilitation of Expression by Mechanical Stretch. In: Goldman RD, Spector DL, editors. Live Cell Imaging: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratories Press; 2005. pp. 51–66. [Google Scholar]
- 34.Grummt I, Roth E, Paule MR. Ribosomal RNA transcription in vitro is species specific. Nature. 1982;296:173–174. doi: 10.1038/296173a0. [DOI] [PubMed] [Google Scholar]
- 35.Miesfeld R, Arnheim N. Identification of the in vivo and in vitro origin of transcription in human rDNA. Nucleic Acids Res. 1982;10:3933–3949. doi: 10.1093/nar/10.13.3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopp K, Gasiorowski JZ, Chen D, Gilmore R, Norton JT, Wang C, Leary DJ, Chan EK, Dean DA, Huang S. Pol I transcription and pre-rRNA processing are coordinated in a transcription-dependent manner in mammalian cells. Mol Biol Cell. 2007;18:394–403. doi: 10.1091/mbc.E06-03-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hediger F, Neumann FR, Van Houwe G, Dubrana K, Gasser SM. Live imaging of telomeres: yKu and Sir proteins define redundant telomere-anchoring pathways in yeast. Curr Biol. 2002;12:2076–2089. doi: 10.1016/s0960-9822(02)01338-6. [DOI] [PubMed] [Google Scholar]
- 38.Chuang CH, Carpenter AE, Fuchsova B, Johnson T, de Lanerolle P, Belmont AS. Long-range directional movement of an interphase chromosome site. Curr Biol. 2006;16:825–831. doi: 10.1016/j.cub.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 39.Anderson JL, Hope TJ. Intracellular trafficking of retroviral vectors: obstacles and advances. Gene Ther. 2005;12:1667–1678. doi: 10.1038/sj.gt.3302591. [DOI] [PubMed] [Google Scholar]
- 40.Platani M, Goldberg I, Swedlow JR, Lamond AI. In vivo analysis of Cajal body movement, separation, and joining in live human cells. J Cell Biol. 2000;151:1561–1574. doi: 10.1083/jcb.151.7.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Vegvar HE, Lund E, Dahlberg JE. 3' end formation of U1 snRNA precursors is coupled to transcription from snRNA promoters. Cell. 1986;47:259–266. doi: 10.1016/0092-8674(86)90448-4. [DOI] [PubMed] [Google Scholar]
- 42.Gallagher JE, Dunbar DA, Granneman S, Mitchell BM, Osheim Y, Beyer AL, Baserga SJ. RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev. 2004;18:2506–2517. doi: 10.1101/gad.1226604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xing Y, Johnson CV, Moen PT, Jr, McNeil JA, Lawrence J. Nonrandom gene organization: structural arrangements of specific pre-mRNA transcription and splicing with SC-35 domains. J Cell Biol. 1995;131:1635–1647. doi: 10.1083/jcb.131.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xing Y, Johnson CV, Dobner PR, Lawrence JB. Higher level organization of individual gene transcription and RNA splicing. Science. 1993;259:1326–1330. doi: 10.1126/science.8446901. [DOI] [PubMed] [Google Scholar]
- 46.Misteli T, Spector DL. The cellular organization of gene expression. Curr Opin Cell Biol. 1998;10:323–331. doi: 10.1016/s0955-0674(98)80007-0. [DOI] [PubMed] [Google Scholar]
- 47.Misteli T, Caceres JF, Spector DL. The dynamics of a pre-mRNA splicing factor in living cells. Nature. 1997;387:523–527. doi: 10.1038/387523a0. [DOI] [PubMed] [Google Scholar]
- 48.Jimenez-Garcia LF, Spector DL. In vivo evidence that transcription and splicing are coordinated by a recruiting mechanism. Cell. 1993;73:47–59. doi: 10.1016/0092-8674(93)90159-n. [DOI] [PubMed] [Google Scholar]
- 49.Bell P, Montaner LJ, Maul GG. Accumulation and intranuclear distribution of unintegrated human immunodeficiency virus type 1 DNA. J Virol. 2001;75:7683–7691. doi: 10.1128/JVI.75.16.7683-7691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dean DA. Import of plasmid DNA into the nucleus is sequence specific. Exp Cell Res. 1997;230:293–302. doi: 10.1006/excr.1996.3427. [DOI] [PubMed] [Google Scholar]