

SUMMARY
Although the use of nonviral vectors for gene therapy offers distinct advantages including the lack of significant inflammatory and immune responses, the levels of expression in vivo remain much lower than those obtained with their viral counterparts. One reason for such low expression is that unlike many viruses, plasmids have not evolved mechanisms to target to the nucleus of the non-dividing cell. In the absence of mitosis, plasmids are imported into the nucleus in a sequence-specific manner and we have shown in cultured cells by transfection and microinjection experiments that the SV40 enhancer mediates plasmid nuclear import in all cell types tested (Dean et al., 1999, Exp Cell Res 253: 713–722). To test the effect of this import sequence on gene transfer in the intact animal, we have recently developed an electroporation method for DNA delivery to the intact mesenteric vasculature of the rat. Plasmids expressing luciferase or GFP from the CMV immediate early promoter/enhancer and either containing or lacking the SV40 enhancer downstream of the reporter gene were transferred to the vasculature by electroporation. When transfected into actively dividing populations of smooth muscle or epithelial cells, the plasmids gave similar levels of expression. By contrast, the presence of the SV40 sequence greatly enhanced gene expression of both reporters in the target tissue. At 2 days post-transfer, plasmids with the SV40 sequence gave 10-fold higher levels of luciferase expression, and at 3 days the difference was over 40-fold. The presence of the SV40 sequence did not simply increase the rate of nuclear import and expression, since expression from the SV40-lacking plasmid did not increase beyond that seen at day 2, the time of maximum expression for either plasmid. In situ hybridization experiments confirmed that the increased gene transfer and expression was indeed due to increased nuclear localization of the delivered SV40 sequence-containing plasmid. Based on these findings, the ability to target DNA to the nucleus can increase gene transfer in vivo and inclusion of the SV40 sequence into plasmids will enhance non-viral gene delivery.
Keywords: nuclear import, non-viral vectors, plasmid DNA, electroporation, vasculature, gene therapy
Currently, gene therapy is limited by two factors: side effects of certain vectors, and inefficiency of others for gene transfer. Over the past decade, numerous viral and non-viral approaches have been proposed and developed for transferring genes to various tissues in vivo, but all have serious limitations. Inefficiency of gene transfer, immunological responses and non-specificity of cell targeting are just a few of the problems. In contrast to viral vectors, non-viral vectors show great potential for gene therapy. Very little immune response or inflammation is generated against DNA, either as naked plasmid or when complexed with liposomes or other polymers such as polyethylenimine1. Plasmid production is simple and yields high levels of vector, and plasmids can be easily purified without contaminants such as wild type or defective virus particles. Thus, multiple administrations of vector can be given and there are fewer safety concerns. Unfortunately, the efficiency of transfer of non-viral vectors in most tissues remains low, and for non-viral vectors to be of significant use, their ability to transfect cells in vivo must be increased.
Two of the areas that are limiting for gene transfer are delivery into the cytoplasm of the cell and targeting to the nucleus2–4. Multiple approaches have been developed to facilitate uptake of plasmids across the plasma membrane. By contrast, relatively few methods have been developed to enhance the delivery of plasmids into the nucleus. In dividing cells, the nuclear envelope breaks down during mitosis and it has been shown that the degree of transfectability of cells correlates well with their progress through the cell cycle5,6. Unfortunately, most of the cells of interest for gene therapy are either very slowly dividing or nondividing, and in these cells, the nuclear envelope is a major barrier to gene transfer7–12.
In non-dividing cells, DNA nuclear entry is not passive, but is an active process in which multiple cellular components have been shown to be involved. Using a variety of approaches, including transfection, microinjection, and permeabilized cell systems, we have observed that plasmid nuclear import in non-dividing cells is a sequence-specific process3,13,14. We have identified several DNA sequences that target plasmids into the nuclei of non-dividing cells3,4,13,15. The common feature to these sequences is that they contain binding sites for transcription factors. One sequence that acts as a DNA nuclear targeting sequence (DTS) is the SV40 enhancer, which is known to bind to over 10 distinct transcription factors. Based on a number of data, we have proposed a model in which the nuclear import of SV40 DTS-containing plasmids is mediated by one or more of these transcription factors which bind to the DNA in the cytoplasm and shuttle the complex into the nucleus14. When this SV40 sequence is placed downstream of a reporter gene driven by the CMV immediate early promoter and enhancer, which does not function as a DTS, gene expression can be detected in either arrested or synchronized cells prior to cell division, while an otherwise identical plasmid that lacks the DTS is incapable of gene expression3,15. Thus, it stands to reason that incorporation of the SV40 DNA targeting sequence would also function to increase nuclear accumulation of DNA and therefore increase gene expression in vivo.
In order to test the efficacy of using such DNA nuclear targeting sequences in vivo, we have taken advantage of our recently developed electroporation technique to deliver DNA to the intact vasculature of the rat16. This technique results in high level gene expression in all cell layers of the vasculature and results in no tissue damage or downstream ischemia16. Because the cells of the vasculature are terminally differentiated and non-dividing, this model serves as an excellent system in which to study the effects of the SV40 DTS. In this study, we demonstrate that incorporation of the SV40 DTS into expression plasmids results in 10–40-fold increases in vascular gene expression, confirming the function of DNA nuclear targeting sequences in vivo.
To determine the effects of the presence of the SV40 DTS in an expression plasmid on in vivo gene transfer and expression, we created two matched pairs of plasmids that expressed different reporter genes (Fig. 1). Because the CMV immediate early promoter (CMViep) does not mediate nuclear import of cytoplasmically delivered plasmids in cultured cells, we were able to use this strong viral promoter to drive either luciferase or GFP expression3,15. The SV40 enhancer was cloned downstream of the reporter gene in two of these constructs. We have shown by microinjecting the GFP plasmids into the nuclei of cells that both plasmids (containing or lacking the SV40 DTS) gave similar levels of gene expression, demonstrating that they are transcriptionally equivalent3. By contrast, when they are injected into the cytoplasm, the plasmid lacking the SV40 DTS cannot localize to the nucleus or allow gene expression until the cells undergo mitosis. As such, these plasmids are efficient reporters for nuclear import.
Figure 1. Cartoon of plasmid constructs.

Matched sets of GFP or luciferase reporter constructs were created3. Reporter genes are driven by the CMV immediate early promoter which does not mediate plasmid nuclear import, and when present, the SV40 DTS is downstream of the reporter gene. All plasmids were prepared using Qiagen Gigaprep kits and greater than 80% were in the supercoiled form as determined by agarose gel electrophoresis.
We have previously used the GFP-expressing plasmid containing the SV40 sequence to develop a method for high level gene transfer to the vasculature using electroporation16. This procedure is rapid, safe, and results in no ischemic damage to the treated vessels or downstream tissues. However, it is possible that the application of the electric field or the procedure itself could promote cell division or neovascularization, thus, complicating our analysis. When electroporated or naïve mesenteric neurovascular bundles were analyzed for proliferating cell nuclear antigen (PCNA) expression, a marker of cell proliferation17, no difference in expression levels were detected between the tissues, whereas significant PCNA nuclear staining can be seen in the small intestine which is known to have a high rate of cell division (Fig. 2). To determine what effect the SV40 DTS has on gene transfer to the vasculature, we transferred the GFP plasmids to the rat mesenteric vasculature and determined the levels of GFP expression two days post-transfer, during the height of gene expression (Fig. 3). Robust GFP expression can be seen in vessels that received pGFP-DTS, but not in a control vessel that received no DNA. In striking contrast, almost no GFP expression over background can be seen in vessels that were electroporated with the same amount of pGFP lacking the SV40 sequence. Multiple vessels from two rats are shown, as are control vessels from each animal. These results demonstrate that the SV40 DTS enhances gene transfer and expression in vivo.
Figure 2. Evaluation of cellular proliferation in electroporated rat mesenteric vessels.

PCNA expression was detected by immunohistochemistry in naïve (A) and electroporated (B) rat mesenteric vessels or in rat small intestine (C). Vessels were electroporated as previously described16. Briefly, male Sprague-Dawley rats (200–400 g) were anesthetized with isoflurane, a midline incision was made, and the small intestine was exteriorized. Approximately 1 cm of each neurovascular bundle was placed individually in the electrode and covered with a solution of 10 mM Tris, pH 8, 1 mM EDTA, 140 mM NaCl. Based on the design of the electrode, approximately 55 µl of solution were used to bathe the vessel. Vessels (8–10 per animal) were electroporated with 8 square wave pulses lasting 10 milliseconds each at a field strength of 200V/cm using a BTX830 electroporator (Genetronics, San Diego CA). After all vessels were electroporated within a given animal, the incision was closed and the animal was allowed to recover and returned to the vivarium. All experiments were conducted in accordance with institutional guidelines in compliance with the recommendations of the Guide for Care and Use of Laboratory Animals. One or two days post-transfer, vessels were removed from the animals which were then euthanized. Vessels were rinsed extensively with cold PBS, fixed in 10% buffered formalin, and embedded in paraffin for thin section preparation. Immunohistochemistry was performed on 6µm sections using a mouse monoclonal antibody directed against PCNA (Santa Cruz Biotechnology, Santa Cruz, CA), and detected with Alkaline Phosphatase ABC reagent and Vector Blue substrate (Vector Laboratories, Burlingame, CA). Sections were counterstained with eosin and photographed. Images are representative of multiple vessels (n= 3 animals, 6 naïve and 6 electroporated vessels from each). Bar = 100 µm.
Figure 3. Effect of the SV40 DTS on GFP gene expression in electroporated rat mesenteric arteries.

Plasmid solutions containing DNA at 2 mg/ml in 10 mM Tris, pH 8, 1 mM EDTA, 140 mM NaCl were delivered to rat mesenteric vessels by electroporation as described in Figure 2. Vessels in panel A received pGFP and those in panel B received pGFP-DTS (or no DNA, “Control”). Two days post-transfer, vessels were removed from the animals which were then euthanized, rinsed extensively with cold PBS and the vessel was dissected away from the surrounding adipose tissue. GFP was excited at 488 nm and visualized at 515 nm using the appropriate filter cubes with a low power objective on an upright Leica DMRX fluorescence microscope. Fluorescent images were collected, all with the same exposure time and gain settings, using a Hamamatsu ORCA-2 cooled CCD camera and OpenLab3.0 software (Improvision, Lexington MA). The vessels shown are representative of those from multiple animals for each plasmid (n = 5 rats for pGFP; n = 11 rats for pGFP-DTS).
To quantify the effect of the nuclear targeting sequence, luciferase expressing plasmids were transferred to the vasculature (Fig. 4). A total of 79 vessels within ten individual animals were electroporated with pCMV-Lux-DTS and harvested at 2 days post-electroporation. In this group of animals we observed a mean luciferase expression of 365 pg ± 75 pg (SEM) per vessel segment (~ 1 cm). By contrast, vessels receiving pCMV-Lux (65 vessels in 10 rats), expressed 28.7 pg ± 8.6 pg luciferase per vessel. Thus, the presence of the SV40 DTS enhanced gene expression by greater than 10-fold.
Figure 4. Quantitative evaluation of the effect of the SV40 DTS on vascular gene transfer.

Rat mesenteric vessels were electroporated with pCMV-Lux (n = 65 vessels in 10 rats) or pCMV-Lux-DTS (n = 79 vessels in 10 rats), as described in Fig. 2. Two days post-transfer, vessels were removed, immediately snap frozen in liquid nitrogen on a prefrozen bed of Promega lysis buffer containing 1mM dithiothreitol (400 µl; Promega, Madison WI), and ground into a fine powder28. Lysates were assayed for luciferase activity, standardized against purified recombinant luciferase protein (Promega) and expressed as pg luciferase per vessel16. Mean expression levels were calculated and standard error of the mean (SEM) was determined using Instat software (GraphPad Inc., San Diego CA). Mann-Whitney U-test revealed a statistically significant difference in expression between the two groups, p<0.001 (*).
One possible reason for the decreased expression from the plasmids lacking an SV40 DTS is that the timing of gene expression has shifted. Inclusion of a nuclear targeting sequence in a plasmid results in rapid transport into the nucleus, whereas plasmids lacking such a sequence may take longer to accumulate using other means. In cell culture we cannot exclude this possibility because the time frame of our experiments is relatively short. However, electroporation is known to deposit large amounts of DNA into the cytoplasm, and if given enough time, it is possible that a few of the plasmids lacking a DTS may enter the nucleus. To test this, we followed gene expression of the luciferase expressing plasmids over time (Fig. 5). The plasmid carrying the SV40 DTS showed a level of expression 100-fold over background in electroporated vessels at days 2 and 3 post-delivery, and then decreased by days 5 and 7 to slightly above background. Similarly, the peak of expression of the DTS-lacking plasmid was at day 2, but expression was only 7-fold over background, and never increased at later times to levels that were statistically significant over background. Thus, the timing of expression of the two plasmids is similar, suggesting that nuclear import of the DTS-lacking plasmids is simply inefficient.
Figure 5. Time course of expression of plasmids containing or lacking import sequences.

Plasmids were transferred to vessels using electroporation and at the indicated times, harvested for measurement of luciferase expression as described in Figs. 2 through 4. Between 2 and 10 animals were used at each time point (n ≥ 12 vessels per time point per construct). Time 0 represents vessels that received no DNA, and accounts for the level of sensitivity of the assay. * p<0.01 versus time 0; # p<0.05 versus pCMV-Lux at the same time point.
Based on all of our present and previous data, the most straight-forward explanation for the increased gene expression seen in vessels electroporated with plasmids carrying the SV40 DTS is that more plasmids are targeting to the nucleus leading to increased transcription. To directly address this, in situ hybridizations to detect the input DNA were performed on tissue sections from animals electroporated with SV40-containing or lacking plasmids (Fig. 6). Eight hours post-electroporation, considerable amounts of signal are seen in tissue sections from animals that received mixtures of pCMV-Lux and pGFP plasmids containing or lacking the DTS (Fig. 6A–D). None of the signal in DTS-lacking sections co-localize with nuclei (Fig. 6A and B), while a portion of the DTS-containing plasmids localize to the nuclei even at this early time (Fig. 6C and D). By 24 hours post-electroporation, most of the DNA signal in sections from vessels electroporated with the DTS-lacking plasmids has disappeared, presumably due to DNA degradation (Fig. 6E and F), although most of the DNA signal is nuclear at the same time point in vessels that received the DTS plasmids (Fig. 6G and H). Very little, if any, signal was detected in tissues that had received a mixture of pCMV-Lux and pGFP plasmids by 48 hours (Fig. 6I and J). By contrast, a significant amount of input DNA could still be detected in tissue sections from animals that were electroporated with the SV40-containing plasmids pCMV-Lux-DTS and pGFP-DTS (Fig. 6K and L). At higher magnification, the presence of input DTS-containing DNA can be detected clearly in the nuclei of cells (Fig 6M and N).
Figure 6. Plasmid localization in rat mesenteric vessels as analyzed by in situ hybridization.
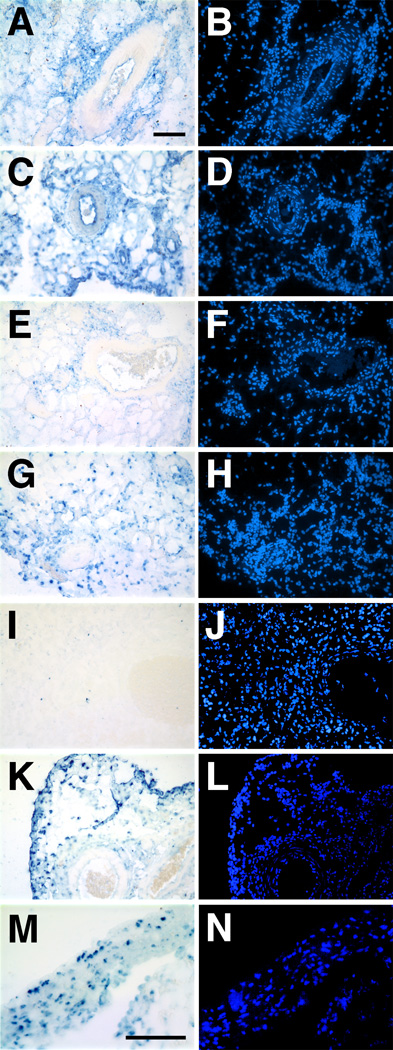
Plasmid solutions containing pCMV-Lux and pGFP (2mg/ml each; A, B, E, F, I, and J) or pCMV-Lux-DTS and pGFP-DTS (2 mg/ml each; C, D, G, H, and K – N) were transferred to the rat mesenteric vasculature by electroporation as described in Figure 2. Vessels were harvested at 8 (A – D), 24 (E – H), or 48 hours (I – N) post-electroporation, rinsed with cold PBS, fixed overnight in 10% buffered formalin and embedded in paraffin for thin sections. In situ hybridizations were performed on 10µm sections as described by Moorman29, using biotin-labeled, nick translated pCMV-Lux-DTS and pGFP-DTS as probe. Following hybridization and washes, sections were treated with RNase H to eliminate detection of mRNA. Electroporated DNA was detected using the TSA Biotin System (Perkin-Elmer, Boston, MA) and Alkaline Phosphatase-Vector Blue ABC system (A, C, E, G, I, K, and M). Nuclei were counterstained with DAPI (B, D, F, H, J, L, and N). Panels A – L are at the same magnification; Bars = 100 µm.
In the present study, we have extended our findings on the role of plasmid nuclear targeting sequences in cultured cells to the intact vasculature and have shown that the presence of a DTS greatly increases gene transfer and expression. Previously, we have demonstrated that in the absence of cell division, plasmids lacking an import sequence do not localize to the nucleus or allow gene expression in cultured cells. Injection of 10,000–20,000 copies of a plasmid that lacks the SV40 sequence into the cytoplasm of arrested or synchronized cells does not allow nuclear import or gene expression, whereas as few as 10 plasmids containing the SV40 sequence lead to gene expression in the same cells when cytoplasmically injected3,13. Thus, in cultured cells, the need for an import sequences appears absolute in the absence of cell division. By contrast, plasmids lacking a DTS are able to enter the nuclei of cells in vivo, at least at some very low level, based on the fact that low level gene expression was detected in vivo (Figs. 4 and 5), but was below the level of detection in our in situ hybridization experiments (Fig. 6). However, the present results demonstrate that incorporation of a nuclear targeting sequence can improve in vivo gene transfer. These results confirm those obtained in skeletal muscle where inclusion of the SV40 sequence into expression plasmids increased gene expression 20–40 fold18.
The data presented here suggest that the SV40 DTS increases gene expression by increasing import of the plasmids into the nucleus. Microinjection followed by in situ hybridization has shown that plasmids lacking the SV40 DTS, including pCMV-Lux and pGFP, fail to localize to the nucleus in the absence of cell division13,19. This confirms that the SV40 enhancer has two activities: the ability to stimulate nuclear import of DNA and the more traditional transcriptional enhancer function9. Another property of the SV40 enhancer is its ability to direct nucleosome phasing in the SV40 genome. In infected cells, the SV40 genome contains approximately 22 host nucleosomes phased at intervals radiating away from the enhancer and late promoter, which remain nucleosome-free20,21. While there is a short palindromic sequence of DNA in the origin of replication (180 bp from the enhancer repeats), it is not necessary for nucleosome phasing or nuclear import, and to date, there have been no other data to support a role for DNA secondary structure in the enhancer that may account for phasing. Rather, it is generally assumed that the higher affinity binding of transcription factors to this region initiates phasing of the nucleosomes. It is possible that such chromatin organization may contribute to the increased gene expression seen with the SV40 DTS-containing plasmids, but it is unlikely since increased nuclear localization and/or gene expression has been detected in vitro and in vivo when the DTS is placed either upstream or downstream of the promoter and reporter gene, suggesting that the effects of the SV40 sequence are position-independent3,13,18. Thus, it is possible that the SV40 sequence is displaying all three activities in vivo to account for increased gene expression, but based on transfection and microinjection studies in vitro and in dividing tumor tissue in vivo where it was found that matched plasmids containing and lacking this SV40 sequence were transcriptionally equivalent within a factor of two, it is likely that the import activity is predominant3,18,22. Indeed, when the presence and location of the electroporated plasmids were analyzed by in situ hybridization, a DNA signal, nuclear or otherwise, was detected only in tissues that received the SV40-containing plasmids.
Based on the sequences required for nuclear import in both SV40 and several other nuclear targeting sequences we predict that DNA nuclear uptake is mediated by specific transcription factors that form a complex with the DNA. Experiments in cultured and permeabilized cells from our laboratory and others support this model3,14,15,23,24. Unlike the SV40 enhancer, many other strong viral promoters such as the CMViep or the RSV LTR appear not to mediate nuclear import even though they bind many of the same transcription factors as the SV40 sequence3,15. The likely reason for this is that it is the overall organization and structure of the transcription factor-DNA complex that is important and that more than one specific transcription factor is needed for nuclear localization. Protein-mediated DNA nuclear import also has been suggested for the nuclear localization of the HIV pre-integration complex, although in the case of HIV, the NLS-containing proteins that are essential for nuclear import are viral proteins that are supplied by infecting virions25. An alternative mechanism for HIV DNA nuclear import has also been proposed to involve a displaced DNA strand “flap” at the 2-LTR junction26. How this “flap” mediates nuclear import has yet to be established mechanistically, but since the SV40 enhancer has no apparent secondary or triplex structure, it is unlikely that a similar mechanism plays a role in the import activity of the SV40 sequence.
Although we have demonstrated in cultured cells that not all promoters, enhancers, and transcription factor binding sites promote nuclear import, it is possible that we have not been able to detect low levels of nuclear import in our systems or that we have not allowed sufficient time for import to occur. Thus, similar but weaker protein and transcription factor-DNA complexes could form on other regions of the plasmids to mediate low level nuclear import. By introducing large quantities of DNA into cells in vivo by electroporation, the low level expression seen from plasmids lacking import sequences could become detectable over time. Because we see low levels of gene expression in vivo from plasmids lacking a DTS (Figs. 4 and 5), at least a few plasmids must be gaining entry into the nucleus for transcription to take place. This suggests that in the absence of nuclear targeting sequences, import is low, but if enough DNA is delivered to the tissue, this limitation can be overcome.
In this study we have demonstrated that incorporation of the SV40 universal DNA targeting sequence into expression plasmids acts to enhance gene expression in the intact rat mesenteric vasculature and in combination with electroporation gives high level gene expression in vivo. Recently, electroporation has been used in the rabbit carotid and has shown similar high level gene expression and therapeutic efficacy when DNA was delivered intraluminally and the electric field was applied from the adventitial surface27. Thus, the use of electroporation for gene transfer to the vasculature is a safe and powerful tool that can be utilized for the study of numerous physiological and cellular processes within the context of the whole animal as well as investigating and exploiting the process of exogenous DNA nuclear import for the development and improvement of non-viral gene therapy vectors.
ACKNOWLEDGMENTS
This work was supported by grants HL59956 from the NHLBI (DAD) and DK51430 from the NIDDK (JNB) and an award from the American Heart Association (JLY).
REFERENCES
- 1.Parker SE. Cancer gene therapy using plasmid DNA: safety evaluation in rodents and non-human primates [see comments] Hum Gene Ther. 1995;6:575–590. doi: 10.1089/hum.1995.6.5-575. [DOI] [PubMed] [Google Scholar]
- 2.Zabner J, et al. Cellular and molecular barriers to gene transfer by a cationic lipid. J. Biol. Chem. 1995;270:18997–19007. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]
- 3.Dean DA, Dean BS, Muller S, Smith LC. Sequence requirements for plasmid nuclear entry. Exp. Cell Res. 1999;253:713–722. doi: 10.1006/excr.1999.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dean DA. Nucleocytoplasmic trafficking. In: Mahato RI, editor. Pharmaceutical perspectives of nucleic acid-based therapeutics. Harwood Academic Publishers. 2002. in press. [Google Scholar]
- 5.Fasbender A, Zabner J, Zeiher BG, Welsh MJ. A low rate of cell proliferation and reduced DNA uptake limit cationic lipid-mediated gene transfer to primary cultures of ciliated human airway epithelia. Gene Therapy. 1997;4:1173–1180. doi: 10.1038/sj.gt.3300524. [DOI] [PubMed] [Google Scholar]
- 6.Brunner S, et al. Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Therapy. 2000;7:401–407. doi: 10.1038/sj.gt.3301102. [DOI] [PubMed] [Google Scholar]
- 7.Capecchi MR. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell. 1980;22:479–488. doi: 10.1016/0092-8674(80)90358-x. [DOI] [PubMed] [Google Scholar]
- 8.Graessman M, Graessman A. Regulation of SV40 gene expression. Adv. Cancer Res. 1981;35:111–149. doi: 10.1016/s0065-230x(08)60910-0. [DOI] [PubMed] [Google Scholar]
- 9.Graessman M, et al. Helper activity for gene expression, a novel function of the SV40 enhancer. Nucleic Acids Res. 1989;17:6603–6612. doi: 10.1093/nar/17.16.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kopchick JJ, Ju G, Skalka AM, Stacey DW. Biological activity of cloned retroviral DNA in microinjected cells. Proc Natl Acad Sci U S A. 1981;78:4383–4387. doi: 10.1073/pnas.78.7.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirzayans R, Remy AA, Malcom PC. Differential expression and stability of foreign genes introduced into human fibroblasts by nuclear versus cytoplasmic microinjection. Mutation Res. 1992;281:115–122. doi: 10.1016/0165-7992(92)90045-j. [DOI] [PubMed] [Google Scholar]
- 12.Thornburn AM, Alberts AS. Efficient expression of miniprep plasmid DNA after needle micro-injection into somatic cells. Biotechniques. 1993;14:356–358. [PubMed] [Google Scholar]
- 13.Dean DA. Import of plasmid DNA into the nucleus is sequence specific. Exp. Cell Res. 1997;230:293–302. doi: 10.1006/excr.1996.3427. [DOI] [PubMed] [Google Scholar]
- 14.Wilson GL, Dean BS, Wang G, Dean DA. Nuclear import of plasmid DNA in digitonin-permeabilized cells requires both cytoplasmic factors and specific DNA sequences. J. Biol. Chem. 1999;274:22025–22032. doi: 10.1074/jbc.274.31.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vacik J, Dean BS, Zimmer WE, Dean DA. Cell-specific nuclear import of plasmid DNA. Gene Therapy. 1999;6:1006–1014. doi: 10.1038/sj.gt.3300924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin JB, Young JL, Benoit JN, Dean DA. Gene transfer to intact mesenteric arteries by electroporation. J Vasc Res. 2000;37:372–380. doi: 10.1159/000025753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohr F, Wenz F, Haas S, Flentje M. Comparison of proliferating cell nuclear antigen (PCNA) staining and BrdUrd-labelling index under different proliferative conditions in vitro by flow cytometry. Cell Prolif. 1995;28:93–104. doi: 10.1111/j.1365-2184.1995.tb00058.x. [DOI] [PubMed] [Google Scholar]
- 18.Li S, et al. Muscle-specific enhancement of gene expression by incorporation of the SV40 enhancer in the expression plasmid. Gene Therapy. 2001;8:494–497. doi: 10.1038/sj.gt.3301419. [DOI] [PubMed] [Google Scholar]
- 19.Dean DA, Kasamatsu H. Signal- and energy-dependent nuclear transport of SV40 Vp3 by isolated nuclei. Establishment of a filtration assay for nuclear protein import. J. Biol. Chem. 1994;269:4910–4916. [PubMed] [Google Scholar]
- 20.Shelton ER, Wassarman PM, DePamphilis ML. Structure, spacing, and phasing of nucleosomes on isolated forms of mature simian virus 40 chromosomes. J Biol Chem. 1980;255:771–782. [PubMed] [Google Scholar]
- 21.Friez M, Hermansen R, Milavetz B. Chromatin structure of the simian virus 40 late promoter: a deletional analysis. J Virol. 1999;73:1990–1997. doi: 10.1128/jvi.73.3.1990-1997.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reddy JA, Dean D, Kennedy MD, Low PS. Optimization of Folate-Conjugated Liposomal Vectors for Folate Receptor-Mediated Gene Therapy. J. Pharm. Sci. 1999;88:1112–1118. doi: 10.1021/js990169e. [DOI] [PubMed] [Google Scholar]
- 23.Langle-Rouault F, et al. Up to 100-fold increase of apparent gene expression in the presence of Epstein-Barr virus oriP sequences and EBNA1: implications of the nuclear import of plasmids. J Virol. 1998;72:6181–6185. doi: 10.1128/jvi.72.7.6181-6185.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mesika A, Grigoreva I, Zohar M, Reich Z. A regulated, NFkappaB-assisted import of plasmid DNA into mammalian cell nuclei. Mol Ther. 2001;3:653–657. doi: 10.1006/mthe.2001.0312. [DOI] [PubMed] [Google Scholar]
- 25.Sherman MP, Greene WC. Slipping through the door: HIV entry into the nucleus. Microbes Infect. 2002;4:67–73. doi: 10.1016/s1286-4579(01)01511-8. [DOI] [PubMed] [Google Scholar]
- 26.Zennou V, et al. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell. 2000;101:173–185. doi: 10.1016/S0092-8674(00)80828-4. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto T, et al. Successful and optimized in vivo gene transfer to rabbit carotid artery mediated by electronic pulse. Gene Ther. 2001;8:1174–1179. doi: 10.1038/sj.gt.3301502. [DOI] [PubMed] [Google Scholar]
- 28.Manthorpe M, Hartikka J, Vahlsing HL, Sawdey M. Quantification of plasmid DNA expression in vivo. In: Ferre F, editor. Gene Quantification. Burhaüser: Cambridge, MA; 1996. [Google Scholar]
- 29.Moorman AF, Houweling AC, de Boer PA, Christoffels VM. Sensitive nonradioactive detection of mRNA in tissue sections: novel application of the whole-mount in situ hybridization protocol. J Histochem Cytochem. 2001;49:1–8. doi: 10.1177/002215540104900101. [DOI] [PubMed] [Google Scholar]