

Abstract
For non-viral gene delivery to be successful, plasmids must move through the cytoplasm to the nucleus in order to be transcribed. While the cytoskeletal meshwork acts as a barrier to plasmid DNA movement in the cytoplasm, the microtubule network is required for directed plasmid trafficking to the nucleus. We have shown previously that plasmid-microtubule interactions require cytoplasmic adapter proteins such as molecular motors, transcription factors, and importins (Vaughan and Dean, 2006, Mol Ther 13;422). However, not all plasmid sequences support these interactions to allow movement to the nucleus. We now demonstrate that microtubule-DNA interactions can show sequence-specificity with promoters containing binding sites for cyclic AMP response-element binding protein (CREB), including the Cytomegalovirus immediate early promoter (CMViep). Plasmids containing CREB binding sites showed stringent interactions in an in vitro microtubule-binding assay. Using microinjection and real-time particle tracking, we show that the inclusion of transcription factor binding sites within plasmids permits cytoplasmic trafficking of plasmids during gene transfer. We found that CREB binding sites are bound by CREB in the cytoplasm during transfection, and allow for enhanced rates of movement and subsequent nuclear accumulation. Moreover, siRNA knockdown of CREB prevented this enhanced trafficking. Therefore, transcription factor binding sites within plasmids are necessary for interactions with microtubules and enhance movement to the nucleus.
Keywords: trafficking, microtubules, CREB, transfection, non-viral
Introduction
Non-viral gene therapy has the potential to be a promising therapeutic intervention for various diseases due to its low toxicity and safety concerns. However, to make this a reality, gene transfer efficiency needs to be improved for non-viral vectors. Regardless of the method of gene delivery, the genetic material must move through the cytoplasm to reach the nucleus for gene expression. In order to enhance the movement of these vectors, we must elucidate how plasmid DNA navigates through the cytoplasm to the nucleus and exploit these mechanisms to develop non-viral treatment approaches.
An intact microtubule network and motor proteins such as dynein are necessary for transfected naked DNA and some viruses to traverse the cytoplasm and reach the nucleus1–3. However, the mechanism by which DNA is able to interact with microtubules is unknown. Since DNA does not bind directly to dynein, it is most likely that a multi-protein complex mediates this interaction by bridging both the DNA and dynein. This complex could contain either non-specific or sequence-specific DNA binding proteins as well as additional proteins that play a role in protein and/or DNA nuclear import, such as importin-β, transportin, and RAN4–7. If sequence-specific DNA binding proteins are involved, only plasmids with these sequences for these DNA binding proteins would be moved toward the nucleus.
The SV40 enhancer acts as a DNA nuclear targeting sequence (DTS), which binds many nuclear localization sequence (NLS)-containing transcription factors to facilitate the entry of plasmids into the nucleus8,9. Whether this enhancer sequence and/or other eukaryotic elements within the plasmid bind the proteins responsible for interactions with dynein for cytoplasmic trafficking is unknown.
In previous studies from our lab, we used a microtubule spin-down assay to show in vitro that plasmids could interact with microtubules only in the presence of additional proteins present in cell extract2. In these studies, the plasmids that carried both the CMViep and the SV40 enhancer showed strong interactions with microtubules. Whether either of these specific sequences, or any eukaryotic transcriptional regulatory sequence for that matter, is needed for or play any role in cytoplasmic plasmid trafficking is unknown. However, a large body of data from our lab and others demonstrates that many plasmids lacking the CMV promoter are capable of efficient transfection and nuclear import8,9.
In the current study, a microtubule spin-down assay was used to determine which, if any, element(s) of the plasmid allow for interactions with microtubules in the presence of cell extract. Plasmids containing binding sites for CREB showed strong microtubule binding while those lacking CREB binding sites failed to interact. Although CREB indeed formed complexes with CREB binding site-containing plasmids within 15 minutes of electroporation-mediated transfection in living cells, it was not required for movement. Microinjection and real-time particle tracking of labeled plasmids found that plasmids containing CREB binding sites appear to have faster rates of movement and more rapid nuclear localization than promoter-containing plasmids lacking these sequences. Further, siRNA-mediated knock down of CREB in cells confirmed that the enhanced movement was indeed due to CREB binding. These results suggest that transcription factors such as CREB are important components of the plasmid trafficking complex, which mediate DNA movement to the nucleus during gene transfer.
Results
Plasmid interactions with microtubules are sequence-specific in vitro
We have previously shown that plasmids interact with microtubules in the presence of cell extract in an in vitro spin-down assay and use microtubules and associated motors for trafficking to the nucleus2,10. In these experiments, the plasmids used contained the SV40 DTS, the CMViep, and the GFP or luciferase gene. Using the microtubule in vitro spin-down assay, a variety of constructs were used to determine which, if any, of these sequence elements were required for microtubule interactions in the presence of cell extract. Using quantitative PCR it was found that pBR322, a plasmid with no eukaryotic sequences, did not fractionate into the pellet, indicating that it was unable to associate with microtubules, whereas pCMV-Lux-DTS, a plasmid that expresses luciferase from the CMViep and also contains the SV40 DTS, showed robust binding (Fig. 1a). When the DTS or the luciferase gene was the only eukaryotic sequence present in the pBR322 plasmid backbone, the plasmids did not associate with microtubules. By contrast, plasmids containing the CMViep, with or without other eukaryotic elements were found in the pellet fraction, suggesting that it is the CMV promoter that mediates the microtubule-plasmid interaction.
Figure 1. Microtubule spin-down assays showing in vitro interaction of DNA with microtubules.

(a) Quantitative analysis of plasmid elements that associate with microtubules. Plasmids containing different sequence elements (the CMV promoter, the luciferase gene, and/or the DTS) were incubated for 30 minutes with cell extract and taxol-stabilized microtubules and subsequently separated over a glycerol cushion by centrifugation. The plasmid contents of the pellets and supernatants were determined by quantitative PCR, and percentage of DNA in the pellet was determined by comparing DNA content in pelleted fractions versus total DNA in both supernatant and pellet fractions combined. (b) Increased incubation times do not change the ability of the DTS to mediate microtubule interactions. pBR322-DTS was incubated with microtubules in the presence of cell extract for 30, 45, 60, and 75 minutes and subsequently centrifuged and quantified by quantitative PCR as in a. Mean DNA concentrations from three independent experiments, preformed in duplicate, are shown ± st. dev. CMV, Cytomegalovirus; Lux, luciferase gene; DTS, Simian Virus 40 DNA nuclear targeting sequence.
Due to the large number of transcription factors that bind the SV40 DTS11–13, it was a somewhat surprising observation that the DTS did not also facilitate microtubule interaction. One possibility is that the incubation times were not sufficient to capture all interactions between the plasmid and microtubules. To address this, the plasmid containing only the DTS was incubated with cell extract and microtubules for increasing amounts of time. However, even after 75 minutes in the presence of an ATP regenerating system, the DTS-containing plasmid did not associate with microtubules (Fig. 1b).
CREB-containing promoters mediate microtubule-plasmid interactions
To determine whether other eukaryotic promoters were able to mediate binding of plasmids to microtubules in this assay, a number of different RNA polymerase I and polymerase II promoters were tested for binding (Fig. 2a). While several strong viral, general, and cell-specific promoters failed to interact more than did the backbone pBR322 alone, both the CMViep and Cauliflower Mosaic virus (CaMV) 35S promoter-containing plasmids were able to bind microtubules and pellet following centrifugation. All plasmids varied in size, but there was no correlation between the size of the plasmid and the ability to pellet with the microtubules. These results suggest that there may be a common sequence or sequences shared by these promoters that the others lack that is responsible for microtubule interaction in this assay. Using the TF Search (ver1.3) (http://www.cbrc.jp/reseach/db/TFSEARCH.html) program, the transcription factor consensus binding sites within these promoters were determined using a threshold score of 85.0. The most obvious candidate was CREB: the CMV promoter has 22 binding sites and the CaMV 35S promoter has 9 binding sites for CREB, while none of the other promoters tested contained CREB binding sites.
Figure 2. Microtubule binding by plasmids containing various eukaryotic promoters.

(a) Not all promoters mediate microtubule-DNA interactions. The following promoters were tested in the microtubule spin-down assay as in Figure 1: pBR322 (backbone plasmid, no promoter), CMViep, the Rous sarcoma virus LTR (RSV), the endothelian I promoter (endo), the VEGF receptor promoter (FLK-1), the alpha integrin promoter (αint), the human ubiquitin C promoter (UbC), the 45S RNA polymerase I promoter (Pol I), the human collagen A promoter (Col), and the 35S promoter of Cauliflower Mosaic virus (CaMV). (b) A single CREB-binding site is sufficient to mediate microtubule-plasmid interactions. Plasmids (pBR322; CREB, a plasmid with just one CREB binding site; or CMV, with just the CMViep) were used in the spin-down assay as in Figure 1a. Mean DNA concentrations from three independent experiments, preformed in duplicate, are shown ± st. dev. *, p < 0.0001.
To test whether CREB binding sites were responsible for these interactions, a single CREB binding site was introduced into pBR322 and was used in the spin-down assay. A single CREB-binding site was sufficient to mediate a plasmid-microtubule interaction (Fig. 2b). Although the interaction is strong, having a single CREB site did not appear to provide interactions as robust as those mediated by the full length CMV or CaMV promoters, suggesting that the presence of multiple CREB binding sites within these two promoters provides for even better binding. Alternatively, other transcription factors that bind to the full-length promoters may provide an additive or a synergistic effect to increase microtubule-plasmid interactions.
The CMV promoter binds CREB during gene transfer
Although inclusion of CREB-binding sites in the plasmid allowed enhanced interactions with microtubules in the cell-free system, we wanted to know if these plasmid constructs bind CREB in cells. To examine this, several biotinylated plasmid constructs were electroporated into cells. The constructs included plasmids containing or lacking CREB binding sites in the regulatory sequences (e.g., CMViep or SV40 enhancer, respectively) as well as plasmids carrying either elements or neither. At certain times post-electroporation (15, 30, 60, and 120 minutes), DNA-protein complexes were cross-linked, cells were lysed, and the biotin-DNA-protein complexes were pulled down using streptavidin-coated beads and analyzed by Western blots using antibodies against CREB. As seen in Figure 3, only those plasmids containing CREB binding sites (i.e., with the CMViep) are able to bind this transcription factor at any of the time-points. Additionally, CREB bound the plasmids at early times after electroporation, before significant nuclear localization of plasmids occurs10. This suggests that this binding occurs during cytoplasmic trafficking, before nuclear entry of DNA.
Figure 3. Binding of CREB by plasmids containing the CMViep during gene transfer.

Biotinylated plasmids were electroporated into A549 cells, and at the indicated times post transfection, formaldehyde was added to cross-link the DNA-protein complexes, cells were lysed, complexes were pulled down with streptavidin-coated beads, cross-links were reversed by boiling beads with Laemmli Sample buffer, and the resulting lysates were run in Western blots using antibodies against CREB. “Lysate” represents crude lysates for each sample, with no bead precipitation. The “No DNA” lane contains pulled down lysates from untransfected cells, showing only background CREB detection. Experiments were performed in duplicate and repeated four times.
Plasmids containing the SV40-DTS or CMV promoter traffic in the cytoplasm
The apparent CREB dependency seems peculiar since plasmids containing only the SV40 DTS (and no CREB binding sites) have been shown to allow efficient nuclear import in many cell types8,9,14, but did not interact with microtubules in the microtubule-binding assay (Fig. 1a, b). Since plasmids require microtubules for movement to the nucleus, these in vitro and in vivo results appear at odds and must be reconciled. Therefore, real-time plasmid tracking was carried out in live cells using different plasmid constructs. Fluorescently labeled plasmids were microinjected into the cytoplasm of A549 cells and cytoplasmic trafficking was monitored via fluorescence microscopy and time-lapse imaging (Fig 4). The same plasmids as in Figure 3 that contain or lack CREB binding sites were used in these experiments. When pBR322 was injected and followed for up to 1 hour after microinjection, most particles failed to show any significant bulk movement and remained largely at the site of injection. Figure 4a shows representative trajectories of 3 such particles. By contrast, plasmids carrying the CMViep, the SV40 enhancer, or both sequences showed significant directed movement (Fig. 4a). When their frequency distribution histograms are plotted, those plasmids containing the CMViep, SV40 DTS, or both show most particles moving at average (net) velocities of around 0.05–0.14 µm/second with individual plasmids moving at rates of up to 0.38 µm/second (Fig. 4b). By contrast, the majority of pBR322 plasmids display rates of less than 0.05 µm/second, which is likely diffusive movement15. Additionally, when the net velocities of all tracked plasmids were averaged, the data shows that inclusion of the CMV promoter mediates enhanced plasmid velocity compared to those plasmids containing only the SV40 DTS (Fig. 4c). This enhanced cytoplasmic trafficking may allow for greater movement to the nucleus over a shorter time. This suggests that movement along microtubules is greater for plasmids that contain the CMV promoter and bind CREB during trafficking, and this may explain why only the CREB-binding plasmids pelleted with microtubules in the spin-down assay. To confirm that the plasmids are moving on microtubules, cells were microinjected with labeled plasmids and twenty minutes later, the cells were fixed and immunofluorescence was used to visualize tubulin (Fig. 4d). Three-dimensional reconstructions of cells imaged by deconvolution microscopy showed that greater than 78% of the intracellular QD-labeled plasmids colocalized with microtubules with a mean Manders overlap coefficient of 0.85±0.07.
Figure 4. Requirement of promoter or enhancer sequences for plasmid movement in the cytoplasm.

(a) Representative traces for individual plasmid trajectories. Quantum dot-labeled plasmids were cytoplasmically microinjected into adherent A549 cells and imaged at 1-second intervals over 5–10 minutes. The traces of representative negative control (pBR322) and positive control (pCMV-DTS) plasmids in injected cells are shown. Plasmid trajectories were created using the PolyParticleTracker software downloaded for use in MATLAB, and all plot areas are mapped to 25 × 25 pixel areas. (b) Average cytoplasmic velocity of individual microinjected plasmids. Movement of individual DNA particles, shown in a, were tracked for up to 10 minutes using time-lapse imaging (1 frame/second). The average velocity of each was determined using particle tracking software (PolyParticleTracker, MATLAB), and the frequency distribution histograms are plotted as the number of plasmids moving at certain velocities for each construct. At least 50 particles were tracked per construct in 3–5 separate experiments. (c) Individual microinjected plasmid velocities were averaged for each of the four constructs. Error bars represent means + st. dev. ‡, p<0.05 compared to pBR-DTS; *, p<0.001 compared to pBR322. (d) Colocalization of Quantum dot-labeled plasmids with microtubules. Cells were microinjected with biotin-PNA-pCMV-DTS plasmids, fixed 20 minutes post-injection, and labeled with anti-tubulin antibodies and streptavidin-conjugated Quantum dots to label plasmids. Cells were imaged using a 100x objective by deconvolution microscopy and a representative deconvolved Z slice is shown. Scale bar, 10 µm.
CREB binding is required for the enhanced rate of movement
The particle tracking experiments show that the CMViep permits enhanced cytoplasmic trafficking over the SV40 DTS enhancer sequence alone following microinjection (Figs. 4b, c). Additionally, DNA-microtubule interactions are strongest for plasmids that contain the CMV promoter (i.e., bind CREB) as determined with the spin-down assay (Figs. 1 and 2). Therefore, net plasmid velocities were examined when CREB was knocked down in microinjected cells. Using siRNA against CREB, between 66% and 85% knockdown was achieved by 48 hours post transfection (Fig. 5a). At this time, cells were cytoplasmically microinjected with quantum dot-labeled DNA and real-time particle tracking was carried out, as in Figure 4. The frequency distribution histograms for the four plasmid constructs in the negative control, scramble RNA-transfected cells were very similar to those seen in the untransfected cells (Fig. 4b), indicating that liposome-mediated transfection did not affect the cytoplasmic trafficking of plasmids (Fig. 5b). However, when CREB is knocked down, CMViep-containing plasmids have histograms that are shifted towards slightly slower velocities, whereas constructs lacking CREB binding sites (pBR322 and pBR-DTS) have histograms that are relatively unchanged (Fig. 5c). When all plasmid net velocities are plotted together in a scatter plot, the pBR-CMViep plasmids show an overall shift in velocity toward slower speeds for individual particles when CREB has been knocked down versus when scrambled siRNA was used (Fig. 5d). This shift produced a significantly lower median velocity for pBR-CMViep plasmids in the knockdown condition compared to the control (p<0.001). Interestingly, the pCMV-DTS plasmids did not show any difference in mean velocity when CREB was knocked down. This is likely due to the presence of multiple binding sites in the SV40 DTS which bind to transcription factors other than CREB to mediate microtubule binding and movement. These results indicate that CREB binding to plasmids facilitates the faster movement seen with CMViep-containing plasmids.
Figure 5. Enhanced plasmid intracellular movement is prevented with siRNA against CREB.

(a) CREB knockdown via RNA interference. Western blot images show CREB protein levels are reduced around 75% in cell lysates after 48 hours incubation with siRNA against CREB (Ambion, Austin, TX) compared to cells transfected with a negative control scramble RNA. (b) Average cytoplasmic velocity of individual microinjected plasmids in scrambled RNA-transfected cells. Movement of individual DNA particles were tracked for 10 minutes and velocities analyzed as in Figure 4. (c) Average cytoplasmic velocity of individual microinjected plasmids in CREB siRNA-transfected cells. Plasmids were imaged and velocities recorded as in b. (d) Scatter plot showing the range of the average velocities for each of the four constructs. Bars represent median velocity for each construct. *, p<0.001. KD, CREB knockdown cells; SC, scrambled control.
Plasmids displaying greater cytoplasmic velocity also display greater levels of nuclear localization
The particle tracking experiments show that CMV promoter-containing plasmids have faster net velocities than those plasmids lacking CREB binding sites (Fig. 4c). Therefore, one would expect faster nuclear accumulation of nuclear import-competent plasmids containing these sequences. To address this, cells were microinjected with fluorescently-labeled plasmids that contain no eukaryotic sequences (pBR322) as a control, or the SV40 DTS alone (pBR-DTS) or both CMViep and DTS sequences (pCMV-DTS). Cells were subsequently scored for the accumulation of nuclear plasmids at the indicated times. Starting at 60 minutes post-injection, the CMViep-containing plasmid localizes to the nuclei of a greater number of cells than does the plasmid carrying the SV40 DTS alone (Fig. 6). While not demonstrating a direct relationship, these results are consistent with a model in which increased directed movement of plasmids through the cytoplasm may lead to more rapid arrival at the nuclear envelope and greater subsequent levels of nuclear accumulation.
Figure 6. CREB-binding plasmids with a DTS sequence have enhanced nuclear localization at earlier time points than plasmids containing the DTS alone.

Negative control plasmid pBR322, pBR-DTS, or pCMV-DTS plasmids labeled with CY3-PNA were injected into A549 cells then incubated for the indicated times. Injected cells were imaged and scored for nuclear import. Experiments were performed in triplicate and at least 50–100 cells were scored per time point. *, p<0.01 compared to pCMV-DTS and pBR-DTS for each time point. Error bars represent means + st. dev.
Discussion
The mechanisms by which plasmid DNA crosses the cytoplasm to the nucleus for successful gene expression are still unknown. However, it is known that motor proteins and an intact microtubule network are required1,2. Since non-viral gene therapy and transfections are relatively inefficient, these fundamental cell processes must be elucidated if we are to improve gene delivery and achieve more DNA in the nucleus. In this study we used a cell-free microtubule-binding assay, plasmid microinjections, and real-time particle tracking to examine whether plasmid interaction with microtubules and intracellular trafficking are sequence-specific events. We found that this is the case, and specifically, transcription factor binding sites in the plasmid play an important role in cytoplasmic movement. However, not all transcription factor binding may result in the same efficiency of movement; binding of some factors may allow greater or lesser cytoplasmic trafficking. Moreover, by enhancing plasmid movement through the cytoplasm, they may be more likely to reach the nucleus faster for subsequent import across the nuclear envelope, which could result in decreased cytoplasmic degradation of plasmids and ultimately greater gene transfer efficiency.
The in vitro microtubule spin-down assay suggested that only those plasmids containing CREB binding sites are able to interact with microtubules (Fig. 2). However, this is a cell-free system and may only show the strongest interactions. We know from previous work that plasmids containing the SV40 DTS, which does not bind CREB, are able to traverse the cytoplasm to the nucleus8,9, and this requires interaction with microtubules1,2. Therefore, there is a discrepancy between what we have seen in vivo and in vitro. Several possibilities could account for this: first, the in vitro assay may be highly stringent and only detect the strongest interactions; second, conditions of the assay were such that not all interactions were allowed to form (e.g., not enough time for binding, or non-optimized buffers and protein concentrations); or third, the in vitro assay does not reflect what actually happens in cells. All of our results, including the presence of CREB in precipitated plasmid complexes during transfection and its ability to enhance, but not be required for, plasmid trafficking in cells, suggests that the first possibility is most likely.
In the DNA precipitation experiments, the transcription factor CREB bound to plasmids containing the CMV promoter during early time points and likely links the plasmids to microtubules (Fig. 3). Because DNA is located in the cell nucleus, and it is where transcription factors function, it is not obvious to think about transcription factor binding to DNA as something that occurs in the cytoplasm. However, like all proteins, transcription factors are translated in the cytoplasm and are regulated via cytoplasmic sequestration until activation, upon which the protein translocates to the nucleus16. Although CREB is activated by phosphorylation and acts in concert with p300 and CBP in the nucleus, even bacterially produced CREB which is unphosphorylated (similar in this respect to classic cytoplasmic CREB) is able to bind to its consensus sequence on DNA17,18, suggesting that cytoplasmic CREB may indeed be capable of binding to DNA. Our experiments show that this is the case. This is, to our knowledge, the first demonstration of transcription factor binding to plasmids during transfection.
It has been shown that plasmids begin to localize to the nucleus within 30 to 45 minutes following cytoplasmic microinjection10. The fact that CREB can be pulled down as part of the plasmid complex within 15 minutes of electroporation-mediated introduction into the cell suggests that this factor binds to the DNA while it is still in the cytoplasm, before nuclear entry. While a number of other reports have shown either that inclusion of transcription factor binding sites to plasmids can increase nuclear localization1,6,19–23 or that knockdown of specific transcription factors or use of dominant negatives can inhibit nuclear localization of the plasmids1,20, none have shown that the factors do indeed bind to the DNA in cells during the transfection process. Further, that knockdown of CREB reduces cytoplasmic movement of the plasmids suggests it binds and plays a functional role in cytoplasmic trafficking. However, the fact that plasmids containing only the SV40 DTS, which lacks CREB binding sites but has binding sites for a number of other transcription factors, are able to effectively move following microinjection suggests that CREB is not solely responsible for this plasmid movement. Indeed, there are a number of other cytoplasmic proteins that likely mediate intracellular movement as components of the “trafficking complex,” such as importin-β, RAN, transportin, dynein, and transcription factors other than CREB (Fig. 7)6,7,22. What roles each of these play in cytoplasmic trafficking remains to be seen.
Figure 7. Possible protein interactions in the plasmid trafficking complex.
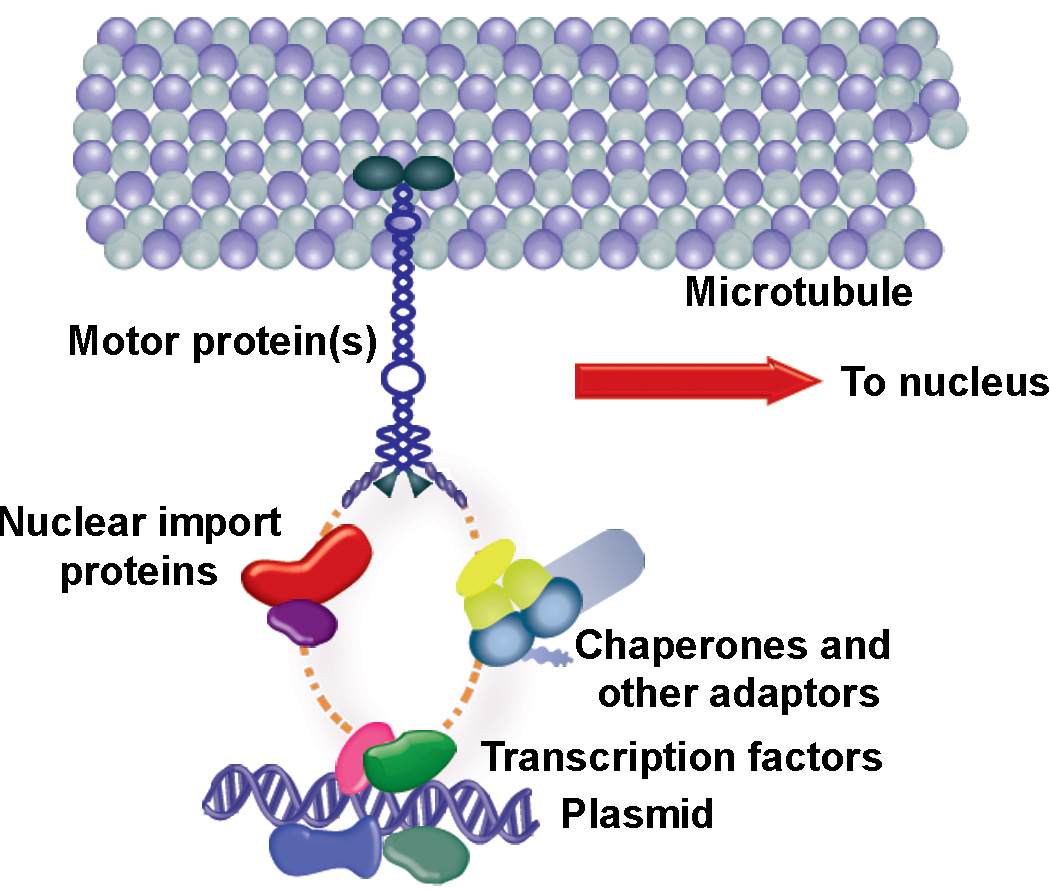
Our model of plasmid interaction with microtubules is not a direct one, and involves transcription factor binding to unique sequences on the plasmid (i.e., CREB binding to CMViep). One or more of the transcription factor NLSs are bound by importins, which are bound by the motor protein dynein and can move along microtubules toward the nucleus for import. Other adaptor molecules, such as chaperones, MAPs (microtubule-associated proteins), and nuclear import proteins may be part of this larger complex as well. TF, transcription factor; NLS, nuclear localization signal; MT, microtubule.
It is interesting in the CREB pull down experiments that the plasmid carrying only the CMViep forms CREB-DNA complexes that can be pulled down at relatively equal abundance at all times up to 2 hours after transfection. By contrast, the plasmid carrying both the CMViep and the SV40 DTS forms complexes within 15 minutes of transfection but by 60 min, very little CREB is pulled down with the plasmid and by 2 hours, no CREB remains complexed with the DNA. Since the latter plasmid carries the SV40 DTS and can be transported into the nucleus by 60 min, while pBR-CMV does not support nuclear import9, it is possible that different plasmid-protein complexes exist in the cytoplasm and the nucleus or that the complexes are dynamic. This is likely, since it has been shown that transcription factor localization and interactions with DNA in the nucleus can be transient or form highly stable protein aggregates24,25. Perhaps plasmid-bound transcription factors such a CREB redistribute to areas of higher affinity so that they move off the plasmids and onto genomic DNA. Alternatively, the nuclei of transfected cells may not have been effectively lysed, leaving the nuclear fractions of CREB in the membranous pellet following centrifugation of lysates. This would explain why plasmids containing the SV40 DTS, which localizes to the nucleus in about an hour, do not show CREB binding at the later time points.
Inclusion of the CMViep, which was shown to bind CREB as early as 15 minutes post-transfection, enabled plasmids to move faster compared to plasmids that lack the sequence. The SV40 DTS allowed significantly increased cytoplasmic velocity compared to the pBR322 plasmid, showing that this sequence alone is sufficient for plasmid movement during gene transfer (Fig. 4). However, CREB-binding plasmids have greater trafficking, which is highly favorable in gene transfer where the plasmid DNA needs to rapidly reach the nucleus. In fact, if unable to successfully move through the cytoplasm, much of the internalized DNA may be degraded by nucleases present in the cytoplasm within several hours26,27. When plasmids either containing or lacking CREB binding sites were compared for their ability to accumulate in nuclei over longer time intervals, there was a higher percentage of cells with nuclear plasmid from 1 to 4 hours post-injection when the plasmid carried the CMViep and SV40 DTS compared to the DTS sequence alone (Fig. 6). This observation is important, since the plasmid tracking experiments only analyze movement over short periods of time (5–10 minutes), and may not reflect bulk plasmid movement that is directed towards the nucleus over longer time intervals.
The difference in average DNA velocity between those plasmids containing the CMV promoter and lacking it may be attributable to the affinity of the bound transcription factors for microtubule motors. For example, if CREB binds more efficiently or tightly to microtubule motors than do other transcription factors, this would allow the bound cargo (i.e., plasmids) to also have enhanced trafficking. Indeed, it has been shown that many cargoes including virus particles and organelles bind multiple motor proteins and that the number of bound motors may be proportional to the rates of movement and processivity28. Further, the multiple CREB binding sites within the CMViep could enable more motors to bind to the plasmid, thereby accounting for the enhanced movement observed. Alternatively, CREB may interact with higher-order regulatory factors that regulate directed microtubule-mediated trafficking, such as dynactin or microtubule-associated proteins29,30.
The rates of movement for the plasmid constructs containing promoter or enhancer sequences (0.05 to 0.4 µm/sec) are consistent with those measured for various components in the cytoplasm that also move on microtubule-associated motor proteins. For example, the motors kinesin and dynein are reported to move at similar average rates of around 0.5 to 1.5 µm/second29–31. Organelles such as peroxisomes are moved via motors around the cytoplasm at rates of 0.1–0.3 µm/second32, but the rates vary depending on cell types. Viral particles such as Adenovirus and Herpes Simplex Virus have been recorded moving at rates of about 0.5 µm/second in cells33–35, which is similar to vesicular movement in neurites36, suggesting that endocytosed or lipid encapsulated particles may move at similar rates. In one study of liposome-mediated transfection, it was observed that microtubule-associated lipoplexes moved with an average velocity of just 0.02 µm/sec, a rate much slower than that seen for motor protein-mediated movement, suggesting that this movement may have been more restricted and diffusive in nature37.
In this study we have shown that plasmid-microtubule interactions are sequence-specific, much like plasmid DNA nuclear import8. While specific binding of transcription factors such as DNA-microtubule interactions CREB may enhance this movement, binding of a number of other transcription factors, including NF-kB, are likely needed for movement. Moreover, these studies show that increasing the rates of movement through the cytoplasm results in greater accumulation of import-competent plasmids in the nucleus, and therefore greater gene expression. By determining the various protein adaptors required for DNA cytoplasmic trafficking and nuclear import, we can further explore ways to modulate and enhance transfection and gene therapy.
Materials and Methods
Plasmids, PNA and Quantum dot labeling
Plasmids pBR322, pBR-RSV, pBR-CMV, and pBR-DTS are based on the pBR322 backbone and carry no eukaryotic sequences, the Rouse Sarcoma Virus Long Terminal Repeat promoter, the CMV immediate early promoter/enhancer, and the SV40 DTS, respectively9. Plasmids pCMV-Lux-DTS and pCMV-Lux express the luciferase gene from the CMV immediate early promoter/enhancer and contain or lack the SV40 DTS9. Plasmid pTA-flk1, and pTA-endothelin are TA cloning vectors (Invitrogen, CA) that carry the flk-1 promoter (−285 to +268) or the endothelin promoter (−204 to +4)38. The 35S promoter from Cauliflower Mosaic Virus is carried on the plasmid pUC8-35S, which was a generous gift from Cathy Radenbaugh (Colorado Sate University)39. The human alpha-integrin promoter40 (1,100 bp, −505 to +455) was PCR amplified from human genomic DNA and cloned into the TA cloning vector (Invitrogen, Carlsbad, CA). The human type I alpha 2 procollagen (hCol1a2) promoter (−267 to +45) was PCR amplified from human genomic DNA and was subcloned into the pGL3 vector from Promega (Madison, WI). The plasmid pHr-BES contains the human polymerase I (pol I) promoter and the first 700 base pairs of the transcribed 45S pre-ribosomal RNA (rRNA) sequence inserted into a pBR322 backbone41. Plasmid pUB6/V5-His/LacZ carries the Ubiquitin C promoter (Invitrogen, Carlsbad, CA). The CREB plasmid was created using the TOPO cloning kit (Invitrogen, CA) by inserting the CREB binding site (5’-TGACGTCAAGATCTA -3’), using the annealed primers (5’-TGACGTCAAGATCTA-3’) and (3’AACTGCAGTTCTAGA-5’), into the multiple cloning site. All plasmids were purified from Escherichia coli using Qiagen Gigaprep kits as described by the manufacturer (Qiagen, Chatsworth, CA). Plasmids used in the CREB pull-down and microinjection experiments contain the GeneGrip1 PNA binding site (Gene Therapy Systems, San Diego, CA). Briefly, plasmids were labeled with biotinylated PNA in Tris EDTA buffer at 37°C for 2 hours, followed by isopropanol precipitation to remove unbound biotin-PNA. Microinjected plasmids were labeled with fluorescent quantum dots (Qdot 605, Invitrogen, Carlsbad, CA) by incubating the streptavidin-conjugated nanocrystals with biotin-labeled plasmids (0.5 mg/ml) at 37°C for 1 hour followed by washing in 0.5x phosphate-buffered saline three times, then filtering through Microsep 1000K filters (Pall Life Sciences, Ann Arbor, MI) to remove unbound quantum dots.
Cell culture, electroporation, and siRNA transfections
Human adenocarcinoma A549 cells (#CCL-185; American Type Culture Collection, Rockville, MD) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1x antimycotic/antibiotic solution (Invitrogen, Carlsbad, CA).
For electroporations, cells were grown to confluency in 6-well dishes, and rinsed twice with 1x phosphate-buffered saline. Ten micrograms of plasmid in 750 µl of phosphate buffered saline were added to each well, and one 165 mV square wave electric pulse was applied for 35 miliseconds using a PetriPulser electrode (BTX, San Diego, CA).
For siRNA transfections, cells were grown in 6-well dishes and washed twice with phosphate-buffered saline. Each well received 50 nM siRNA against CREB (CAUUAGCCCAGGUAUCUAUtt, ID#s4389, lot ASO0JJZ9) or scramble control (Negative control #1, lot ASO0J3TO) (Ambion Inc, Austin, TX) in Transfection Medium (Santa Cruz, Santa Cruz, CA), and cells were transfected according to the manufacturer’s instructions. Two days after transfection, cells were first microinjected for analysis of DNA trafficking, then lysed in Promega lysis buffer (Promega, Madison, WI). Lysates were boiled for 5 minutes in Laemmli sample buffer, separated by SDS-PAGE, and proteins were transferred to nitrocellulose membranes and anti-CREB antibodies (1:200, ab31387 Abcam, Cambridge, MA) were used to probe the membrane. Chemiluminescence detection was used to determine CREB knockdown with siRNA. CREB knockdown was determined relative to actin levels by stripping the nitrocellulose membrane and re-probing with anti-actin antibodies (1:1000, #C5838, Sigma Aldrich, St. Louis, MO).
Microinjections and particle tracking
For microinjections, A549 cells were grown on coverslips in Mattek dishes (Ashland, MA), and Hoechst 33242 nuclear stain (Invitrogen, Carlsbad, CA) was added to the medium prior to injections. For siRNA experiments, microinjections were done 2 days post-transfection. Cells were cytoplasmically microinjected with Quantum dot-labeled plasmid constructs (0.5 mg/ml) using an Eppendorf Femtojet system as previously described42. Immediately after injections, fluorescent plasmids were imaged in the cytoplasm using a Leica DMI 6000 B inverted microscope with a 100x objective (N.A. 1.47) and a Hamamatsu EM- CCD camera (Hamamatsu, Japan). Series of images were collected at 1 frame per second for 5 to 10 minutes using Volocity software (Improvision, Waltham, PA). This was repeated for different fields of injected cells for up to 1 hour post-injection. The collected series of images were converted to movie files using ImageJ, and a modified algorithm written in MATLAB was used to track DNA particles and create representative particle trajectories43. Briefly, after choosing the region of interest in the images with fluorescent plasmids, particles were fitted to a fourth order polynomial weighted by a two dimensional Gaussian distribution. The center of the particle was used as the maximum point in the fit. The average velocity of individual tracked particles was determined for 35 to 50 plasmids per condition, using a minimum of 20 frames in focus as a cutoff.
For the nuclear localization experiments, cells were microinjected with CY3-PNA labeled plasmids (pBR322, pBR-DTS, and pCMV-DTS at 0.5 mg/ml) and imaged as described above. Culture dishes were placed in a 37°C, 5% CO2 incubator for 30, 60, 90, 120, or 240 minutes and then cells were viewed and imaged to determine the number of injected cells that had plasmids in the nucleus at each time point. Typically, 100 cells were injected for each plasmid and time point and the experiment was repeated a minimum of three times.
Immunofluorescence
A549 cells were grown on coverslips and microinjected as above with PNA-biotin labeled plasmids (pCMV-DTS) and then incubated for 20 minutes at 37°C. Cells were fixed for 10 minutes in 100% ice-cold methanol, coverslips were washed with 1x phosphate-buffered saline, and immunofluorescence was carried out using primary antibodies against β-tubulin (1:1000, Sigma-Aldrich Corp, St. Louis, MO) and secondary Alexa 488 antibodies (1:200, Invitrogen, Carlsbad, CA) to stain microtubules. Injected biotinylated plasmids were labeled with fluorescent Quantum Dots post-cell-fixation (30 nM Qdot 605, Invitrogen, Carlsbad, CA) by incubating the coverslips with streptavidin-conjugated nanocrystals for 1 hour at room temperature followed by washing with 1x phosphate-buffered saline. The coverslips were then mounted onto glass slides using Qmount Qdot mounting medium (Invitrogen, Carlsbad, CA). Multiple z-series of cells were acquired using a Leica DMI 6000 B inverted microscope with a 100x objective (N.A. 1.47) and a Hamamatsu EM- CCD camera (Hamamatsu, Japan). Volocity software iterative restoration deconvolution (PerkinElmer, Waltham, MA) was used on the resulting images and colocalization quantified on the 3 dimensional reconstructions by determining the mean Manders overlap coefficient of 13 different cells with over 100 particles.
Microtubule spin-down assay
Purified bovine brain tubulin, 5 mg/ml (Cytoskeleton Inc., Denver, CO) was placed on ice and 2.5 µl of PEM buffer (80 mM PIPES pH 7.0, 1 mM MgCl2, 1 mM EGTA, and 50% glycerol) was added. The tubulin was allowed to polymerize for 20 min at 35°C and then stabilized by the addition of 20 µM taxol (Cytoskeleton, Inc., Denver, CO). Tubulin (10 µg) was incubated with DNA (20 ng) and/or cell extract (24 µg) in PEM buffer containing 20 µM taxol for 30 minutes, placed over cushion buffer (PEM with 50% volume/volume glycerol and 20 µM taxol) and centrifuged at 100,000 × g for 40 min in an Airfuge (Beckman Instruments Inc., Palo Alto, CA). The pellet was re-suspended in cushion buffer for PCR. Cell extract was prepared as described44.
Real-time quantitative PCR
Quantitative, real-time PCR was performed in a 20 µl reaction volume, using the DyNAmoTM SYBRR Green qPCR Kit as described by the manufacturer (Finnzymes, Espoo, Finland). Reactions were carried out and quantified with the MJ Research Opticon 2. The supernatants and pellets from the microtubule spin-down assays were diluted 1:1 in water and 4 µl were used for the reactions. All samples were run in duplicate. The primers amplified a 116 base-pair region of the beta-lactamase gene present in the plasmids. Standard curves were generated using seven 10-fold dilutions of pCMV-Lux-DTS, and the threshold was set manually by determining the best-fit line for the quantitation of the standards. A melting curve analysis was performed to ensure reaction specificity. The amount of plasmid present in supernatant and pellet of each sample was determined based on the standard curve. The percentage of DNA present in the pellet was determined by quantifying the amount of DNA in the pellet compared to the total DNA present in the combined supernatant and pellet fractions of each sample. All experiments were preformed at least three times.
Live cell plasmid pull-downs
Biotinylated plasmid DNA constructs (pBR322, pBR-CMV, pBR-DTS, and pCMV-DTS) were electroporated into adherent cells, and at desired times post-electroporation, plasmid DNA-protein complexes were cross-linked with formaldehyde, cells were lysed, and the resulting biotin-plasmid-protein complexes were pulled down as previously described (Miller, AM et al. 2009). Resulting lysate proteins were run in SDS-PAGE and transferred to nitrocellulose membranes. Crude lysates were run as a positive control on each gel. The membranes were probed with anti-CREB antibodies (1:200, Abcam, Cambridge, MA), and chemiluminescence detection was used to determine levels of bound CREB. All experiments were performed in duplicate wells and repeated at least four times.
Acknowledgments
We would like to thank doctors Aaron Miller, Rui Zhou, and Josh Gasiorowski, along with Mootaz Eldib for insightful discussions and technical advice. This work was supported in part by predoctoral fellowships from the Founders (MAB) and the Midwest (EEV) Affiliates of the American Heart Association, and grants EB9903, HL71643, ES07026, and center grant ES01247 from the National Institutes of Health.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Mesika A, Kiss V, Brumfeld V, Ghosh G, Reich Z. Enhanced intracellular mobility and nuclear accumulation of DNA plasmids associated with a karyophilic protein. Hum Gene Ther. 2005;16:200–208. doi: 10.1089/hum.2005.16.200. [DOI] [PubMed] [Google Scholar]
- 2.Vaughan EE, Dean DA. Intracellular trafficking of plasmids during transfection is mediated by microtubules. Mol Ther. 2006;13:422–428. doi: 10.1016/j.ymthe.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leopold PL, Kreitzer G, Miyazawa N, Rempel S, Pfister KK, Rodriguez-Boulan E, et al. Dynein- and microtubule-mediated translocation of adenovirus serotype 5 occurs after endosomal lysis. Hum Gene Ther. 2000;11:151–165. doi: 10.1089/10430340050016238. [DOI] [PubMed] [Google Scholar]
- 4.Vaughan EE, DeGiulio JV, Dean DA. Intracellular trafficking of plasmids for gene therapy: mechanisms of cytoplasmic movement and nuclear import. Curr Gene Ther. 2006;6:671–681. doi: 10.2174/156652306779010688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson GL, Dean BS, Wang G, Dean DA. Nuclear import of plasmid DNA in digitonin-permeabilized cells requires both cytoplasmic factors and specific DNA sequences. J Biol Chem. 1999;274:22025–22032. doi: 10.1074/jbc.274.31.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller AM, Munkonge FM, Alton EW, Dean DA. Identification of Protein Cofactors Necessary for Sequence-specific Plasmid DNA Nuclear Import. Mol Ther. 2009 doi: 10.1038/mt.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lachish-Zalait A, Lau CK, Fichtman B, Zimmerman E, Harel A, Gaylord MR, et al. Transportin mediates nuclear entry of DNA in vertebrate systems. Traffic. 2009;10:1414–1428. doi: 10.1111/j.1600-0854.2009.00968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dean DA. Import of plasmid DNA into the nucleus is sequence specific. Exp Cell Res. 1997;230:293–302. doi: 10.1006/excr.1996.3427. [DOI] [PubMed] [Google Scholar]
- 9.Dean DA, Dean BS, Muller S, Smith LC. Sequence requirements for plasmid nuclear import. Exp Cell Res. 1999;253:713–722. doi: 10.1006/excr.1999.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughan EE, Geiger RC, Miller AM, Loh-Marley PL, Suzuki T, Miyata N, et al. Microtubule acetylation through HDAC6 inhibition results in increased transfection efficiency. Mol Ther. 2008;16:1841–1847. doi: 10.1038/mt.2008.190. [DOI] [PubMed] [Google Scholar]
- 11.Mercurio F, Karin M. Transcription factors AP-3 and AP-2 interact with the SV40 enhancer in a mutually exclusive manner. EMBO J. 1989;8:1455–1460. doi: 10.1002/j.1460-2075.1989.tb03528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner WJ, Woodworth ME. DNA replication efficiency depends on transcription factor-binding sites. J Virol. 2001;75:5638–5645. doi: 10.1128/JVI.75.12.5638-5645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark L, Pollock RM, Hay RT. Identification and purification of EBP1: a HeLa cell protein that binds to a region overlapping the 'core' of the SV40 enhancer. Genes Dev. 1988;2:991–1002. doi: 10.1101/gad.2.8.991. [DOI] [PubMed] [Google Scholar]
- 14.Kalderon D, Richardson WD, Markham AF, Smith AE. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311:33–38. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- 15.Suh J, Wirtz D, Hanes J. Efficient active transport of gene nanocarriers to the cell nucleus. Proc Natl Acad Sci U S A. 2003;100:3878–3882. doi: 10.1073/pnas.0636277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whiteside ST, Goodbourn S. Signal transduction and nuclear targeting: regulation of transcription factor activity by subcellular localisation. J Cell Sci. 1993;104(Pt 4):949–955. doi: 10.1242/jcs.104.4.949. [DOI] [PubMed] [Google Scholar]
- 17.Park EA, Roesler WJ, Liu J, Klemm DJ, Gurney AL, Thatcher JD, et al. The role of the CCAAT/enhancer-binding protein in the transcriptional regulation of the gene for phosphoenolpyruvate carboxykinase (GTP) Mol Cell Biol. 1990;10:6264–6272. doi: 10.1128/mcb.10.12.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muchardt C, Li C, Kornuc M, Gaynor R. CREB regulation of cellular cyclic AMP-responsive and adenovirus early promoters. J Virol. 1990;64:4296–4305. doi: 10.1128/jvi.64.9.4296-4305.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 20.Miller AM, Dean DA. Cell-specific nuclear import of plasmid DNA in smooth muscle requires tissue-specific transcription factors and DNA sequences. Gene Ther. 2008;15:1107–1115. doi: 10.1038/gt.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mesika A, Grigoreva I, Zohar M, Reich Z. A regulated, NFkappaB-assisted import of plasmid DNA into mammalian cell nuclei. Mol Ther. 2001;3:653–657. doi: 10.1006/mthe.2001.0312. [DOI] [PubMed] [Google Scholar]
- 22.Munkonge FM, Amin V, Hyde SC, Green AM, Pringle IA, Gill DR, et al. Identification and functional characterization of cytoplasmic determinants of plasmid DNA nuclear import. J Biol Chem. 2009;284:26978–26987. doi: 10.1074/jbc.M109.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breuzard G, Tertil M, Goncalves C, Cheradame H, Geguan P, Pichon C, et al. Nuclear delivery of NFkappaB-assisted DNA/polymer complexes: plasmid DNA quantitation by confocal laser scanning microscopy and evidence of nuclear polyplexes by FRET imaging. Nucleic Acids Res. 2008;36:e71. doi: 10.1093/nar/gkn287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hager GL, Elbi C, Becker M. Protein dynamics in the nuclear compartment. Curr Opin Genet Dev. 2002;12:137–141. doi: 10.1016/s0959-437x(02)00278-2. [DOI] [PubMed] [Google Scholar]
- 25.Misteli T. Protein dynamics: implications for nuclear architecture and gene expression. Science. 2001;291:843–847. doi: 10.1126/science.291.5505.843. [DOI] [PubMed] [Google Scholar]
- 26.Barry ME, Pinto-Gonzalez D, Orson FM, McKenzie GJ, Petry GR, Barry MA. Role of endogenous endonucleases and tissue site in transfection and CpG-mediated immune activation after naked DNA injection. Hum Gene Ther. 1999;10:2461–2480. doi: 10.1089/10430349950016816. [DOI] [PubMed] [Google Scholar]
- 27.Lechardeur D, Sohn KJ, Haardt M, Joshi PB, Monck M, Graham RW, et al. Metabolic instability of plasmid DNA in the cytosol: a potential barrier to gene transfer. Gene Ther. 1999;6:482–497. doi: 10.1038/sj.gt.3300867. [DOI] [PubMed] [Google Scholar]
- 28.Gazzola M, Burckhardt CJ, Bayati B, Engelke M, Greber UF, Koumoutsakos PA. stochastic model for microtubule motors describes the in vivo cytoplasmic transport of human adenovirus. PLoS Comput Biol. 2009;5:e1000623. doi: 10.1371/journal.pcbi.1000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King SJ, Schroer TA. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol. 2000;2:20–24. doi: 10.1038/71338. [DOI] [PubMed] [Google Scholar]
- 30.Paschal BM, Shpetner HS, Vallee RB. MAP 1C is a microtubule-activated ATPase which translocates microtubules in vitro and has dynein-like properties. J Cell Biol. 1987;105:1273–1282. doi: 10.1083/jcb.105.3.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science. 2005;308:1469–1472. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]
- 32.Rapp S, Saffrich R, Jakle U, Ansorge W, Gorgas K, Just WW. Microtubule-mediated peroxisomal saltations. Ann N Y Acad Sci. 1996;804:666–668. doi: 10.1111/j.1749-6632.1996.tb18659.x. [DOI] [PubMed] [Google Scholar]
- 33.Suomalainen M, Nakano MY, Boucke K, Keller S, Greber UF. Adenovirus-activated PKA and p38/MAPK pathways boost microtubule-mediated nuclear targeting of virus. EMBO J. 2001;20:1310–1319. doi: 10.1093/emboj/20.6.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee GE, Murray JW, Wolkoff AW, Wilson DW. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J Virol. 2006;80:4264–4275. doi: 10.1128/JVI.80.9.4264-4275.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suomalainen M, Nakano MY, Keller S, Boucke K, Stidwill RP, Greber UF. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J Cell Biol. 1999;144:657–672. doi: 10.1083/jcb.144.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lochner JE, Kingma M, Kuhn S, Meliza CD, Cutler B, Scalettar BA. Real-time imaging of the axonal transport of granules containing a tissue plasminogen activator/green fluorescent protein hybrid. Mol Biol Cell. 1998;9:2463–2476. doi: 10.1091/mbc.9.9.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ondrej V, Lukasova E, Falk M, Kozubek S. The role of actin and microtubule networks in plasmid DNA intracellular trafficking. Acta Biochim Pol. 2007;54:657–663. [PubMed] [Google Scholar]
- 38.Dean DA. In: Nucleocytoplasmic trafficking, in Pharmaceutical perspectives of nucleic acid-based therapeutics. Mahato RI, editor. London: Harwood Academic Publishers; 2002. pp. 229–260. [Google Scholar]
- 39.Guilley H, Dudley RK, Jonard G, Balazs E, Richards KE. Transcription of Cauliflower mosaic virus DNA: detection of promoter sequences, and characterization of transcripts. Cell. 1982;30:763–773. doi: 10.1016/0092-8674(82)90281-1. [DOI] [PubMed] [Google Scholar]
- 40.Obata H, Hayashi K, Nishida W, Momiyama T, Uchida A, Ochi T, et al. Smooth muscle cell phenotype-dependent transcriptional regulation of the alpha1 integrin gene. J Biol Chem. 1997;272:26643–26651. doi: 10.1074/jbc.272.42.26643. [DOI] [PubMed] [Google Scholar]
- 41.Gasiorowski JZ, Dean DA. Intranuclear Trafficking of Episomal DNA Is Transcription-dependent. Mol Ther. 2007 doi: 10.1038/sj.mt.6300275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gasiorowski JZ, Dean DA. Postmitotic nuclear retention of episomal plasmids is altered by DNA labeling and detection methods. Mol Ther. 2005;12:460–467. doi: 10.1016/j.ymthe.2005.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rogers SS, Waigh TA, Zhao X, Lu JR. Precise particle tracking against a complicated background: polynomial fitting with Gaussian weight. Phys Biol. 2007;4:220–227. doi: 10.1088/1478-3975/4/3/008. [DOI] [PubMed] [Google Scholar]
- 44.F M Ausubel B R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. New York: 1994. [Google Scholar]