

Abstract
Dihydroartemisinin-piperaquine is an effective drug in the treatment of Plasmodium falciparum and P. vivax malaria. The objective of this study was to evaluate the population pharmacokinetics and pharmacodynamics of piperaquine in patients with P. vivax malaria in Thailand after a standard regimen of dihydroartemisinin-piperaquine to determine whether residual piperaquine prevents or delays the emergence of P. vivax relapse. Sparse blood samples were collected from 116 patients. Piperaquine pharmacokinetics were described well by a three-compartment distribution model. Relapsing P. vivax malaria was accommodated by a constant baseline hazard (8.94 relapses/year) with the addition of a surge function in a fixed 3-week interval and a protective piperaquine effect. The results suggest that a large proportion of the first relapses were suppressed completely by residual piperaquine concentrations and that recurrences resulted mainly from emergence of the second or third relapse or from reinfection. This suggests a significant reduction in P. vivax morbidity when using dihydroartemisinin-piperaquine compared with other antimalarial drugs with shorter terminal postprophylactic effects.
Plasmodium vivax is the most difficult of the five species of human malaria parasites to eliminate. It is the most geographically widespread human malaria parasite, causing an estimated 130–390 million cases every year.1 P. vivax is transmitted within 95 countries in America, Africa, and Asia.2,3 In total, 2.85 billion people were estimated to be at risk of P. vivax transmission in 2009, especially in Central and Southeast Asia, where more than 90% of the world's P. vivax transmission occurs.3 Acute P. vivax malaria is rarely life threatening, but it causes substantial morbidity in higher transmission areas as a result of multiple relapses from latent liver-stage parasites.4 These repeated infections may result in life-threateningly severe anemia due to destruction of both infected and noninfected red blood cells. P. vivax is an important cause of low birth weight.4,5,6,7
Chloroquine, a blood schizonticide, is used in first-line treatment and prophylaxis of P. vivax malaria in most endemic areas. However, P. vivax resistance to chloroquine, which was first observed in Papua New Guinea in 1989,8 is increasing in many parts of the world.9 A combination of two drugs with different mechanisms of action is preferred for the treatment of malaria to reduce the risk of the development of resistant parasite strains. The fixed combination of dihydroartemisinin-piperaquine (DHA-PQ) is recommended by the World Health Organization as the first-line treatment of uncomplicated P. falciparum malaria10 and has replaced chloroquine in the first-line treatment of resistant P. vivax malaria in Indonesia. DHA-PQ is currently available as a fixed-dose combination, administered once daily over 3 days. A meta-analysis pooled from 12 different studies in 6 countries between 2003 and 2006 showed high efficacy against P. falciparum malaria (polymerase chain reaction-corrected cure rates of 98.7% at day 28) and good tolerance (4.8% total incidence of early vomiting) of DHA-PQ.11 DHA-PQ also has excellent efficacy against P. vivax malaria.12,13,14
Piperaquine is distributed extensively into tissues, resulting in multiphasic distribution and a slow elimination from the systemic circulation (elimination half-life (t1/2) of ~18–28 days). Similar pharmacokinetic properties have been reported in children but these pediatric studies suggest that small children need a higher weight-based dose to achieve exposure comparable to that in older children.15 Most piperaquine pharmacokinetic studies have sampled venous plasma. However, capillary sampling is a desirable alternative for field pharmacokinetic studies, especially in children. The population pharmacokinetics and pharmacodynamics of piperaquine have not been reported previously in patients with P. vivax malaria.
In South East Asia and Oceania, P. vivax relapses at 3-week intervals. Treatment with efficacious slowly eliminated antimalarials results in substantially delayed emergence of the relapse, but it has been unclear whether the first relapse is prevented (and so the second relapse is observed) or delayed. The aim of this study was to characterize the population pharmacokinetics and antimalarial pharmacodynamics of piperaquine in the treatment of P. vivax malaria and to use this information to determine whether piperaquine prevents or delays the first P. vivax relapse.
Results
Safety and efficacy
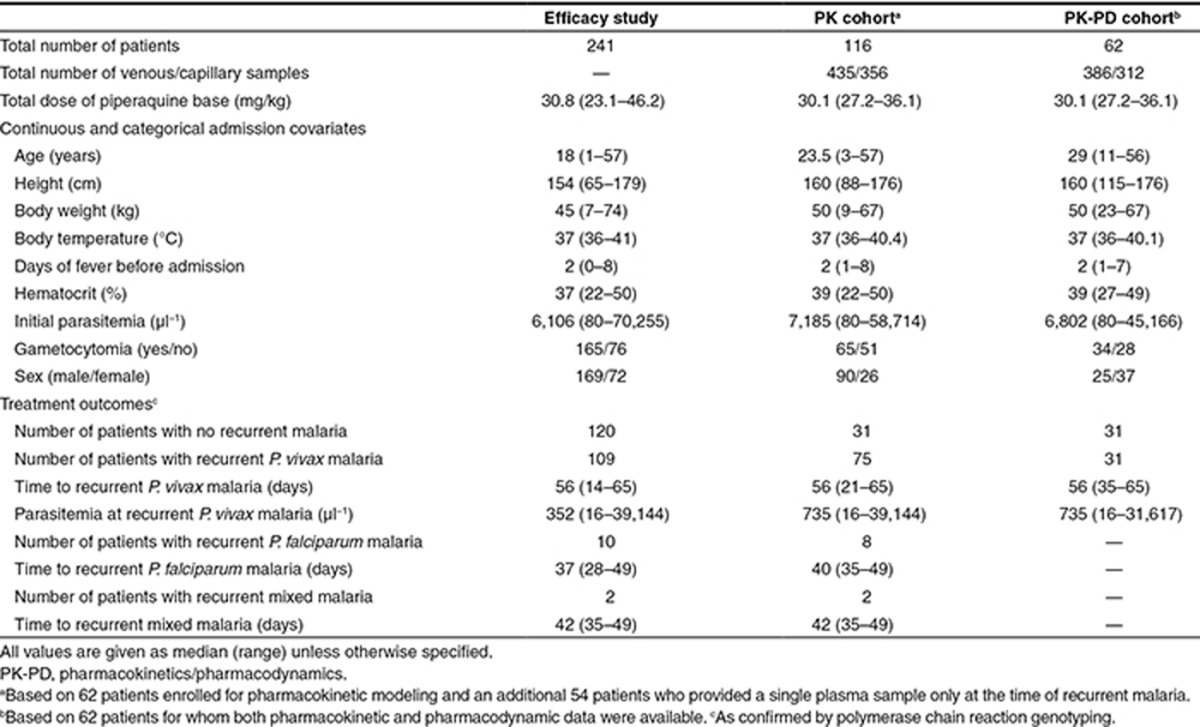
This study was conducted at Shoklo Malaria Research Unit clinics, Mae Sot, Thailand, an area of low and seasonal malaria transmission located at the Thailand–Myanmar border. This pharmacokinetic study was nested into a larger efficacy trial in 500 patients with P. vivax malaria infections. Full clinical details have been published elsewhere and DHA-PQ was found to be an effective alternative treatment with a lower cumulative risk of recurrent P. vivax malaria at 9 weeks compared with chloroquine (54.9 vs. 79.1%).16 Two-hundred and fifty Karen and Burmese patients with P. vivax malaria infections were enrolled in the DHA-PQ-treated arm of the efficacy study (Table 1). All patients received DHA-PQ (Duo-cotecxin, Beijing Holley-Cotec Pharmaceuticals, China) tablets containing 40 mg of dihydroartemisinin and 320 mg of piperaquine phosphate. A standard weight-based regimen (2.2 mg/kg/day of dihydroartemisinin and 17.8 mg/kg/day of piperaquine phosphate) was administered with milk once daily for 3 days. The fixed combination of DHA-PQ was well tolerated with no serious adverse events reported during follow-up. In this population modeling, nine patients were excluded: two patients did not meet the inclusion criteria, five patients had an incomplete course of medication, one patient self-medicated with other drugs, and one patient withdrew consent. Overall, 45% (109/241) of patients presented with recurrent P. vivax malaria during the follow-up period at a median (range) of 56 (14–65) days (Table 1).
Table 1. Clinical outcomes and patient demographics of patients with Plasmodium vivax malaria enrolled in this study.
Pharmacokinetics of piperaquine
A total of 791 plasma concentrations (435 venous and 356 capillary samples) from 116 patients were quantified and used in the pharmacokinetic modeling. Venous and capillary plasma concentrations were transformed into their natural logarithms and modeled simultaneously using nonlinear mixed-effects modeling. Substantial differences in matched venous and capillary blood concentrations have been reported in a previous clinical study.17 A proportional venous-capillary transformation factor was, therefore, used to compensate for the difference between sampling matrices, resulting in 41% higher capillary concentrations compared with venous concentrations.
The initial structural base models were parameterized as elimination clearance (CL/F), intercompartment clearance(s) (Q/F), apparent volume of distribution of the central compartment (VC/F), apparent volume of distribution of the peripheral compartment(s) (VP/F), and absorption rate constant. Several absorption, distribution, and covariate models were fitted to the data to construct the best performing model. Piperaquine pharmacokinetics in patients with P. vivax malaria were best described by a three-compartment distribution model (Figure 1). A three-compartment disposition model resulted in a significant improvement in model fit compared with a two-compartment model (difference in objective function value (ΔOFV) of 108) with no additional benefit of an additional peripheral compartment (ΔOFV = 0.57). A transit-compartment model with a fixed number of transit compartments (n = 3, transit rate constant was set to be identical to absorption rate constant) described the absorption phase better than all other absorption models (ΔOFV > 54.2). A relative bioavailability parameter (i.e., fixed to 100% for the population) was implemented to allow quantification of the interindividual variability in the absorption of piperaquine and resulted in a significant improvement in the model fit (ΔOFV = 81.8).
Figure 1.

Final piperaquine population pharmacokinetic-pharmacodynamic model in patients with P. vivax malaria. KTR is the transit absorption rate constant (KTR = (n + 1)/mean absorption transit time), CL is the elimination clearance, VC is the apparent central volume of distribution, VP is the apparent peripheral volume of distribution, Q is the intercompartment clearance, and F is the relative oral bioavailability.
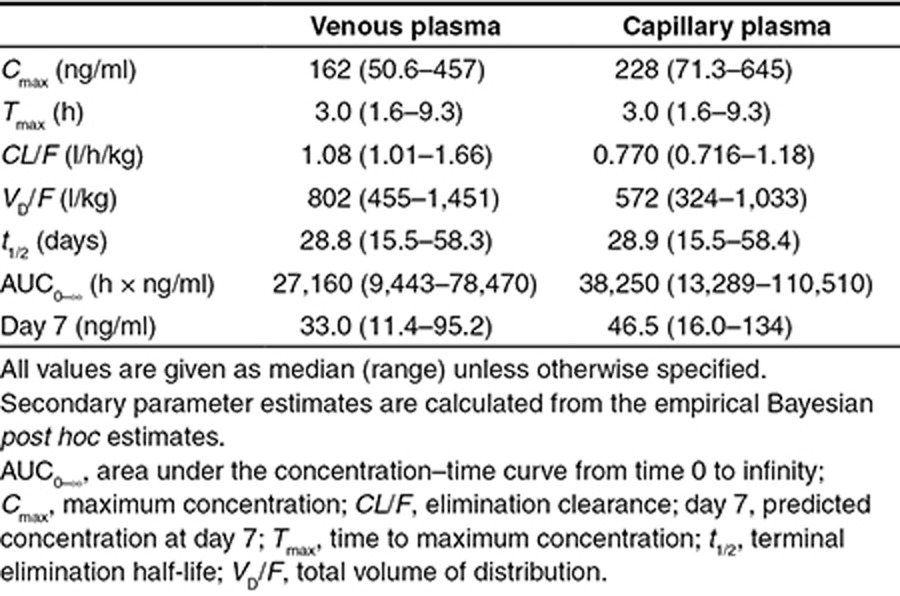
Body weight, incorporated as a fixed allometric function, improved the model fit significantly (ΔOFV = 31.0) and resulted in better precision and decreased interindividual variability in clearance and volume parameters. Body temperature on admission on VP2/F (ΔOFV = 11.5) and age on Q1/F (ΔOFV = 5.46) were selected in the forward covariate selection but could not be retained in the backward deletion. The estimated interindividual variability was small for the transformation factor, CL/F, Q2/F, and VC/F, and could, therefore, be removed with no significant impact in the final model (ΔOFV = 2.40). Final pharmacokinetic parameter estimates and secondary parameter estimates are reported in Tables 2 and 3.
Table 2. Population pharmacokinetic-pharmacodynamic parameters of the final model describing piperaquine in patients with Plasmodium vivax malaria.

Table 3. Secondary parameters of the final piperaquine model in patients with Plasmodium vivax malaria.
The mean parameter estimates from the simultaneous model with both venous and capillary data were also comparable with estimates when capillary and venous data were modeled separately. The final pharmacokinetic model displayed satisfactory goodness-of-fit diagnostics (Supplementary Figure S1 online) with a small epsilon-shrinkage of 15.0%. However, a relatively high η-shrinkage (up to 43.5%) could be seen for certain parameters in the final model because of the sparse data. The numerical and prediction-corrected visual predictive checks resulted in 4.6% (95% CI: 2.7–8.1%) and 3.7% (95% CI: 2.5–8.2%) of piperaquine observations below and above the simulated 90% prediction interval, respectively (Figure 2).
Figure 2.

Prediction-corrected visual predictive check of the final model describing piperaquine pharmacokinetics in P. vivax malaria. Open circles represent the observed piperaquine concentrations. Solid black line represents the 50th percentile of the observations, and dash lines represent the 5th and 95th percentiles of the observations. Gray areas represent the 95% confidence intervals of the simulated 5th, 50th, and 95th percentiles from 1,000 simulations. The inset shows a prediction-corrected visual predictive check for the first 3 days of piperaquine treatment.
Pharmacodynamics of piperaquine
Piperaquine inhibits asexual parasite multiplication but has no effect on liver-stage parasites. The influence of piperaquine exposure on the risk of recurrent P. vivax malaria was investigated in 62 patients for whom both pharmacokinetic and pharmacodynamic data were available. However, efficacy data were available from all the 241 patients included in the study. Pharmacodynamic parameters were evaluated using an interval-censoring time-to-event model,18 implemented with the Laplacian estimation method. The pharmacokinetic parameter and variability estimates were fixed to that of the final pharmacokinetic model and used in the pharmacokinetic-pharmacodynamic model. P. vivax malaria in tropical regions commonly displays frequent relapses which emerge at three-week intervals when the infections are treated with rapidly eliminated antimalarials.19 A multiple surge function was, therefore, implemented to characterize this periodicity in the risk of recurrent malaria. The constant hazard function with a periodically increased risk of relapses described the data well. Piperaquine had a significant (ΔOFV = 20.3) protective effect on the risk of recurrent malaria infections with a required in vivo venous piperaquine concentration of 6.92 ng/ml for a 50% relative reduction in baseline hazard (PC50). Sex was selected as a significant covariate (P < 0.05) on baseline hazard in the forward selection step, but it was removed during the backward elimination resulting in a covariate-free pharmacodynamic model. Final pharmacodynamic parameters were well estimated with high precision (Table 2) and simulation-based diagnostics showed good agreement between simulated and observed recurrent malaria infections (Figure 3).
Figure 3.

Visual predictive check of the final time-to-event model describing piperaquine pharmacodynamics in P. vivax malaria. Gray area represents the 95% prediction interval of the time to recurrent infections from 500 simulations. Solid line represents the observed Kaplan–Meier plot for (a) patients in the pharmacokinetic and pharmacodynamic cohort (62 patients, Table 1) and (b) all patients in the efficacy study (241 patients, Table 1).
The individual patient parasite burden (Table 1) corresponded approximately to a total of 1011 parasites in a typical adult at admission. This assumes that P. vivax does not sequester substantially. DHA-PQ is administered for 3 consecutive days, covering two asexual cycles. Assuming a parasiticidal effect (parasite reduction ratio) of a 10,000-fold parasite reduction per cycle by dihydroartemisinin (without any additional effect by piperaquine) results in a 100,000,000-fold reduction in total parasite biomass during the first two cycles.20 At an initial parasite burden of 1011 parasites, this would leave ~1,000 parasites to be eliminated by residual piperaquine concentrations to prevent recrudescent malaria. Piperaquine alone has not been adequately assessed in P. vivax malaria, but if it is similar to chloroquine, it should provide parasite reduction ratios of ~100 to 1,000 per cycle, so most patients should be parasite free 6–8 days post-treatment if piperaquine levels are sufficient to sustain a maximum parasiticidal effect for this time. If the parasite burden is not eliminated completely, then these remaining parasites can multiply and cause a recrudescent infection. However, a high cure rate of the asexual infection is likely in this study as concentrations of piperaquine after the second asexual cycle (i.e., ≥ 4 days) are highly likely to be able to eliminate ~1,000 parasites. In the first relapse of tropical P. vivax malaria (unaffected by drugs and assuming 1–10 schizonts and a subsequent parasite multiplication rate of ~10 per cycle), the hypnozoite-derived hepatic schizonts must liberate 13–17 days after the onset of the primary illness to produce a symptomatic infection at day 21. However, if residual piperaquine concentrations are sufficiently high, they could suppress and eliminate the asexual parasites derived from 10,000 to 100,000 released merozoites and thereby prevent the first relapse completely. If residual piperaquine concentrations are not sufficiently high for complete elimination, they could cause an initial suppression, resulting in a delayed symptomatic first relapse. Only 14% (15 out of 109) of recurrent P. vivax infections occurred before day 39, which suggests that DHA-PQ treatment prevents a large proportion of the first relapses (Figure 4). The majority of infections (64%, 70 out of 109) occurred between day 42 and 56, suggesting that these are mainly delayed second relapses (Figure 4). There will be some contribution from reinfections, but as entomological inoculation rates are probably well below 0.5/person/year for P. vivax in this area, this contribution is likely to be small.
Figure 4.

Relationship between piperaquine treatment and risk of relapsing P. vivax malaria infections. (a) Mean hazard rate of relapsing P. vivax malaria versus time in the presence (solid line) and absence (dashed line) of a standard piperaquine treatment (primary y-axis). The frequency of observed recurrent P. vivax infections for all patients in the efficacy study (241 patients, Table 1) is displayed on the secondary y-axis. The hazard of the first relapse (3 weeks) is completely suppressed and the hazard of the second relapse (6 weeks) is almost completely suppressed after piperaquine treatment. The model-predicted hazard rate after piperaquine treatment is in agreement with the observed frequency of observed recurrent P. vivax infections. (b) Simulated day-7 venous piperaquine plasma concentrations and chance of remaining malaria free for 30 days. Open gray circles represent simulated patients. Solid black line represents the 50th percentile and dash lines represent the 5th and 95th percentiles of the prediction interval. The dashed vertical line indicates the predicted day-7 concentration that results in 99% chance of remaining malaria free for 30 days for a typical patient.
Discussion
The fixed combination of DHA-PQ is a highly efficacious antimalarial drug with an excellent safety profile. Antimalarial treatment with DHA-PQ resulted in lower cumulative risk (54.9%) of recurrent P. vivax malaria compared with chloroquine (79.1%) in Thailand.16 This difference could be a result of the very potent artemisinin derivative which generally has a rapid onset of action against P. vivax malaria,21 but it is likely to result mainly from increasing chloroquine resistance and hence the superiority of piperaquine. In fully sensitive P. vivax malaria, recrudescence rates are close to zero with chloroquine treatment. Previous studies in this region have shown high relapse rates of P. vivax within one month of treatment with a short acting artemisinin derivative.19 Thus, both slowly eliminated drugs prevent the first relapse. Whether the lower cumulative risk of recurrent P. vivax malaria, following DHA-PQ, results from a higher cure rate of the primary infection or a more effective suppression of the first or the second relapse has been unclear. As relapses can arise from parasites which are either genetically similar or genetically distinct to those causing the primary infection, it is not possible to distinguish with certainty between relapse, recrudescence, or reinfection in patients with P. vivax malaria who remain in the endemic area. Recrudescence can occur many weeks after treatment with slowly eliminated drugs, but the pharmacokinetic-pharmacodynamic data modeled in this study suggest that only a negligible proportion (<2%) of the recurrent P. vivax infections could be recrudescent.
Pharmacokinetics of piperaquine
The population pharmacokinetic properties of piperaquine have been described previously in patients with uncomplicated P. falciparum malaria.15,22,23,24 This is the first study reporting the population pharmacokinetics of piperaquine in patients with P. vivax malaria.
The structural three-compartment model described in this study was similar to that reported recently in young children and in pregnant and nonpregnant women with P. falciparum malaria.15,22,25 Simplified two-compartment structural models in children and adult patients have also been reported reflecting differences in follow-period and/or sampling strategies.23,24 A transit-compartment absorption model allowed more flexibility in the absorption phase and provided a clear advantage over other absorption models, although there were not enough data in the absorption phase to estimate both KTR and KA separately. Large interindividual variability was seen in the absorption of piperaquine as in previous studies.15,22,25 Data collected here were too sparse to allow an estimation of the variability between dose occasions, which is likely to have inflated the inter-individual variability. Indeed interindividual variability was high in this study with values up to 138%. Venous and capillary plasma concentrations were successfully modeled simultaneously with a transformation factor of 1.41 (relative standard error of 2.11%). This factor was similar to that from a linear regression between observed venous and capillary plasma concentrations (slope = 1.29), which further supports the appropriateness of the simultaneous model. However, the exact mechanism of this matrix-dependent difference cannot be elucidated from the data collected in this study. The advantage of a simultaneous modeling approach is that the model can be used to simulate drug exposures from any sampling technique and therefore enables literature comparisons.
Body weight was the only significant covariate in this model. This has also been reported in previous studies15,24 and was not unexpected, considering the strong biological prior of body weight as a covariate on pharmacokinetic disposition and elimination parameters.26 All pharmacokinetic parameters were estimated with good precision (relative standard error < 20%) and the simulation-based diagnostics indicated excellent predictive performance of the final pharmacokinetic model.
Piperaquine CL/F in this P. vivax study was 1.08 l/h/kg, total volume of distribution (VD/F) was 802 l/kg, and t1/2 was 28.8 days, which were comparable with previously published reports in patients with an uncomplicated P. falciparum malaria (mean (range) CL/F of 1.18 (0.90–1.4) l/h/kg, VD/F of 789 (574–877) l/kg, and t1/2 of 23.7 (18–28) days).22,23,24,27,28
Pharmacodynamics of piperaquine
The observed high relapse rate of P. vivax malaria was expected as DHA-PQ eliminates only blood-stage parasites and does not provide cure of the latent liver-stage parasites. It was expected that piperaquine would delay the time to relapse, as does chloroquine, and this was supported by the pharmacodynamic results. Tropical frequent relapsing strains of P. vivax typically relapse at 3–4-week intervals, and the median time to relapse approximates three weeks (21 days) in Thailand after treatment with rapidly eliminated antimalarial drugs (such as artesunate, quinine, and halofantrine).19 In an area of much higher transmission in Papua, Indonesia, there were significantly more early recurrences of P. vivax malaria following artemether-lumefantrine treatment of P. vivax malaria compared with DHA-PQ (cumulative risk of 38 vs. 10% at day 42)13 reflecting the failure of the more rapidly eliminated lumefantrine to suppress the first relapse. However, with longer follow-up in a high transmission area, the cumulative reinfection rates converge as everyone is reinfected eventually. A critical and unanswered question has been whether the slowly eliminated drugs prevented, or simply delayed, the relapse. Explicitly, prevention would mean that the relapse emerged but was eliminated by the residual drug levels, and thus, the first observed recurrence is in fact the second relapse. Delay would mean that the residual drug levels reduced multiplication of the relapse, so it reached patency after a substantial delay. In the former explanation, overall relapse numbers might be reduced as this would exhaust the liver burden of hypnozoites more rapidly. A pharmacometric time-to-event approach explaining relapsing P. vivax malaria, first presented for amodiaquine treatment in pregnant women,29 described the data observed in this study well. An interval-censoring time-to-event model was successfully used since the exact times of relapses were unknown. The risk of relapsing P. vivax malaria in patients was explained by a constant baseline hazard (i.e., 8.94 relapses/year) with a 123% increased risk every third week. The pharmacodynamic model was significantly improved by adding a protective drug effect, indicating that piperaquine cured the primary blood stage P. vivax infection and fully suppressed a large proportion of the first relapses that would have occurred three weeks after the initial infection. The pharmacodynamic model also indicated that the observed high recurrence rate is likely to be explained mainly by delayed second relapses, although it cannot be excluded that some recurrences are reinfections. This can only be mechanistically evaluated by an individual parasite-data-driven pharmacokinetic-pharmacodynamic model. Simulations using the final pharmacokinetic-pharmacodynamic model (Figure 4) demonstrated that minimum day-7 piperaquine venous and capillary concentrations of 27 and 38 ng/ml, respectively, were needed to suppress the risk of relapse for 30 days (i.e., for 99% of simulated patients). This is in close agreement with a previously defined day-7 threshold venous concentration of 30 ng/ml in patients with P. falciparum malaria to minimize the risk of recrudescent infections.30
In conclusion, the population pharmacokinetic properties of piperaquine in patients with P. vivax malaria were described successfully by modeling venous and capillary plasma concentrations simultaneously. Body weight was the only significant covariate and resulted in increasing clearance and volumes with increasing body weight. Times to recurrent P. vivax infections were successfully modeled with a time-to-event approach and resulted in a significantly delayed time to recurrent infections during the post-treatment phase of piperaquine treatment. This was explained by prevention of the first P. vivax malaria relapse by residual piperaquine concentrations. Piperaquine is a good candidate for the treatment of P. vivax malaria and the modeling conducted here demonstrated the added benefit of reduced morbidity compared with other more rapidly eliminated antimalarial drugs.
Methods
Ethics approval.
Ethics approval was granted by the Faculty of Tropical Medicine Ethics Committee, Mahidol University, Bangkok, Thailand, and the Oxford Tropical Research Ethics Committee, Oxford University, UK. This study was registered in the ISRCTN Register (ISRCTN87827353). Details on the clinical study design and outcomes have been presented in full elsewhere.16
Drug regimen and blood collection.
All patients received a standard regimen of DHA-PQ. Patients who vomited the dose were excluded from the pharmacokinetic study. A full or half replacement dose was administered if vomiting occurred within 30 min of or between 30 min to 1 h after administration, respectively.
Venous blood samples (3 ml) and capillary blood samples (200 μl) were drawn from each patient for piperaquine plasma measurements. Samples were collected randomly over 69 days (4–7 venous samples and 1–11 capillary samples) from 62 patients and an additional venous sample (5 ml) was drawn from these patients at the time of recurrent P. vivax malaria. This random sampling allowed of an adequate coverage of the entire concentration–time profile. An additional 54 patients provided a single plasma sample only at the time of recurrent malaria. Patients were excluded from the study after recurrent malaria. Plasma samples were shipped on dry ice to the Department of Clinical Pharmacology, Mahidol-Oxford Tropical Medicine Research Unit, Thailand, for drug quantification.31
Population modeling.
Piperaquine pharmacokinetics and pharmacodynamics were evaluated using nonlinear mixed-effects modeling (NONMEM version VII; Icon Development Solutions, Ellicott City, MD). Postprocessing and automation were performed using Pearl-Speaks-NONMEM version 3.5.3, Census version 1.2b2,32 and Xpose version 4.033 in the programming language R version 2.13.2 (The R Foundation for Statistical Computing). Measurements below the lower limit of quantification were less than 0.5% of the total data and therefore omitted. OFV, calculated by NONMEM as minus twice the log-likelihood up to constant, was used for model selection during the model-building process. A difference in the OFV (ΔOFV) of >3.84 was considered significant (P < 0.05) when comparing two nested models with 1 degree of freedom difference. Full details of the pharmacokinetic and pharmacodynamic modeling methodology can be found in supplementary material.
Author Contributions
J.T., P.T., N.P.J.D., and N.J.W. wrote the manuscript. A.P.P., K.M.L., E.A.A., N.P.J.D., F.N., and N.J.W. designed the research. J.T., A.P.P., K.M.L., E.A.A., and F.N. performed the research. J.T., P.T., and W.H. analyzed the data.
Conflict of Interest
The Wellcome Trust is a UK-based charity that supports medical research and is independent of any drug companies. It has no financial links with the manufacturers of either the diagnostic tests or the drugs used in this study. The authors declared no conflict of interest.
Study Highlights

Acknowledgments
The authors gratefully thank all the patients who participated in this study. They also thank the staff at the Shoklo Malaria Research Unit, Mae Sot, Thailand, and all the staff involved in taking and processing the samples. This study was part of the Mahidol-Oxford Tropical Medicine Research Unit, funded by the Wellcome Trust of Great Britain.
Supplementary Material
References
- Price R.N., Tjitra E., Guerra C.A., Yeung S., White N.J., Anstey N.M. Vivax malaria: neglected and not benign. Am. J. Trop. Med. Hyg. 2007;77:79–87. [PMC free article] [PubMed] [Google Scholar]
- Guerra C.A., et al. The limits and intensity of Plasmodium falciparum transmission: implications for malaria control and elimination worldwide. PLoS Med. 2008;5:e38. doi: 10.1371/journal.pmed.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C.A., et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl. Trop. Dis. 2010;4:e774. doi: 10.1371/journal.pntd.0000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstey N.M., Russell B., Yeo T.W., Price R.N. The pathophysiology of vivax malaria. Trends Parasitol. 2009;25:220–227. doi: 10.1016/j.pt.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Poespoprodjo J.R., et al. Vivax malaria: a major cause of morbidity in early infancy. Clin. Infect. Dis. 2009;48:1704–1712. doi: 10.1086/599041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosten F., et al. Effects of Plasmodium vivax malaria in pregnancy. Lancet. 1999;354:546–549. doi: 10.1016/s0140-6736(98)09247-2. [DOI] [PubMed] [Google Scholar]
- Collins W.E., Jeffery G.M., Roberts J.M. A retrospective examination of anemia during infection of humans with Plasmodium vivax. Am. J. Trop. Med. Hyg. 2003;68:410–412. [PubMed] [Google Scholar]
- Rieckmann K.H., Davis D.R., Hutton D.C. Plasmodium vivax resistance to chloroquine. Lancet. 1989;2:1183–1184. doi: 10.1016/s0140-6736(89)91792-3. [DOI] [PubMed] [Google Scholar]
- Baird J.K. Chloroquine resistance in Plasmodium vivax. Antimicrob. Agents Chemother. 2004;48:4075–4083. doi: 10.1128/AAC.48.11.4075-4083.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . World Health Organization Guidelines for the Treatment of Malaria. WHO, Geneva, Switzerland; 2010. [Google Scholar]
- Zwang J., et al. Safety and efficacy of dihydroartemisinin-piperaquine in falciparum malaria: a prospective multi-centre individual patient data analysis. PLoS ONE. 2009;4:e6358. doi: 10.1371/journal.pone.0006358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas N.M., Anstey N.M., Angus B.J., Nosten F., Price R.N. Artemisinin combination therapy for vivax malaria. Lancet Infect. Dis. 2010;10:405–416. doi: 10.1016/S1473-3099(10)70079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliff A., et al. Two fixed-dose artemisinin combinations for drug-resistant falciparum and vivax malaria in Papua, Indonesia: an open-label randomised comparison. Lancet. 2007;369:757–765. doi: 10.1016/S0140-6736(07)60160-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunajeewa H.A., et al. A trial of combination antimalarial therapies in children from Papua New Guinea. N. Engl. J. Med. 2008;359:2545–2557. doi: 10.1056/NEJMoa0804915. [DOI] [PubMed] [Google Scholar]
- Tarning J., et al. Population pharmacokinetics and pharmacodynamics of piperaquine in children with uncomplicated falciparum malaria. Clin. Pharmacol. Ther. 2012;91:497–505. doi: 10.1038/clpt.2011.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phyo A.P., et al. Dihydroartemisinin-piperaquine versus chloroquine in the treatment of Plasmodium vivax malaria in Thailand: a randomized controlled trial. Clin. Infect. Dis. 2011;53:977–984. doi: 10.1093/cid/cir631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley E.A., et al. Comparison of plasma, venous and capillary blood levels of piperaquine in patients with uncomplicated falciparum malaria. Eur. J. Clin. Pharmacol. 2010;66:705–712. doi: 10.1007/s00228-010-0804-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox E.H., Veyrat-Follet C., Beal S.L., Fuseau E., Kenkare S., Sheiner L.B. A population pharmacokinetic-pharmacodynamic analysis of repeated measures time-to-event pharmacodynamic responses: the antiemetic effect of ondansetron. J. Pharmacokinet. Biopharm. 1999;27:625–644. doi: 10.1023/a:1020930626404. [DOI] [PubMed] [Google Scholar]
- White N.J. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar. J. 2011;10:297. doi: 10.1186/1475-2875-10-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White N.J. Pharmacokinetic and pharmacodynamic considerations in antimalarial dose optimization. Antimicrob. Agents Chemother. 2013;57:5792–5807. doi: 10.1128/AAC.00287-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukrittayakamee S., et al. Therapeutic responses to different antimalarial drugs in vivax malaria. Antimicrob. Agents Chemother. 2000;44:1680–1685. doi: 10.1128/aac.44.6.1680-1685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarning J., et al. Population pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and non-pregnant women with uncomplicated malaria. Antimicrob. Agents Chemother. 2012; 56:1997–2007 . doi: 10.1128/AAC.05756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung T.Y., et al. Population pharmacokinetics of piperaquine in adults and children with uncomplicated falciparum or vivax malaria. Br. J. Clin. Pharmacol. 2004;57:253–262. doi: 10.1046/j.1365-2125.2003.02004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarning J., et al. Population pharmacokinetics of piperaquine after two different treatment regimens with dihydroartemisinin-piperaquine in patients with Plasmodium falciparum malaria in Thailand. Antimicrob. Agents Chemother. 2008;52:1052–1061. doi: 10.1128/AAC.00955-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund R.M., et al. A population pharmacokinetic model of piperaquine in pregnant and non-pregnant women with uncomplicated Plasmodium falciparum malaria in Sudan. Malar. J. 2012;11:398. doi: 10.1186/1475-2875-11-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holford N.H. A size standard for pharmacokinetics. Clin. Pharmacokinet. 1996;30:329–332. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- Rijken M.J., et al. Pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated falciparum malaria. Antimicrob. Agents Chemother. 2011;55:5500–5506. doi: 10.1128/AAC.05067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.V., et al. Pharmacokinetics and ex vivo pharmacodynamic antimalarial activity of dihydroartemisinin-piperaquine in patients with uncomplicated falciparum malaria in Vietnam. Antimicrob. Agents Chemother. 2009;53:3534–3537. doi: 10.1128/AAC.01717-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarning J., et al. Population pharmacokinetic and pharmacodynamic modeling of amodiaquine and desethylamodiaquine in women with Plasmodium vivax malaria during and after pregnancy. Antimicrob. Agents Chemother. 2012;56:5764–5773. doi: 10.1128/AAC.01242-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price R.N., et al. Clinical and pharmacological determinants of the therapeutic response to dihydroartemisinin-piperaquine for drug-resistant malaria. Antimicrob. Agents Chemother. 2007;51:4090–4097. doi: 10.1128/AAC.00486-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindegardh N., Annerberg A., White N.J., Day N.P. Development and validation of a liquid chromatographic-tandem mass spectrometric method for determination of piperaquine in plasma stable isotope labeled internal standard does not always compensate for matrix effects. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008;862:227–236. doi: 10.1016/j.jchromb.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Wilkins J.J. NONMEMory: a run management tool for NONMEM. Comput. Methods Programs Biomed. 2005;78:259–267. doi: 10.1016/j.cmpb.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Jonsson E.N., Karlsson M.O. Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.