

Abstract
Background
The identification of protein-protein interaction sites is a computationally challenging task and important for understanding the biology of protein complexes. There is a rich literature in this field. A broad class of approaches assign to each candidate residue a real-valued score that measures how likely it is that the residue belongs to the interface. The prediction is obtained by thresholding this score.
Some probabilistic models classify the residues on the basis of the posterior probabilities. In this paper, we introduce pairwise conditional random fields (pCRFs) in which edges are not restricted to the backbone as in the case of linear-chain CRFs utilized by Li et al. (2007). In fact, any 3D-neighborhood relation can be modeled. On grounds of a generalized Viterbi inference algorithm and a piecewise training process for pCRFs, we demonstrate how to utilize pCRFs to enhance a given residue-wise score-based protein-protein interface predictor on the surface of the protein under study. The features of the pCRF are solely based on the interface predictions scores of the predictor the performance of which shall be improved.
Results
We performed three sets of experiments with synthetic scores assigned to the surface residues of proteins taken from the data set PlaneDimers compiled by Zellner et al. (2011), from the list published by Keskin et al. (2004) and from the very recent data set due to Cukuroglu et al. (2014). That way we demonstrated that our pCRF-based enhancer is effective given the interface residue score distribution and the non-interface residue score are unimodal.
Moreover, the pCRF-based enhancer is also successfully applicable, if the distributions are only unimodal over a certain sub-domain. The improvement is then restricted to that domain. Thus we were able to improve the prediction of the PresCont server devised by Zellner et al. (2011) on PlaneDimers.
Conclusions
Our results strongly suggest that pCRFs form a methodological framework to improve residue-wise score-based protein-protein interface predictors given the scores are appropriately distributed. A prototypical implementation of our method is accessible at http://ppicrf.informatik.uni-goettingen.de/index.html.
Background
Protein-protein interactions are constitutive of almost every biological process. The ability to identify the residues that form the interaction sites of these complexes is necessary to understand them. In particular, it is the basis for new therapeutic approaches to treat diseases [1, 2].
A great deal of work has been done on developing in-silico prediction methods. As already observed by Zhou et al. [3], these methods can be subdivided with respect to the kind of mathematical foundation invoked and with respect to the features or characteristics of the protein used.
Residue-wise score-based prediction methods
Let xr be the data relevant for a residue r in a given protein chain. These methods then employ a function f(xr,λ), where λ are some coefficients which have been learned through the training. The value of f(xr,λ) then determines, whether r is rated as an interface or not. The linear regression method [4, 5], the scoring function method [6–11], the neural network method [12–17], and the support vector machine method [18–25] are of this kind.
Probabilistic methods
Let X be the data relevant for a protein chain, where these data are assumed to stem from a random source thus obeying a random distribution. X, which alternatively is called the observation, typically includes the structure. The label sequence of the residues Y that classifies each individual residue either as interface or as non-interface is assumed to be random, too. Typically, probabilistic methods use the conditional probability distribution 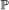 (Y | X) to determine a classification y* of the residues of maximal posterior probability
(Y | X) to determine a classification y* of the residues of maximal posterior probability 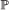 (y* | x). Naive Bayesian methods [26], Bayesian network methods [27], hidden Markov models (HMMs) [26], and linear-chain Conditional Random Fields taking the backbone as underlying graphical structure [28] fall in this category. Using posterior decoding on the basis of the forward-backward algorithm, both HMMs and CRFs are residue-wise score-based prediction methods, where the binary decision is made by thresholding the posterior probabilities of classifying the residues as interface.
(y* | x). Naive Bayesian methods [26], Bayesian network methods [27], hidden Markov models (HMMs) [26], and linear-chain Conditional Random Fields taking the backbone as underlying graphical structure [28] fall in this category. Using posterior decoding on the basis of the forward-backward algorithm, both HMMs and CRFs are residue-wise score-based prediction methods, where the binary decision is made by thresholding the posterior probabilities of classifying the residues as interface.
Notations
We use Latin uppercase letters when referring to random aspects of the objects denoted by them. In contrast, lowercase letters denote arbitrarily chosen but fixed objects. In this context boldface letters indicate vectors, the corresponding non-boldface letters their coefficients.
The vast majority of methods use the 3D structure of the target protein chain in form of a PDB file as input [4–13, 15, 17–21, 23–25]. However, a few methods are not requiring a 3D structure and rather use sequences only [14, 16, 22]. We here consider the problem with a given 3D structure of the target protein chain. Sequence-based input may include a multiple sequence alignment of related proteins from which, for example, sequence conservation can be inferred. When the 3D structure of an unbound binding partner is also available, protein-protein docking methods can be applied. This has also been exploited to provide feedback from docking to the more specific problem of interface prediction [29]. We here consider the case where the binding partner’s 3D structure is not given. Nor requires the presented method the sequence of the binding partner. Albeit, we tested on homodimers only as we here rather focus on our new method rather than on features or types of proteins. The protein features used for interface prediction in the literature are reviewed in the Methods section as far as we make use of them in this article.
Most of the current studies for predicting interaction sites of proteins that use a probabilistic method are restricted by treating the residues of the proteins as independent vertices. Li et al. have taken the backbone neighborhood into account thus modeling the protein as a sequence [28] using what can be called a line CRF or linear-chain CRF. The features they define on the label pair of two backbone neighbors have the effect of smoothing the predicted labels along the protein sequence. Decisive is, however, that they were the first who used conditional random fields (CRFs) for interface prediction. CRFs in turn have come into use for solving sequence labeling problems due to Lafferty et al.
[30]. See [31] for an overview. From the mathematical point of view they take advantage of the fact that they model the conditional probability 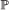 (Y | X) rather than the joint probability
(Y | X) rather than the joint probability 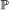 (Y, X). Recently there has been an explosion of interest in conditional random fields (CRFs) with successful applications. It has been shown that CRFs have the abilities for solving sequence labeling problems like part-of-speech tagging (POST) [32] and natural language processing [33]. Furthermore in the web extraction problem, in which the web-sites are modeled as two dimensional grid graphs, CRFs perform well [34]. One of their outstanding benefits over many other statistical models is that a CRF can easily describe the dependencies of observations.
(Y, X). Recently there has been an explosion of interest in conditional random fields (CRFs) with successful applications. It has been shown that CRFs have the abilities for solving sequence labeling problems like part-of-speech tagging (POST) [32] and natural language processing [33]. Furthermore in the web extraction problem, in which the web-sites are modeled as two dimensional grid graphs, CRFs perform well [34]. One of their outstanding benefits over many other statistical models is that a CRF can easily describe the dependencies of observations.
As proteins are folded into three dimensional structures, spatial relationships create dependencies between residues. For example, we find on the test data described below that the correlation coefficient between spatial neighbors that are not also sequence neighbors (distance ≤3.5 Å) is 0.45. This is only slightly lower than the correlation coefficient between residues that are sequence neighbors (0.49). As there are more than three times as many spatial pairs of neighbors than sequence neighbors at this threshold it is reasonable from a modeling standpoint to use a model that respects all dependencies induced by spatial proximity, not only the dependencies induced by proximity along the backbone.
There are many papers using spatial neighborhood information of residues to predict-protein interaction sites (see e.g. [2, 13, 21, 28]). However, the spatial information of proteins was only integrated into the feature functions, but not represented in the model. For probabilistic models, the difference between the two ways to integrate spacial information is that in previous models the label of the i-th residue Yi is conditional independent from the labels of other residues given data X and – in the case of linear CRFs or HMMs – given the labels of Yi−1 and Yi+1. Even when neighborhood information is only used for spatial smoothing of the labels, the intuitive advantage over, say, an SVM classifier that uses spatial neighborhood in the features but classifies each residue independently, is that not-patch-like candidate labelings are explicitly punished. In contrast, such an independent classifier-approach may have a tendency to predict individual interface residues ‘sprinkled’ around the protein surface [28].
For this reason, a general CRF seems to be more suitable for the task. However, inference for general CRFs is intractable. In this paper, pairwise conditional random fields (pCRFs) are utilized. Specializing general CRFs, only node cliques and edge cliques are taken into consideration in pCRFs. A pCRF retains most spatial information of proteins, can be specified with the same number of parameter as a line CRF and approximate inference remains feasible with the generalization of the Viterbi algorithm introduced here. Taking pattern from piecewise training methods [35], we disentangled the labels of nodes and edges to train the model.
In order to take advantage of a residue-wise score-based predictor, we model the protein surface by means of a pCRF, where the observation is solely a sequence of surface residue scores between 0 and 1 output by the predictor. We then utilize a generalized Viterbi algorithm and piecewise training. The resulting tool tries to enhance the predictor chosen on the surface of the protein under study. It is the aim of this paper to demonstrate effectiveness of this approach provided that the interface residue scores and the non-interface residue scores are appropriately distributed.
Methods
We address the problem of improving residue-wise score-based predictors for protein interface residues as a node labeling problem for undirected graphs using the model class of conditional random fields (CRFs). Lafferty et al. [30] were the first who applied CRFs to the problem of labeling sequence data. Li et al. [28] used line CRFs to address the interaction site prediction. They have the advantage that the Viterbi algorithm well-known from decoding HMMs can be used to efficiently infer the most likely labeling sequence. Very useful and illustrative presentations on CRFs are given in [31, 32, 36, 37]. Above CRF-based models make the assumption that the label of one residue is conditionally independent of the labels of all other residues given the labels of the two adjacent residues in the protein sequence. To the best of our knowledge, we are the first to employ a graphical model that takes the spatial neighborhood of residues located on the protein surface into account.
This section is subdivided into three parts. We first explain how we model protein surfaces by pairwise CRFs. Then we introduce our new inference method. Finally, we elucidate our training method.
Using conditional random fields to model protein surfaces
For every protein under study that has n surface residues, a pair of random vectors (X, Y) is considered. The vector X is the observation that represents the knowledge about this protein that is utilized in the prediction, e.g. the 3D structure of the target protein and a multiple sequence alignment together with homologs.
The vector Y is a random sequence of length n over the alphabet {I,N} that labels the index set {1,2,…,n}, which in turn is called the set of positions (of the surface residues). The label I represents interface residues, whereas the label N represents non-interface residues. {I,N}n is the set of all label sequences of length n over {I,N}. We will also call them assignments as the term ‘label sequence’ may lead to confusion when applied below to subsets of {1,2,…,n} that are not contiguous sequences.
Let 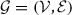 be the neighborhood graph, where
be the neighborhood graph, where 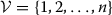 is the set of positions,
is the set of positions, 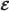 is the set of edges that typically results from an atom-distance-based neighborhood definition for positions. We assume for convenience in notation that
is the set of edges that typically results from an atom-distance-based neighborhood definition for positions. We assume for convenience in notation that 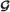 has no isolated nodes. Cases with isolated nodes could trivially be reduced to cases without isolated nodes. Let
has no isolated nodes. Cases with isolated nodes could trivially be reduced to cases without isolated nodes. Let 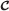 be the set of
be the set of 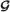 ’s cliques, which we refer to as node cliques. For a node clique
’s cliques, which we refer to as node cliques. For a node clique 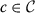 and an assignment y we denote by yc the restriction of y to the positions belonging to the node clique c. For c={i} and c={i,j} we write yi and
and an assignment y we denote by yc the restriction of y to the positions belonging to the node clique c. For c={i} and c={i,j} we write yi and 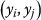 rather than y{i} and y{i,j}.
rather than y{i} and y{i,j}.
The preceding notation is also used in the slightly more general case of partial label assignments to arbitrarily chosen subsets 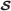 of the set of positions
of the set of positions 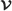 . Formally, let
. Formally, let 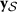 denote
denote 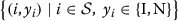 . Given two partial assignments
. Given two partial assignments 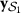 and
and 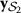 are identical on
are identical on 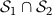 , the union
, the union 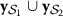 is well-defined.
is well-defined.
The conditional distribution function of our pCRF (X, Y) with respect to the neighborhood graph 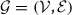 is defined as follows:
is defined as follows:
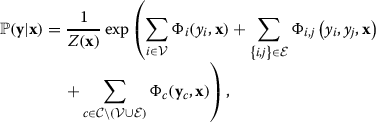 |
1 |
where x and y are arbitrarily chosen instances of the random observation X and the random label sequence Y, respectively, Φc(yc,x)∈∙ (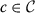 ) is the feature of the CRF located at the node clique c (again Φi and Φi,j simplify notation for Φ{i} and Φ{i,j}), and Z(x) is the observation-specific normalization factor defined by
) is the feature of the CRF located at the node clique c (again Φi and Φi,j simplify notation for Φ{i} and Φ{i,j}), and Z(x) is the observation-specific normalization factor defined by
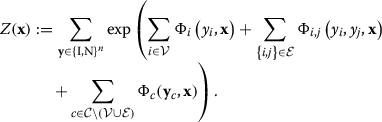 |
2 |
Let us call 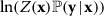 the score of the label sequence y given the observation x.
the score of the label sequence y given the observation x.
A CRF is called a pairwise CRF (pCRF) if Φc≡0, for all node cliques c larger than two. The remaining features Φi and Φi,j are referred to as node features and edge features, respectively. Thus, every position 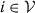 and every edge
and every edge 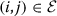 is represented by the pair (Φi(N,x),Φi(I,x)) and by the quadruplet (Φ{i,j} (N,N,x),Φ{i,j}(I,N,x),Φ{i,j}(N,I,x),Φ{i,j}(I,I,x)).
is represented by the pair (Φi(N,x),Φi(I,x)) and by the quadruplet (Φ{i,j} (N,N,x),Φ{i,j}(I,N,x),Φ{i,j}(N,I,x),Φ{i,j}(I,I,x)).
Following [30], we assume moreover that each node feature and each edge feature is a sum of weighted base features. More precisely, for every position 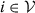 and every edge
and every edge 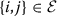 we assume representations
we assume representations
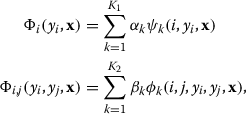 |
where y∈{I,N}n and x is an observation. The two real vectors
| 3 |
need to be calculated in a training phase.
In the most general sense, protein characteristics are real-valued evaluations of positions and pairs of adjacent positions (edges of the neighborhood graph), respectively, that are correlated with our position labeling problem. We use a standard step function technique to obtain base features from protein characteristics, rather than taking the raw values of the characteristics. To make our paper self-contained, let us describe this technique for short.
A protein characteristic depends on the observation and either a node or an edge. Each protein characteristic, such as e.g. the relative solvent-accessible surface area of a residue, is transformed into several binary features by binning, i.e. we distinguish only a few different cases rather than the whole range of the characteristic. Assuming the common case of real-valued characteristics, the bins are a partition of the reals into intervals. The use of this discretization allows to approximate any shape of dependency of the labels on the characteristics, rather than assuming a fixed shape such as linear or logarithmic.
From protein characteristics for positions to node features. We subdivide the range of the characteristics C into say γ intervals, where γ is at least two. Let s1<s2<…<sγ−1 be the corresponding interval boundaries. It is reasonable to take sι as the ι/γ-quantile of the empirical distribution of C for non-interface residues, where C(i,x)∈(s0,sγ]. Then we define for each position 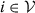 the following 2γ base features associated with the position characteristics C.
the following 2γ base features associated with the position characteristics C.
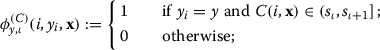 |
4 |
where y=N,I, and ι=0,1,…,γ−1 and s0:=−∞,sγ:=∞.
From protein characteristics for edges to edge features. Let D be the characteristics. Analogous to the previous case, we then obtain for each edge 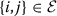 the following 4γ base features associated with D, where y,y′∈{N,I} and ι=0,1,…,γ−1.
the following 4γ base features associated with D, where y,y′∈{N,I} and ι=0,1,…,γ−1.
| 5 |
In both cases we set γ=5.
Devising a generalized Viterbi algorithm for pCRFs
The problem of finding a most probable label sequence y∗ given an observation x is NP-hard for general pCRFs [31]. In this subsection we present a heuristic that approximately solves this problem.
To this end, we first devise an algorithm, which we call generalized Viterbi algorithm. It computes an optimal label sequence, where the posterior probability of y∗ given x is maximized. Unfortunately, its run-time is in too many cases not acceptable. That is why we transform it in a second step into a feasible, time-bounded approximation algorithm.
The generalized Viterbi algorithm
Let 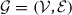 be the neighborhood graph underlying the protein under study. For any assignment (label sequence) y and any subset
be the neighborhood graph underlying the protein under study. For any assignment (label sequence) y and any subset 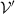 of
of 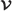 , let
, let 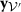 denote the partial assignment of y with respect to
denote the partial assignment of y with respect to 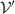 . (This is in line with the notation yc (c a position clique) introduced earlier in this study).
. (This is in line with the notation yc (c a position clique) introduced earlier in this study).
If 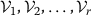 are pairwise disjoint position sets, the assignment for
are pairwise disjoint position sets, the assignment for 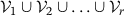 canonically resulting from assignments
canonically resulting from assignments 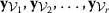 is denoted by
is denoted by 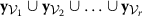 . For
. For 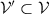 , the score
, the score 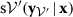 is defined by
is defined by
Then the problem of determining a most probable label sequence y∗ given an observation x can be reformulated as
This is the case, because it suffices to consider the score.
To put this into practice, we devised an algorithm we call generalized Viterbi. On the one hand, it is analogous to the classical Viterbi algorithm. On the other hand, there is a major difference. In our case there is no canonical order in which the positions of 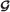 are traversed. Having explained our algorithm for any order, we show how to calculate a fairly effective one. In what follows, we assume that the positions not yet touched are held in a dynamic queue. Those positions having already left the queue form the history set
are traversed. Having explained our algorithm for any order, we show how to calculate a fairly effective one. In what follows, we assume that the positions not yet touched are held in a dynamic queue. Those positions having already left the queue form the history set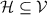 .
.
Assume that the subgraph of 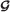 induced by
induced by 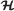 has connected components
has connected components 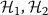 , …,
, …, 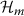 . For μ=1,2,…,m, let
. For μ=1,2,…,m, let 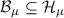 be the so-called boundary component associated with
be the so-called boundary component associated with 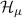 defined by
defined by 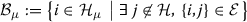 . The complement
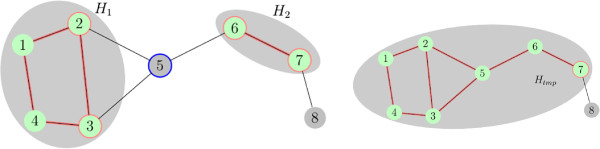
. The complement 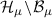 is the interior of the μ-th history component. See Figure 1 for an example.
is the interior of the μ-th history component. See Figure 1 for an example.
Figure 1.
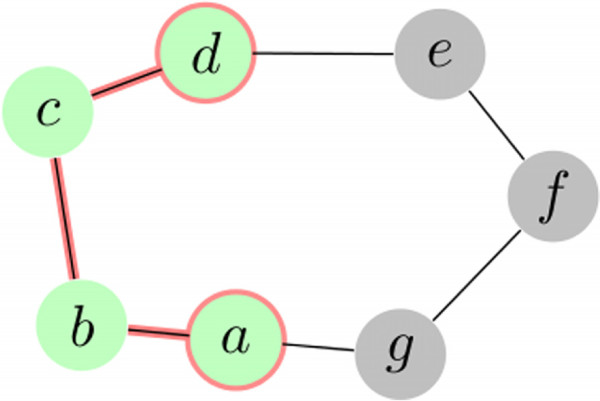
Example history set
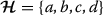 having boundary
having boundary
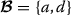 .
.
For assignments 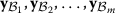 of the boundary components
of the boundary components 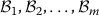 , the Viterbi variables
, the Viterbi variables 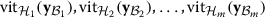 are defined as
are defined as
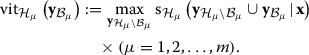 |
6 |
The Viterbi variables can be represented as a set of tables, one table of size 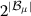 for each boundary component
for each boundary component 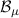 . In the case where a boundary component is empty the table reduces to a single number.
. In the case where a boundary component is empty the table reduces to a single number.
At any stage, the algorithm stores the connected components 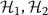 , …,
, …, 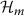 of the current history set
of the current history set 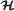 , the corresponding boundary components
, the corresponding boundary components 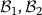 , …,
, …, 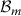 , and Viterbi variable values
, and Viterbi variable values 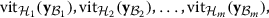 where
where 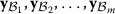 range over all possible assignments of corresponding boundary component. We store for every assignment on the boundary, a maximizing interior assignment. This assignment is the argmax of (6) but is determined with the dynamic programming recursions defined below. Let us call these data the current state of the algorithm. It mainly consists of record sets indexed by the boundary labelings.
range over all possible assignments of corresponding boundary component. We store for every assignment on the boundary, a maximizing interior assignment. This assignment is the argmax of (6) but is determined with the dynamic programming recursions defined below. Let us call these data the current state of the algorithm. It mainly consists of record sets indexed by the boundary labelings.
At the very beginning the queue contains all positions, the history set 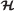 and the corresponding boundary component
and the corresponding boundary component 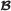 are empty. As long as the position queue is not empty, the top element v is extracted and the state is updated as follows.
are empty. As long as the position queue is not empty, the top element v is extracted and the state is updated as follows.
Adjoining v to the history set  , there are two cases to distinguish. Either position v is not adjacent to any other position of any old boundary component (see Figure 2) or adjoining position v to
, there are two cases to distinguish. Either position v is not adjacent to any other position of any old boundary component (see Figure 2) or adjoining position v to  results in adding it to some connected component of the old history set or even merging together two or more of them (see Figure 3).
results in adding it to some connected component of the old history set or even merging together two or more of them (see Figure 3).
Figure 2.
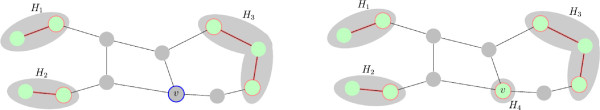
Computing the connected components of the new history set - case one.
Figure 3.
Computing the connected components of the new history set - case two.
In the first case we simply have to take over all the old connected components, boundary sets and Viterbi variables. Moreover, we perform the instructions
In the second case position v is adjacent to some boundary components, say 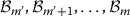 . Then the old history components
. Then the old history components 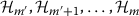 and the current position v are merged together:
and the current position v are merged together:
The other history set components and corresponding Viterbi variables are not affected.
For μ=m′,m′+1,…,m, let 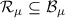 be the set of all positions out of
be the set of all positions out of 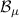 that are no longer boundary nodes after having adjoined v to the history set. The nodes in
that are no longer boundary nodes after having adjoined v to the history set. The nodes in 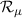 are removed from the boundary
are removed from the boundary 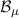 after the iteration. Let
after the iteration. Let 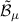 be the complement of
be the complement of 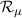 in
in 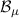 . By inspecting the edges incident to the current position v, all these sets can be computed in linear time.
. By inspecting the edges incident to the current position v, all these sets can be computed in linear time.
The new boundary set 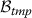 is then either
is then either 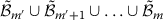 or
or 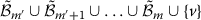 , where it can be checked in linear time whether or not v is a new boundary position.
, where it can be checked in linear time whether or not v is a new boundary position.
We are now in a position to calculate the new Viterbi variables 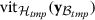 , where
, where 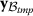 ranges over all assignments of the new boundary set
ranges over all assignments of the new boundary set 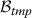 .
.
If 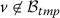 then
then
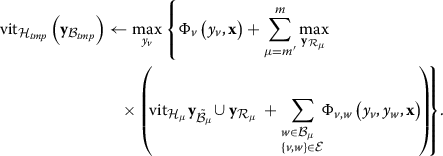 |
Here, any assignment of a node set is assumed to implicitly define assignments for any subset thereof. Figure 4 illustrates this case of the recursion step. If, however, 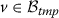 , then
, then
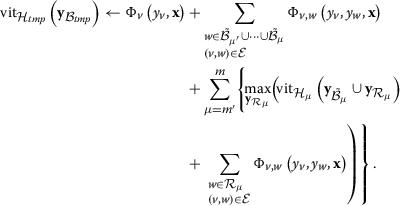 |
Figure 4.
Viterbi recursion step in case
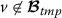 . After adding node v to the history set,
. After adding node v to the history set, 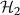 and
and 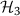 will be replaced by
will be replaced by 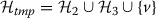 . In this example, for every assignment of the new boundary
. In this example, for every assignment of the new boundary 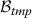 the score of
the score of 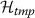 is maximized by varying over the assignments of v and
is maximized by varying over the assignments of v and 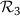 and using the Viterbi variables of
and using the Viterbi variables of 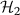 and
and 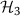 .
.
Finally, the interior labeling is stored, where the maximum is attained. The algorithm terminates after the last node v from 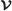 has been processed. In the typical case, where the graph is connected, at termination
has been processed. In the typical case, where the graph is connected, at termination 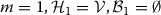 .
.
The running time of the algorithm is 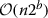 , where b is the size of the largest boundary set and n is the number of surface residues. We call this algorithm generalized Viterbi algorithm as for the case of a graph that is a linear chain 1−2−3−⋯−n of nodes using the node order 1,2,…,n the Viterbi variables we define are the same as in the standard Viterbi algorithm for HMMs. In the case of a graph that is a tree, this algorithm specializes to the Fitch algorithm or an argmax-version of Felsenstein’s pruning algorithm when a leaf-to-root node order is chosen after rooting the tree at an arbitrary node. In both special cases the boundary sets always have size at most 1. The tree example also motivate the use of several history sets at the same time: using a single history set only, one would not be able to achieve a linear running time on trees.
, where b is the size of the largest boundary set and n is the number of surface residues. We call this algorithm generalized Viterbi algorithm as for the case of a graph that is a linear chain 1−2−3−⋯−n of nodes using the node order 1,2,…,n the Viterbi variables we define are the same as in the standard Viterbi algorithm for HMMs. In the case of a graph that is a tree, this algorithm specializes to the Fitch algorithm or an argmax-version of Felsenstein’s pruning algorithm when a leaf-to-root node order is chosen after rooting the tree at an arbitrary node. In both special cases the boundary sets always have size at most 1. The tree example also motivate the use of several history sets at the same time: using a single history set only, one would not be able to achieve a linear running time on trees.
A heuristic based on the generalized Viterbi algorithm
First, it is vital for our generalized Viterbi algorithm to keep the size of the boundary sets small. A good position order is here of great importance. The algorithm starts by choosing a vertex of minimal degree. When determining the next position to be dequeued, the algorithm selects a boundary node such that the number of incident edges leading to nodes not belonging to any current history set is minimal. In an arbitrarily chosen order these nodes are dequeued next.
Second, the space demand is reduced by restricting the number of boundary labelings admitted. Starting from the available labelings of the current history set, the percentage of the reachable boundary labelings of the successor history that will be discarded is calculated. Then the corresponding percentile is estimated. To this end, a sufficiently large sample of possible labelings of the new boundary set is drawn, the Viterbi variables are computed, and the corresponding sample percentile is taken. Finally, only those boundary labelings of the new history set are retained whose Viterbi variables exceed this percentile.
That way we compute near-optimal solutions good enough for our purposes within feasible computation time.
Piecewise training for pCRFs
Let
be the independent identically distributed training sample. For every μ=1,2,…,m, let 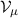 and
and 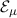 be the set of positions and edges in the neighborhood graph associated with xμ, let
be the set of positions and edges in the neighborhood graph associated with xμ, let 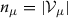 be the number of positions of the μ-th training example and let
be the number of positions of the μ-th training example and let 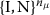 be the set of all possible label sequences of this graph.
be the set of all possible label sequences of this graph.
This data set is unbalanced as there are many more non-interface positions as interface positions. As customary for other machine learning approaches such as support vector machines and artificial neural networks [28], we here manipulated the ratio of positive and negative example positions for training in order to obtain reasonable results.
We have amplified the influence of the positive examples, rather than selecting various sets of training data by deleting negative ones as done in [28].
Let νI, νN, νII and νNN be the number of interface positions, the number of non-interface positions, the number of interface-interface edges, and the number of non-interface-non-interface edges in 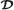 , respectively. Then we define the following two amplifier functions for all positions i and for all edges {i,j} of the m neighborhood graphs resulting from the training data
, respectively. Then we define the following two amplifier functions for all positions i and for all edges {i,j} of the m neighborhood graphs resulting from the training data 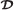 .
.
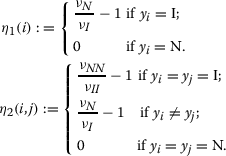 |
To uniformly govern the influence of the amplifiers, we introduce an amplifier control parameterη3∈ [ 0,1].
We set up our two log-likelihood objective function by
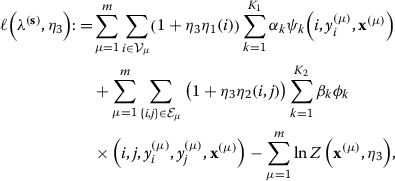 |
where ideally for each μ=1,2,…,m
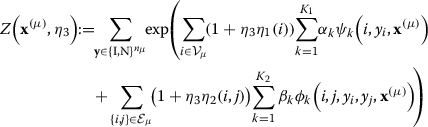 |
is the training-instance-specific normalization factor.
Unfortunately, maximizing this objective function in general is algorithmically intractable. Taking pattern from Sutton et al.
[35] who introduced what they called piecewise training, we deal with this problem by disentangling the labels of nodes and edges. For μ=1,2,…,m, a non-coherent labeling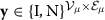 of the neighborhood graph
of the neighborhood graph 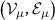 is any mapping that assigns to every position
is any mapping that assigns to every position 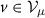 and every edge
and every edge 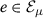 a label yv∈{I,N} and a pair of labels ye∈{I,N}2, respectively.
a label yv∈{I,N} and a pair of labels ye∈{I,N}2, respectively.
We then replace Z(x(μ),η3) by
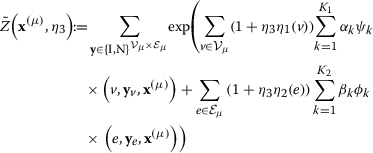 |
as normalization factor. This makes the optimization problem computationally feasible.
The L-BFGS method [38] is used to solve it. That way we obtain the coefficient vectors α and β (see Equations 3), which depend on the amplifier control parameter η3∈ [ 0,1].
To mitigate the negative consequences of disentanglement, we use a correction factorδ≥1. For any characteristics D and ι=0,1,…,γ−1, the weights of the bases edge features 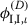 and
and 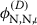 (see Equation 5) are all multiplied by δ. Thus a change in classification along an edge is additionally penalized. The correction factor δ is set best between 1.15 and 1.25.
(see Equation 5) are all multiplied by δ. Thus a change in classification along an edge is additionally penalized. The correction factor δ is set best between 1.15 and 1.25.
For our implementation of the training, we used the Java CRF package from Sunita Sarawagi at http://crf.sourceforge.net/.
Results and discussion
In this section we demonstrate effectiveness of our pCRF-based protein surface model to enhance residue-wise score-based predictions of protein-protein interfaces. For the sake of ensuring reliability of the methods we used three data sets. The first one is PlaneDimers due to Zellner et al. [25], the second one is the list of 1276 two-chain-proteins published by Keskin et al. [39], which was used by Liet al. [28] to test their linear-chain CRF. Third, we used a non-redundant data set containing 22604 unique interface structures very recently compiled by Cukuroglu et al. and published in [40].
The data set PlaneDimers is less known than the data due to Keskin et al.. It consists of redundancy-free homodimers with flat protein-protein interfaces. Zellner et al. [25] developed an SVM, called PresCont, that assigns to each residue on the protein surface a score between 0 and 1, which we refer to as PresCont score in the sequel. The larger the score, the more likely the residue belongs to the interface. Zellner et al. made the prediction by thresholding the score. The PresCont server and the data list PlaneDimers are publicly available (see http://www-bioinf.uni-regensburg.de/).
In the first subsection we describe two sets of experiments performed with synthetic data, one on PlaneDimers
[25], the other one on the list published by Keskin et al.
[39]. In both cases we independently assign to each surface position a random score drawn according to two different parametrized sequences of β-distributions Beta(αI(ς)βI(ς)) and Beta(αN(ς)βN(ς)), one for the interface sites determined by the reference labeling, the other one for the non-interface positions. The parametrized values αI(ς), αN(ς), βI(ς) and βN(ς) determining the two sequences of distributions are chosen such that the following conditions are satisfied. The mean values eI>eN are the average PresCont scores on interface sites and non-interface sites of all chains from PlaneDimers. The variances 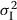 and
and 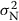 are equal to
are equal to 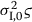 and
and 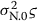 , where
, where 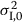 and
and 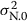 are the corresponding variances of the PresCont score, and ς∈{0.8,0.9,1.0,1.1,1.2} models the precision of the synthetic score. The deciding feature of all these distributions is that they are unimodal. The result of the subsection is that enhancement works for unimodal score distributions.
are the corresponding variances of the PresCont score, and ς∈{0.8,0.9,1.0,1.1,1.2} models the precision of the synthetic score. The deciding feature of all these distributions is that they are unimodal. The result of the subsection is that enhancement works for unimodal score distributions.
The second subsection is about a synthetic data experiment on a new data set due to Cukuroglu [40]. Here we follow the line of the first subsection except for the fact that we restrict ourselves to signal precision ς=1.0.
In the third subsection we study the PresCont scores for two-chain protein complexes from the data set PlaneDimers. According to Figure 5, the PresCont score for non-interface residues is far from being unimodal. However, if one restrict oneself to the part above a threshold in the neighborhood of 0.5 and larger, one may ask whether enhancement restricted to that domain will works. The subsection answers this question in the affirmative. Having chosen a threshold as described above, one can improve the classification with respect to this threshold as follows. Take over the prediction for scores below the threshold and reclassify the residues the scores of which are above by means of the pCRF-based enhancer.
Figure 5.
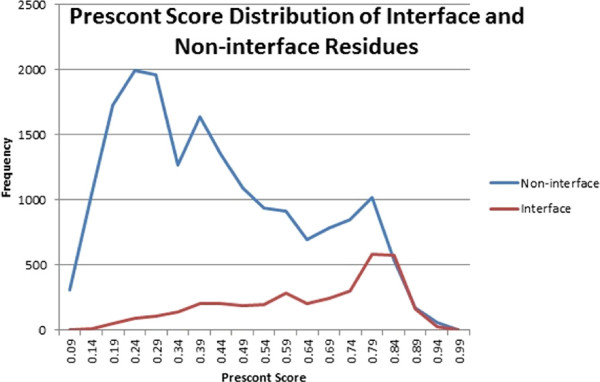
The distribution of the PresCont score for complexes from the data set PlaneDimers .
In general, observations x could encompass a PDB file, which in particular determines the 3D-structure of the protein, together with an MSA that models evolutionary aspects. In our case an observation solely consists of the PresCont score sequence or of the sequence of synthetic scores for the surface residues. Formally, every observation x is equal to a vector (ζ1,ζ2,…,ζn)∈ [ 0,1]n.
There are several neighborhood notions for residues, surface/core definitions and interface determinations in the literature. When studying the data set PlaneDimers, we follow [25]. In the case of the list due to Keskin et al. [39], the definitions according to [28] are used. Finally, when studying complexes taken from the data set published in [40], we take the following definitions. The RASA value of a surface residue is at least 15% (see [28]). Two residues are defined as contacting if the distance between any two of their atoms is less than the sum of the corresponding van der Waals radii plus 0.5 Å (see [40]).
Anyway, according to Keskin et al.
[39] we define the distance of two residues on one and the same chain as the distance of their major carbon atoms. We then say that one residue is nearby another residue, if they are at distance below 6 Å. (Note that usually residues adjacent on backbone are at distance of less than or equal to 3.5 Å). This definition in turn is the basis of the neighborhood graph 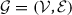 underlying the pCRF. Two surface positions are joined together by an undirected edge if and only if the corresponding residues are nearby ones.
underlying the pCRF. Two surface positions are joined together by an undirected edge if and only if the corresponding residues are nearby ones.
Our pCRF-based enhancer utilizes one position characteristic and two edge characteristics on the basis of the standard step function method explained in the Methods section. If x=(ζ1,ζ2,…,ζn)∈ [ 0,1]n is the observation associated with the protein under study, and if 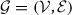 is the neighborhood graph, then for every position
is the neighborhood graph, then for every position 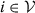 and every edge
and every edge 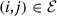 we set
we set
To enhance predictions obtained by thresholding, solely information coming from the residue neighborhood relations on the surface is additionally used.
In order to be able to calculate the performance measure of area under the ROC curve (AUC) for our pCRF-based enhancer on synthetic scores, we proceed as follows. For each edge 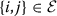 , we replace the local feature value Φi,j(I,I,x) by κΦi,j(I,I,x), where κ∈(0,∞).
, we replace the local feature value Φi,j(I,I,x) by κΦi,j(I,I,x), where κ∈(0,∞).
We enhance residue-wise score-based predictors only on the protein surface. In our synthetic data experiments there is no predictor available for core residues. For proteins taken from the data list published by Keskin et al. [39] it happens that interface sites belong to the core. That is why we use what we call Surface AUC RatioΓ of the enhancer as our performance measure for our synthetic data experiments.
If Γ is greater than 1, the enhancement was successful. The larger Γ, the greater success.
To estimate performance measures, we applied 5-fold cross-validation experiments.
A fully built-out pCRF-based tool box for modeling protein surfaces needs to comprise all the standard algorithms as e.g. forward-backward techniques, marginalization and posterior decoding known for HMMs and linear-chain CRFs. To begin with, in the fourth subsection we explain how to put a variant form of the forward algorithm and posterior decoding for pCRFs into practice.
Simulating unimodal scores of various precisions
We estimated means eI and eN and variances 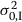 and
and 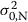 of the PresCont score on interface sites and noninterface positions of PlaneDimers, respectively, as follows.
of the PresCont score on interface sites and noninterface positions of PlaneDimers, respectively, as follows.
| 7 |
We randomly chose 120 instances under the uniform distribution from the data set published by Keskin et al. [39] to perform our experiments. Let us refer to this set as KL-subset in the sequel. (It is accessible at http://ppicrf.informatik.uni-goettingen.de/index.html).
Zellner et al. [25] used the following determinations. A residue is defined to be part of the protein surface, if its relative solvent-accessible surface area is at least 5% [17]. A surface residue is said to constitute an inter-facial contact, if there exists at least one atom of this residue which has a van-der-Waals-sphere at a distance of at most 0.5 Å from the van-der-Waals sphere to any atom from a partner chain residue [39].
Based on [3, 12, 15, 20, 41], Li et al. [28] assume an inter-facial contact of a residue on a chain is assumed to be there, if any heavy atom of this residue is at distance of at most 5 Å from any heavy atom from a partner chain. The relative solvent-accessible surface area of surface residue is at least 15%.
We independently assigned to each interface surface residue of the two data sets a random score between zero and one according to the β-distribution Beta(αI(ς)βI(ς)), and to every non-interface surface residue a score according to Beta(αN(ς)βN(ς)), where the score precision ς satisfies
| 8 |
and the parameters αI(ς),βI(ς),αN(ς),βN(ς) were chosen such that
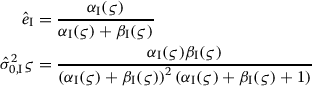 |
9 |
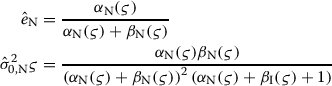 |
10 |
The Surface AUC Ratios of the enhancer compared with the threshold predictor on PlaneDimers and the KL-subset are displayed in Table 1. There is an improvement of 8.4%−9.3% on PlaneDimers and of 3.2%−5.0% on the KL-subset.
Table 1.
Classification results on PlaneDimers and the KL-subset, where the β -distributions according to which the synthetic scores were drawn are defined by Equations 7, 8, 9 and 10
| PlaneDimers | Score precisionς | 0 .8 | 0 .9 | 1 .0 | 1 .1 | 1.2 |
|---|---|---|---|---|---|---|
| Surface AUC ratio Γ | 1.084 | 1.091 | 1.093 | 1.089 | 1.093 | |
| Kl-subset | Signal precision ς | 0.8 | 0.9 | 1.0 | 1.1 | 1.2 |
| Surface AUC ratio Γ | 1.032 | 1.039 | 1.045 | 1.045 | 1.050 |
Depending on the variances determined by ς, the enhancer increases the AUC referred to the protein surface by 8.4%-9.3%. on PlaneDimers, and by 3.2%-5.0% on the KL-subset.
Moreover, we compared individual classification results obtained by thresholding the scores with pCRF-based enhanced predictions. Because of the fact that the specificity of the threshold predictor can be easily changed by manipulating the threshold, we proceeded as follows. For every score precision, the pCRF-based enhancer has a well-defined specificity referred to the surface residues. We then chose the threshold such that the specificity of the threshold predictor is close to that of the enhancer. The results are shown in Table 2. The sensitivity is increased by 53%−67% on the data set PlaneDimers and by 14%−22% on the KL-subset.
Table 2.
Comparing the enhancer with the threshold classifier of approximately equal specificity on synthetic scores assigned to surface residues of protein complexes taken from the data set PlaneDimers and the KL-subset
| Data Set | Score Precisionς | Classifier | Specificity | Sensitivity | MCC |
|---|---|---|---|---|---|
| 0.8 | Threshold Predictor | 0.9672 | 0.2562 | 0.3253 | |
| Enhancer | 0.9666 | 0.4281 | 0.4911 | ||
| PlaneDimer | 0.9 | Threshold Predictor | 0.9618 | 0.2556 | 0.3077 |
| Enhancer | 0.9624 | 0.4086 | 0.4610 | ||
| 1.0 | Threshold Predictor | 0.9611 | 0.2428 | 0.2912 | |
| Enhancer | 0.9612 | 0.3872 | 0.4379 | ||
| 1.1 | Threshold Predictor | 0.9681 | 0.2100 | 0.2753 | |
| Enhancer | 0.9677 | 0.3307 | 0.4045 | ||
| 1.2 | Threshold Predictor | 0.9649 | 0.2100 | 0.2648 | |
| Enhancer | 0.9647 | 0.3213 | 0.3854 | ||
| 0.8 | Threshold Predictor | 0.9568 | 0.2936 | 0.3549 | |
| Enhancer | 0.9577 | 0.3586 | 0.4210 | ||
| KL-subset | 0.9 | Threshold Predictor | 0.9533 | 0.2843 | 0.3369 |
| Enhancer | 0.9531 | 0.3290 | 0.3820 | ||
| 1.0 | Threshold Predictor | 0.9570 | 0.2559 | 0.3152 | |
| Enhancer | 0.9571 | 0.2971 | 0.3591 | ||
| 1.1 | Threshold Predictor | 0.9615 | 0.2279 | 0.2949 | |
| Enhancer | 0.9614 | 0.2743 | 0.3459 | ||
| 1.2 | Threshold Predictor | 0.9604 | 0.2199 | 0.2828 | |
| Enhancer | 0.9599 | 0.2516 | 0.3175 |
Table 1 and Table 2 justify the following conclusion. Enhancing the threshold prediction by our pCRF works provided that the distributions of the interface scores as well as the non-interface scores are unimodal. The enhancement for the data set PlaneDimers is larger than for the KL-subset. This might be caused by the plain interface geometry of the complexes taken from PlaneDimers.
Utilizing the new data set due to Cukuroglu [40]
As in the case of the KL-subset, we randomly chose 60 dimers. We refer to the resulting list as CGNK-subset. Having assigned synthetic scores according to Equations 7, 9 and 10, where ς=1.0, we compared individual classification results obtained by thresholding the scores with pCRF-based enhanced predictions in exactly the same way as we did for the KL-subset. The results are shown in Table 3. The sensitivity is increased by 22%.
Table 3.
Comparing the enhancer with the threshold classifier of approximately equal specificity on synthetic scores assigned to surface residues of protein complexes taken from the CGNK-subset
| Classifier | Specificity | Sensitivity | MCC |
|---|---|---|---|
| Threshold predictor | 0.9399 | 0.3782 | 0.3387 |
| Enhancer | 0.9400 | 0.3104 | 0.2767 |
A main finding of Cukuroglu [40] relevant to protein-protein interface prediction is, that the average interface RASA value is greater than 40%. Since our method is designed to improve performance of a given residue-wise predictor, using this result is not in the scope of this paper. However, a CRF-based predictor integrating features for cliques of size greater than 2 is not beyond the range of current algorithmic capabilities. In such a model a feature set that discretizes the mean RASA value of cliques is promising.
Enhancing the PresContserver prediction on PlaneDimers
For the sake of completeness, we shortly review the residue characteristics used by PresCont.
Relative solvent-accessible surface area
For any residue a, the solvent-accessible surface areaasa(a) can be computed by e.g. the software library BALL [42]. Most of the classifiers known from the literature utilize this characteristic (see [43]). For PresCont the relative solvent-accessible surface area according to
| 11 |
is taken into operation, where asamax(a) is the maximally possible accessible surface area of residue a [44].
Hydrophobicity
Many interfaces possess a hydrophobic core surrounded by a ring of polar residues [45, 46]. In order to reduce noise, in [25] the contribution of hydrophobic patches rather then the influence of individual residues is utilized.
Residue conservation
Measures of this type utilized in [25] are the Shannon entropy and the relative Shannon entropy of empirical residue distributions in MSA columns. As an alternative, empirical expectations of BLOSUM-based similarities are taken for them.
Scores of local neighborhoods
They are evaluated by means of log-odd ratios of neighboring residue pair frequencies in interfaces as opposed to residue pair frequencies on complementary protein surface areas. The resulting scores are averaged both over the neighborhood of the positions under study and the rows of the MSA associated with the protein.
On the basis of Figure 5 we enhanced PresCont for thresholds θ∈ [ 0.500,0.625]. The decisive factor for this choice is that the PresCont score distributions for interface sites as well as non-interface positions above θ are “sufficiently close to” unimodal distributions. For every such θ, we set all scores less than or equal to θ to zero and then left the classification of all surface residues to the pCRF modified as follows. The residues of score zero are not taken into account when it comes to discretizing the protein characteristics (see Equations 4 and 5). Let us call this enhancing aboveθ.
To evaluate improvements we proceeded as when compiling Table 2. For every threshold θ under consideration another threshold θ′ was chosen such that thresholding at θ′ has the same specificity as enhancing above θ. The results are displayed in Table 4 and visualized for an individual protein by Figure 6. According to Table 4 the increase in sensitivity ranges from 4% to 7%. The true-positive predictions on the surface of the protein with PDB-Entry 1QM4 are compared in Figure 6, where again the specificity of the two classifiers is the same.
Table 4.
Enhancing above various thresholds on PlaneDimers , where PresCont ’s threshold was chosen such that the specificity approximately equals that of enhancing
| tp | tn | fp | fn | Spec. | Sen. | MCC | ||
|---|---|---|---|---|---|---|---|---|
| Enhancing above 0.500 | 2181 | 23182 | 4145 | 1414 | 0.848 | 0.607 | 0.362 | |
| PresCont | 2100 | 23197 | 4130 | 1495 | 0.849 | 0.584 | 0.346 | |
| Enhancing above 0.525 | 2303 | 22917 | 4410 | 1292 | 0.839 | 0.641 | 0.373 | |
| PresCont | 2206 | 22912 | 4415 | 1389 | 0.838 | 0.614 | 0.353 | |
| Enhancing above 0.550 | 2507 | 22103 | 5224 | 1088 | 0.809 | 0.697 | 0.375 | |
| PresCont | 2419 | 22102 | 5225 | 1176 | 0.809 | 0.673 | 0.358 | |
| Enhancing above 0.575 | 2560 | 21992 | 5335 | 1035 | 0.805 | 0.712 | 0.380 | |
| PresCont | 2463 | 21915 | 5412 | 1132 | 0.802 | 0.685 | 0.358 | |
| Enhancing above 0.600 | 2379 | 22685 | 4642 | 1216 | 0.830 | 0.662 | 0.376 | |
| PresCont | 2253 | 22780 | 4547 | 1342 | 0.834 | 0.627 | 0.356 | |
| Enhancing above 0.625 | 2287 | 23044 | 4283 | 1308 | 0.843 | 0.636 | 0.376 | |
| PresCont | 2136 | 23049 | 4278 | 1459 | 0.843 | 0.594 | 0.346 |
The sensitivity increased that way by 4%-7%. For every pair of experiments, the number of true negatives (tn), false negatives (fn), false positives (fp) and true positives (tp) are displayed.
Figure 6.
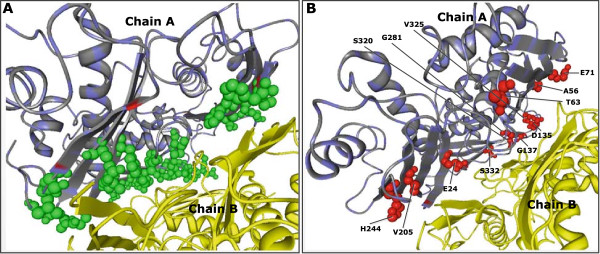
Comparison of enhancer and PresCont service of same specificity on the protein with PDB-Entry 1QM4. (A) Green spheres on the left show the interface surface residues correctly predicted by both tools. (B) Red spheres on the right indicate additional true positives of the enhancer.
Discussing posterior decoding
As in the case of linear-chain CRFs, the generalized Viterbi algorithm can be transformed into a variant form of the forward algorithm. It might be the case that the following additional problem arises.
Let v1,v2,…,vn be the ordering in which the positions of  are traversed by the algorithm, and let
are traversed by the algorithm, and let  denote the set of position indices i<n such that vi is not an element of the boundary
denote the set of position indices i<n such that vi is not an element of the boundary  of the history set
of the history set 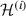 at stage i. If
at stage i. If 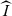 is not empty, we encounter an obstacle when it comes to sampling label sequences. For
is not empty, we encounter an obstacle when it comes to sampling label sequences. For 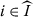 , position vi is not labeled in the course of the sampling procedure. That is why we augment the neighborhood graph
, position vi is not labeled in the course of the sampling procedure. That is why we augment the neighborhood graph 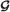 so that those positions no longer exist, all predictions remain unchanged, and the order of magnitude of the running time is not increased. To this end, we complement the ordering v1,v2,…,vn as follows. For every
so that those positions no longer exist, all predictions remain unchanged, and the order of magnitude of the running time is not increased. To this end, we complement the ordering v1,v2,…,vn as follows. For every 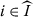 , we insert a new node
, we insert a new node 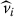 between vi and vi+1. Having extended the neighborhood graph by these nodes not being associated with residue positions of the protein under study and by new edges
between vi and vi+1. Having extended the neighborhood graph by these nodes not being associated with residue positions of the protein under study and by new edges 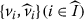 , where for
, where for 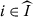 and
and 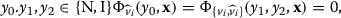 the above mentioned obstacle is eliminated without any influence on the prediction and the order of magnitude of the running time.
the above mentioned obstacle is eliminated without any influence on the prediction and the order of magnitude of the running time.
Proceeding now in a way analogous to the classical case, in every formula that is a building block of the generalized Viterbi algorithm the following two steps of replacement need to be performed.
First, for every position 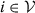 , every edge
, every edge 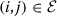 , every label y0∈{I,N}, and every label pair (y1,y2)∈{I,N}2, we replace Φi(y0,x) with exp(Φi(y0,x)), and Φ{i,j}(y1,y2,x) with exp(Φ{i,j}(y1,y2,x)).
, every label y0∈{I,N}, and every label pair (y1,y2)∈{I,N}2, we replace Φi(y0,x) with exp(Φi(y0,x)), and Φ{i,j}(y1,y2,x) with exp(Φ{i,j}(y1,y2,x)).
Second, we replace sums with products and then maxima with sums.
Thus we obtain as analogues of the Viterbi variables 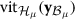 defined by Equation 6 what we call component forward variables
defined by Equation 6 what we call component forward variables 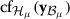 .
.
If 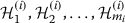 and
and 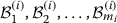 are the connected components of the history set
are the connected components of the history set 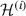 and the corresponding boundary set
and the corresponding boundary set 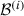 at stage i∈{1,2,…,n}, respectively, then the forward variable at stage i with respect to a boundary assignment
at stage i∈{1,2,…,n}, respectively, then the forward variable at stage i with respect to a boundary assignment 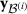 is defined as
is defined as
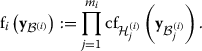 |
For any assignment 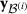 (i>1), the forward variable
(i>1), the forward variable 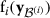 is a nontrivial linear combination of forward variables
is a nontrivial linear combination of forward variables 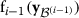 , where
, where 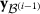 ranges over some assignments of the boundary set
ranges over some assignments of the boundary set 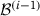 at stage i−1. Analogous to the linear-chain case, a random backward walk through a state graph, with all possible assignments
at stage i−1. Analogous to the linear-chain case, a random backward walk through a state graph, with all possible assignments 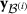 (i=n,n−1,…,1) being the set of nodes, results in a random labeling of the positions, where each labeling is drawn with its posterior probability.
(i=n,n−1,…,1) being the set of nodes, results in a random labeling of the positions, where each labeling is drawn with its posterior probability.
This sampling technique allows the efficient calculation of posterior probabilities at nodes and edges in a straightforward manner.
Conclusions
Residue-wise score-based threshold predictors of protein-protein interaction sites assign to each residue of the protein under study a score. The classification is then made by thresholding the score. In case of using probabilistic data models, the parameters of the threshold predictor have been learned on a training data set in advance.
We have demonstrated that such threshold predictors can be improved by pCRF-based enhancers given the shape of the interface surface score distribution and the non-interface surface score distribution with respect to the training set resemble the shape of unimodal distributions. Besides the surface residue scores, only the spatial neighborhood structure between the surface residues of the protein under study is taken into account. Thus, the improvement can be attributed to our model. In addition to the precision of the scores, the amount of improvement depends on the 3D-complexity of the interfaces to be predicted. To this end, three sets of experiments with synthetic surface residue scores for protein complexes randomly chosen from the data set PlaneDimers compiled by Zellner et al. [25] and from the lists published by Keskin et al. [39] and Cukuroglu et al. [40].
The enhancement is structurally based on the following model property of pCRFs in contrast to residue-wise predictors. Though the scores of near-by residues may be correlated, labeling a position as interface or non-interface by thresholding the score does not influence the classification of its neighbors. When using pCRFs, this is the case.
The pCRF-based enhancer is also applicable, if the score distributions are only unimodal over a certain sub-domain. The improvement is then restricted to that domain. Thus we were able to improve the prediction of the PresCont server devised by Zellner et al. on PlaneDimers [25].
The prediction is made on grounds of a generalized Viterbi inference heuristic. As for training, we developed a piecewise training procedure for pCRFs, where the enhancer needs to be trained on data originating from the same source as the training data of the threshold predictor to be improved.
A prototypical implementation of our pCRF-based method is accessible at http://ppicrf.informatik.uni-goettingen.de/index.html.
Acknowledgements
Zhijie Dong acknowledges financial support by the Deutsche Forschungsgemeinschaft (Research Training Group 1023 “Identification in Mathematical Models: Synergy of Stochastic and Numerical Methods”). Moreover, we thank Rainer Merkl and Hermann Zellner from Regensburg, who supported us in gaining a deeper understanding of the PresCont service. Special thank goes to Moritz Manecke for a first implementation of the generalized Viterbi algorithm. Finally, we acknowledge with thanks the valuable suggestions and comments of the unknown referees.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The model was designed by ZD and KW, who also adapted piecewise training to pCRFs and performed the analysis of the first submission. The tool was designed and implemented by ZD and KW, who were supported by MW and TW. The algorithms were devised by MG, MS, MW and SW. The generalized Viterbi algorithm, however, was conceived by MS. The Web server was set up by MG. The analysis necessary for the revised version was performed by TKLD, who was supported by MW. The manuscript was drafted by ZD and KW, written by MS and SW and revised by TW. SW conceived the study. All authors read and approved the final manuscript.
Contributor Information
Zhijie Dong, Email: zdong@informatik.uni-goettingen.de.
Keyu Wang, Email: kwang@informatik.uni-goettingen.de.
Truong Khanh Linh Dang, Email: t.dang@stud.uni-goettingen.de.
Mehmet Gültas, Email: mehmet.gueltas@bioinf.med.uni-goettingen.de.
Marlon Welter, Email: marlon.welter@cs.uni-goettingen.de.
Torsten Wierschin, Email: torsten.wierschin@uni-greifswald.de.
Mario Stanke, Email: mario.stanke@uni-greifswald.de.
Stephan Waack, Email: waack@informatik.uni-goettingen.de.
References
- 1.Sowa ME, He W, Slep KC, Kercher MA, Lichtarge O, Wensel TG. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat Struct Biol. 2001;8:234–237. doi: 10.1038/84974. [DOI] [PubMed] [Google Scholar]
- 2.Zhou HX. Improving the understanding of human genetic diseases through predictions of protein structures and protein-protein interaction sites. Curr Med Chem. 2004;11:539–549. doi: 10.2174/0929867043455800. [DOI] [PubMed] [Google Scholar]
- 3.Zhou HX, Qin S. Interaction-site prediction for protein complexes: a critical assessment. Bioinformatics. 2007;23(17):2203–2209. doi: 10.1093/bioinformatics/btm323. [DOI] [PubMed] [Google Scholar]
- 4.Li JJ, Huang DS, Wang B, Chen P. Identifying protein-protein interfacial residues in heterocomplexes using residue conservation scores. Int J Biol Macromol. 2006;38(3–5):241–247. doi: 10.1016/j.ijbiomac.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 5.Kufareva I, Budagyan L, Raush E, Totrov M, Abagyan R. PIER: protein interface recognition for structural proteomics. Proteins. 2007;67(2):400–417. doi: 10.1002/prot.21233. [DOI] [PubMed] [Google Scholar]
- 6.Burgoyne NJ, Jackson RM. Predicting protein interaction sites: binding hot-spots in protein-protein and protein-ligand interfaces. Bioinformatics. 2006;22(11):1335–1342. doi: 10.1093/bioinformatics/btl079. [DOI] [PubMed] [Google Scholar]
- 7.de Vries SJ, van Dijk AD, Bonvin AM. WHISCY: what information does surface conservation yield? Application to data driven docking. Proteins. 2006;63(3):479–489. doi: 10.1002/prot.20842. [DOI] [PubMed] [Google Scholar]
- 8.Hoskins J, Lovell S, Blundell TL. An algorithm for predicting protein-protein interaction sites: abnormally exposed amino acid residues and secondary structure elements. Protein Sci. 2006;15(5):1017–1029. doi: 10.1110/ps.051589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33(Web-Server-Issue):299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang SL, Zhang C, Liu S, Zhou Y. Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 2006;34(13):3698–3707. doi: 10.1093/nar/gkl454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murakami Y, Jones S. SHARP²: protein-protein interaction predictions using patch analysis. Bioinformatics. 2006;22(14):1794–1795. doi: 10.1093/bioinformatics/btl171. [DOI] [PubMed] [Google Scholar]
- 12.Zhou HX, Shan Y. Prediction of protein interaction sites from sequence profile and residue neighbor list. Protein Struct Funct Genet. 2001;44:336–243. doi: 10.1002/prot.1099. [DOI] [PubMed] [Google Scholar]
- 13.Fariselli P, Pazos F, Valencia A, Casadio R. Eur J Biochem. 2002. Prediction of protein-protein interaction sites in heterocomplexes with neural networks. [DOI] [PubMed] [Google Scholar]
- 14.Ofran Y, Rost B. Predicted protein-protein interaction sites from local sequence information. FEBS Lett. 2003;544:236–239. doi: 10.1016/S0014-5793(03)00456-3. [DOI] [PubMed] [Google Scholar]
- 15.Chen H, Zhou HX. Prediction of interface residues in protein-protein complexes by a consensus neural network: test against NMR data. Protein Struct Funct Genet. 2005;61:21–35. doi: 10.1002/prot.20514. [DOI] [PubMed] [Google Scholar]
- 16.Ofran Y, Rost B. ISIS: interaction sites identified from sequence. Bioinformatics. 2007;23(2):13–16. doi: 10.1093/bioinformatics/btl303. [DOI] [PubMed] [Google Scholar]
- 17.Porollo A, Meller J. Prediction-based fingerprints of protein-protein interactions. Protein Struct Funct Genet. 2007;66:630–645. doi: 10.1002/prot.21248. [DOI] [PubMed] [Google Scholar]
- 18.Bordner A, Abagyan R. Statistical analysis and prediction of protein-protein interfaces. Protein Struct Funct Genet. 2005;60:353–366. doi: 10.1002/prot.20433. [DOI] [PubMed] [Google Scholar]
- 19.Bradford J, Westhead D. Improved prediction of protein-protein binding sites using a support vector machines approach. Bioinformatics. 2005;21(8):1487–1494. doi: 10.1093/bioinformatics/bti242. [DOI] [PubMed] [Google Scholar]
- 20.Chung JL, Wang W, Bourne PE. Exploiting sequence and structure homologs to identify protein-protein binding sites. Proteins. 2006;62:630–640. doi: 10.1002/prot.20741. [DOI] [PubMed] [Google Scholar]
- 21.Koike A, Takagi T. Prediction of protein-protein interaction sites using support vector machines. Protein Eng Design Selec. 2004;17(2):165–173. doi: 10.1093/protein/gzh020. [DOI] [PubMed] [Google Scholar]
- 22.Res I, Mihalek I, Lichtarge O. An evolution-based classifier for prediction of protein interfaces without using protein structures. Bioinformatics. 2005;21(10):2496–2501. doi: 10.1093/bioinformatics/bti340. [DOI] [PubMed] [Google Scholar]
- 23.Wang B, Wong HS, Huang DS. Inferring protein-protein interaction sites using residue conservation and evolutionary information. Protein Pept Lett. 2006;13(10):999–1005. doi: 10.2174/092986606778777498. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Chen P, Huang DS, Li JJ, Lok TM, Lyu MR. Predicting protein interaction sites from residue spatial sequence profile and evolution rate. FEBS Lett. 2006;580(2):380–384. doi: 10.1016/j.febslet.2005.11.081. [DOI] [PubMed] [Google Scholar]
- 25.Zellner H, Staudigel M, Trenner M, Bittkowski M, Wolowski V, Icking M, Merkl R. PresCont: Predicting Protein-Protein Interfaces Utilizing Four Residue Properties. Proteins: Struct Funct Bioinformatics. 2011;80(1):154–168. doi: 10.1002/prot.23172. [DOI] [PubMed] [Google Scholar]
- 26.Neuvirth H, Raz R, Schreiber G. ProMate: a structure based prediction program to identify the location of protein-protein binding sites. J Mol Biol. 2004;338(1):181–199. doi: 10.1016/j.jmb.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 27.Bradford JR, Needham CJ, Bulpitt AJ, Westhead DR. Insights into protein-protein interfaces using a Bayesian network prediction method. J Mol Biol. 2006;362(2):365–386. doi: 10.1016/j.jmb.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 28.Li MH, Lin L, Wang XL, Liu T. Protein-protein interaction site prediction based on conditional random fields. Bioinformatics. 2007;23(5):597–604. doi: 10.1093/bioinformatics/btl660. [DOI] [PubMed] [Google Scholar]
- 29.Hwang H, Vreven T, Weng Z. Proteins: Struct Funct Bioinformatics. 2013. Binding interface prediction by combining protein-protein docking results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lafferty JD, McCallum A, Pereira FCN. Conditional random fields: probabilistic models for segmenting and labeling sequence data. In: Brodley CE, editor. Proceedings of the Eighteenth International Conference on Machine Learning. San Francisco, CA, USA: Danyluk AP. Morgan Kaufmann Publishers Inc.; 2001. pp. 282–289. [Google Scholar]
- 31.Sutton C, McCallum A. Introduction to Statistical Relational Learning. Cambridge, Massachusetts, USA: MIT Press; 2006. [Google Scholar]
- 32.McCallum A, Li W. Proceedings of the seventh conference on Natural language learning at HLT-NAACL 2003 - Volume 4, CONLL ‘03. Stroudsburg: Association for Computational Linguistics; 2003. Early results for named entity recognition with conditional random fields, feature induction and web-enhanced lexicons; pp. 188–191. [Google Scholar]
- 33.Dietterich TG, Ashenfelter A, Bulatov Y. Training conditional random fields via gradient tree boosting. In: Brodley CE, editor. Proceedings of the Twenty-first International Conference on Machine Learning, Volume 69 of ACM International Conference Proceeding Series. New York, NY, USA: ACM; 2004. pp. 28–28. [Google Scholar]
- 34.Zhu J, Nie Z, Wen JR, Zhang B, Ma WY. 2D Conditional, Random Fields for Web information extraction. In: Raedt LD, Wrobel S, editors. Proceedings of the 22Nd International Conference on Machine Learning, Volume 119 of ACM International Conference Proceeding Series. New York, NY, USA: ACM; 2005. pp. 1044–1051. [Google Scholar]
- 35.Sutton C, McCallum A. UAI ’05, Proceedings of the 21st Conference on Uncertainty in Artificial Intelligence (UAI), Edinburgh, Scotland, July 26-29. 2005. Piecewise training of undirected models; pp. 568–575. [Google Scholar]
- 36.McCallum A, Rohanimanesh K, Sutton C. NIPS-2003 Workshop on Syntax, Semantics and Statistics. 2003. Dynamic conditional random fields for jointly labeling multiple sequences. [Google Scholar]
- 37.Sha F, Pereira F. Shallow parsing with conditional random fields. 2003. [Google Scholar]
- 38.Liu DC, Nocedal J. On the limited memory BFGS method for large scale optimization. Math Program. 1989;45:503–528. doi: 10.1007/BF01589116. [DOI] [Google Scholar]
- 39.Keskin O, Tsai CJ, Wolfson H, Nussinov R. A new, structurally nonredundant, diverse data set of protein-protein interfaces and its implications. Protein Sci. 2004;13:1043–1055. doi: 10.1110/ps.03484604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cukuroglu E, Gursoy A, Nussinov R, Keskin O. Non-redundant unique interface structures as templates for modeling protein interactions. PLoS ONE. 2014;9:e86738. doi: 10.1371/journal.pone.0086738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rost B, Sander C. Conservation and prediction of solvent accessibility in protein families. Proteins: Struct Funct Bioinformatics. 1994;20(3):216–226. doi: 10.1002/prot.340200303. [DOI] [PubMed] [Google Scholar]
- 42.Hildebrandt A, Dehof AK, Rurainski A, Bertsch A, Schumann M, Toussaint NC, Moll A, Stöckel D, Nickels S, Mueller SC, Lenhof HP, Kohlbacher O. BALL - biochemical algorithms library 1.3. BMC Bioinformatics. 2010;11:531. doi: 10.1186/1471-2105-11-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xia JF, Zhao XM, Song J, Huang DS. APIS: accurate prediction of hot spots in protein interfaces by combining protrusion index with solvent accessibility. BMC Bioinformatics. 2010;11:174. doi: 10.1186/1471-2105-11-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller S, Janin J, Lesk AM, Chothia C. Interior and surface of monomeric proteins. J Mol Biol. 1987;196(3):641–656. doi: 10.1016/0022-2836(87)90038-6. [DOI] [PubMed] [Google Scholar]
- 45.Larsen TA, Olson AJ, Goodsell DS. Morphology of protein–protein interfaces. Structure. 1998;6(4):421–427. doi: 10.1016/S0969-2126(98)00044-6. [DOI] [PubMed] [Google Scholar]
- 46.Bouvier B, Grünberg R, Nilges M, Cazals F. Shelling the Voronoi interface of protein–protein complexes reveals patterns of residue conservation, dynamics, and composition. Proteins: Struct Funct Bioinformatics. 2009;76(3):677–692. doi: 10.1002/prot.22381. [DOI] [PubMed] [Google Scholar]