

Abstract
Lung adenocarcinoma is comprised of distinct mutational subtypes characterized by mutually exclusive oncogenic mutations in RTK/RAS pathway members KRAS, EGFR, BRAF and ERBB2, and translocations involving ALK, RET and ROS1. Identification of these oncogenic events has transformed the treatment of lung adenocarcinoma via application of therapies targeted toward specific genetic lesions in stratified patient populations. However, such mutations have been reported in only ∼55% of lung adenocarcinoma cases in the United States, suggesting other mechanisms of malignancy are involved in the remaining cases. Here we report somatic mutations in the small GTPase gene RIT1 in ∼2% of lung adenocarcinoma cases that cluster in a hotspot near the switch II domain of the protein. RIT1 switch II domain mutations are mutually exclusive with all other known lung adenocarcinoma driver mutations. Ectopic expression of mutated RIT1 induces cellular transformation in vitro and in vivo, which can be reversed by combined PI3K and MEK inhibition. These data identify RIT1 as a driver oncogene in a specific subset of lung adenocarcinomas and suggest PI3K and MEK inhibition as a potential therapeutic strategy in RIT1-mutated tumors.
Supplementary information
The online version of this article (doi:10.1038/onc.2013.581) contains supplementary material, which is available to authorized users.
Keywords: lung adenocarcinoma, cancer genetics, RAS pathway, signal transduction, oncogene, GTPase
Subject terms: Mutation, Cancer genetics, Cell signalling, Lung cancer
Introduction
Lung adenocarcinomas contain characteristic, mutually exclusive mutations in receptor tyrosine kinase (RTK) and RAS pathway oncogenes including frequent mutation of KRAS or EGFR in ∼30 or 10% of US cases, respectively.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 Rarely, translocations are seen involving EML4 and ALK (1–4%), RET (1%) or ROS1 (2%)2, 5 (The Cancer Genome Atlas, manuscript submitted). Additional rare oncogenic mutations are seen in HRAS, NRAS, BRAF, ERBB2, MET and MAP2K1.1, 2 Together these oncogenic events are found in ∼55% of lung adenocarcinomas in the US population.2 The importance of identifying such ‘driver’ mutations is exemplified by the success of targeted therapies directed against these events—the prototypical example being the use of the small molecule EGFR inhibitors Iressa (gefitinib) and Tarceva (erlotinib) in EGFR-mutated lung adenocarcinoma.14 Among patients with EGFR mutations, single-line EGFR-targeted therapy significantly extends progression-free survival compared with conventional chemotherapy.7, 8 Patients without EGFR mutations, however, typically do not benefit from targeted therapy, underscoring the need to genotype patient tumor resections and/or biopsies to guide appropriate treatment decisions. Modeling this paradigm, clinical trials of the ALK inhibitor crizotinib for treatment of tumors with EML4-ALK translocation were rapidly reported15 only 3 years after the identification of the EML4-ALK event.5 Therefore, continued identification of oncogenic driver genes in the remaining ∼45% of ‘oncogene-negative’ lung adenocarcinoma cases may allow further patient stratification and targeted therapies.
Recent genomic analyses of lung adenocarcinoma have identified novel mutations in diverse genes, but few additional oncogenic mutations have been identified that display mutual exclusivity with known lung adenocarcinoma driver oncogenes.1, 16 Here we report the genomic and functional characterization of somatic RIT1 mutations that we identify as novel driver mutations in lung adenocarcinoma.
Results
To identify rare oncogenic driver mutations, we and others in The Cancer Genome Atlas (TCGA) Research Network performed a targeted analysis of ‘oncogene-negative’ tumors that lack mutations in the known lung adenocarcinoma driver oncogenes KRAS, EGFR, BRAF, ERBB2, MAP2K1, MET, HRAS, NRAS and/or fusions involving ALK, RET or ROS1 (TCGA, manuscript submitted). In 87 ‘oncogene-negative’ samples lacking mutations/fusions in the known driver genes, RIT1 (Ras-like in all tissues) was among the top significantly mutated genes. 5/87 samples in this group harbored somatic RIT1 mutations. In contrast, no mutations in RIT1 were found in the 143 samples containing known driver oncogenic mutations (P<0.01 by Fisher’s exact test). Moreover, we identified five additional RIT1-mutant samples in sequence data from an independent cohort of 183 lung adenocarcinomas.1
In total, non-synonymous somatic mutations of RIT1 were identified in 10/413 (2.4%) lung adenocarcinomas subjected to whole exome sequencing (Figure 1a, Supplementary Table 1). Mutations in RIT1 consisted of missense mutations and small in-frame insertion/deletions. In all, 7/10 RIT1 mutations were clustered near the switch II domain region (Figures 1a and b), including three recurrent p.M90I mutations. The switch II domain in RIT1 is homologous to the switch II region in RAS genes that contains glutamine 61 (Supplementary Figure 1), which is directly involved in GTP hydrolysis and also a hotspot of mutation in RAS genes.17 This switch II domain hotspot is distinct from the P29 hotspot in RAC genes recently reported in melanoma and other cancers18, 19, 20 (Supplementary Figure 1).
Figure 1.
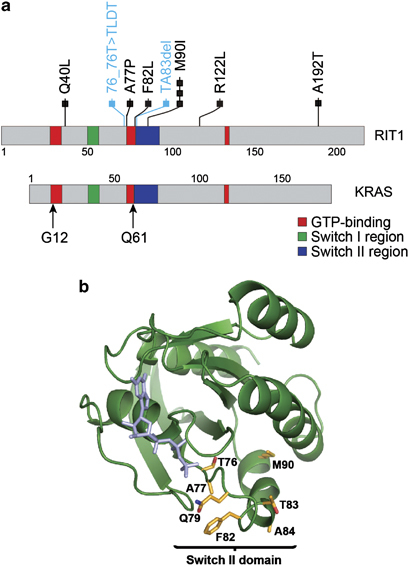
Somatic RIT1 mutations in lung adenocarcinoma. (a) 2D protein structure schematics of RIT1 and KRAS with major protein domains shaded as indicated. Numbers indicate amino-acid positions. Each box represents an independent somatic RIT1 mutation with missense and in-frame indel mutations represented in black or blue, respectively. Arrows indicate two mutational hotspots in KRAS. (b) Predicted protein structure of RIT1. A homology model of RIT1 was generated from an alignment with HRAS (1AGP) using SWISS model and displayed using PYMOL. Amino acids near the switch II domain found mutated in this study are highlighted in yellow, in addition to Q79, which is shown for reference.
Recurrent alteration of RIT1 alanine 77 was also observed; one of the ten mutated samples from this analysis harbored a p.A77P mutation and an additional TCGA sample (TCGA-73-4666) harbored a p.A77S mutation. TCGA-73-4666 was excluded from the 230 tumor TCGA data set due to lack of data from its paired normal sample. We hence cannot rule out that p.A77S represents a rare germline variant, though it is unlikely given its absence in sequence data generated from 1092 normal genomes by the 1000 Genomes Project.21
All seven samples with switch II domain RIT1 mutations lacked oncogenic driver mutations in EGFR, KRAS, BRAF, ERBB2, HRAS, NRAS and MAP2K1. This pattern of mutational mutual exclusivity is consistent with the possibility that RIT1 switch II domain mutations may function as RAS/RTK pathway lung adenocarcinoma oncogenes. However, one of three non-switch II domain mutations, p.R122L, co-occurred with a KRAS mutation (Supplementary Figure 2).
We surveyed recent exome sequencing data from other diverse cancer types to determine whether RIT1 is mutated in malignancies other than lung adenocarcinoma. Most tumor types had no mutations, or rare RIT1 variants of unknown significance (Supplementary Table 2). However, one sample each from acute myelogenous leukemia22 and cervical carcinoma sequencing studies harbored the RIT1 p.M90I variant seen recurrently in lung adenocarcinoma. In line with these observations, somatic RIT1 mutation was recently reported in myeloid malignancies.23 The mutations observed in myeloid malignancies overlap with the mutations we report in lung adenocarcinoma (F82L, M90I) and include additional mutations clustering in the switch II domain.
To test whether mutated RIT1 is capable of inducing cellular transformation, we expressed wild-type or mutated RIT1 cDNA constructs in NIH3T3 cells and assayed the ability of these cells to form colonies in soft agar. A constitutively active form of RIT1, RIT1 Q79L,24 was used as a positive control. With the exception of RIT1 Q40L, all other RIT1 mutations robustly induced colony formation of NIH3T3 cells in soft agar (Figure 2a). To confirm that these cells were indeed transformed, we injected RIT1-transduced cells subcutaneously into nude mice and assessed tumor-forming capability. Six of six switch II domain mutations induced significant tumor formation by 3 weeks post-injection, comparable to transformation induced by KRAS G12V or EGFR L858R (Figure 2b and Supplementary Figure 3), whereas transformation capability of non-switch II domain mutations varied. RIT1 Q40L, although consistently less active than other RIT1 variants, showed intermediate transforming capability in the xenograft assay (Supplementary Figure 3). It remains possible that RIT1 Q40L is weakly activating, and should be noted that RIT1 Q40 is homologous to KRAS Q22, which is found mutated in Noonan syndrome25 and rarely in somatic cancers.
Figure 2.
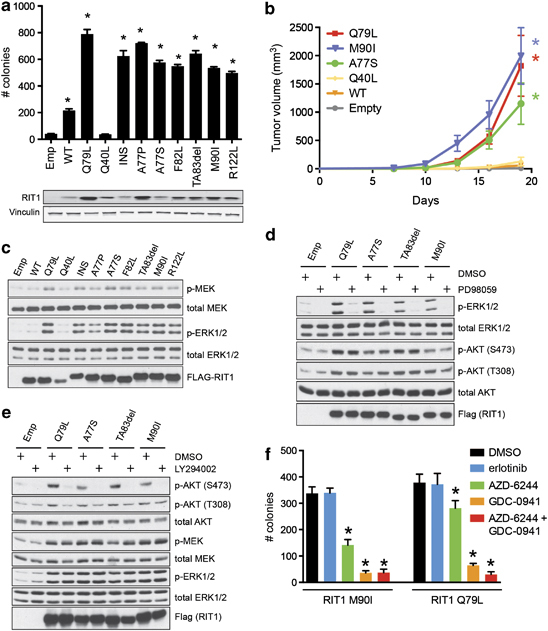
Mutated RIT1 induces cellular transformation via activation of MEK and PI3K. (a) Soft agar transformation assay in NIH3T3 cells. Cells were transduced with retrovirus to ectopically express wild-type (WT) or mutated RIT1 constructs or empty vector (control), then plated in soft agar (Methods). Colonies were visualized at 14 days and quantified using CellProfiler. Top panel, data shown are mean+s.e.m. of triplicate wells. Data shown are representative of at least three independent experiments. *P<0.05 by two-tailed t-test. Bottom panels, western blot showing expression of RIT1 or vinculin (loading control). ‘INS’, T76_insTLDT. (b) Tumor growth of xenografts of NIH3T3 cells with or without expression of RIT1. Data shown is mean+s.e.m. of nine replicates per construct. *P<0.01 by two-tailed t-test. Data shown are representative of at least two independent experiments per construct. (c) Western blot of PC6 cell lysates following transfection of wild-type or mutant RIT1 or vector control (‘Emp’). Data shown is representative of at least three independent experiments. (d) Western blot of PC6 lysates following transfection of FLAG-RIT1 constructs or vector control (‘Emp’) in the presence or absence of 10 μM PD98059. Cells were serum starved for 5 h prior to lysis. (e) Western blot of PC6 cell lysates generated after transfection of FLAG-RIT1 mutant constructs in the presence or absence of 10 μM LY294002. Cells were serum starved for 5 h prior to lysis. (f) Soft agar colony formation of NIH3T3 cells stably expressing RIT1 M90I or RIT1 Q79L in the presence or absence of 1 μM erlotinib, GDC-0941, AZD-6244 or GDC-0941/AZD-6244 or vehicle control (dimethylsulfoxide). 5 × 103 cells were suspended in a top agar solution together with each respective drug to a final concentration of 1 μM in triplicate. After 15 days, colonies were photographed and quantified using CellProfiler. *P<0.05.
In addition to the lung adenocarcinoma mutations, we tested the transforming capability of RIT1 mutations reported in myeloid malignancies23 or curated in COSMIC26 from diverse tumor types (Supplementary Table 3). Consistent with the lung adenocarcinoma data, transforming mutations were mainly clustered in the switch II domain. We identified transforming RIT1 mutations in salivary gland carcinoma, endometrial carcinoma, myeloid malignancies and melanoma. These data indicate that rare RIT1 mutations may have a role in the pathogenesis of diverse cancers.
RIT1 mutation is mutually exclusive with mutations in other RAS/RTK-pathway genes, so we hypothesized that RIT1 may activate PI3K and MEK. To investigate the signaling changes induced by mutated RIT1, we expressed wild-type or mutated RIT1 in PC6 cells and analyzed downstream signaling changes by western blot. Transformation potential of RIT1 mutants correlated with their ability to activate MEK and ERK pathways; all RIT1 mutations with the exception of RIT1 Q40L induced phosphorylation of MEK and ERK (Figure 2c). Treatment of RIT1-expressing cells with the specific MEK inhibitor, PD98059, completely inhibited RIT1-induced ERK phosphorylation, indicating that RIT1 induces ERK phosphorylation via activation of MEK (Figure 2d). In addition to MEK/ERK signaling, we observed robust activation of PI3K/AKT signaling by mutated RIT1. Oncogenic RIT1 mutants induced phosphorylation of AKT (S473 and T308), which could be inhibited by treatment with the PI3K/mTOR inhibitor LY294002 (Figure 2e). RIT1-activated MEK and PI3K signaling independently, as MEK inhibition failed to impair AKT phosphorylation and PI3K/mTOR inhibition failed to impair ERK phosphorylation.
To determine if activation of MEK and PI3K was responsible for RIT1-induced transformation, we assayed the ability of RIT1 mutants to form colonies in soft agar in the presence or absence of different kinase inhibitors. Either MEK inhibition with 1 μM AZD-6244 or PI3K/mTOR inhibition with 1 μM GDC-0941 significantly impaired colony formation (Figure 2f), whereas treatment with the EGFR inhibitor erlotinib (1 μM) showed no effect. Combination MEK and PI3K inhibition almost completely abrogated colony formation (Figure 2f).
Given the prevalence of RIT1 mutation in primary human lung adenocarcinomas, we hypothesized that human lung adenocarcinoma cell lines may also harbor mutations in RIT1 and that identification of these cell lines would facilitate study of RIT1 function in human cancer pathogenesis. On the basis of the pattern of RIT1 mutation mutual-exclusivity with driver mutations in primary samples, we focused our efforts on ‘oncogene-negative’ human non-squamous non-small cell lung cancer cell lines. We curated the mutational status of all known lung adenocarcinoma driver genes from non-squamous non-small cell lung cancer lines in the Cancer Cell Line Encyclopedia.27 Among 35 ‘oncogene-negative’ cell lines, NCI-H2110 showed the highest level of RIT1 mRNA expression (Supplementary Figure 4 and Supplementary Table 4). Next, we sequenced the switch II domain of RIT1 in cDNA from 19 ‘oncogene-negative’ cell lines and 3 ‘oncogene-positive’ cell lines (Supplementary Table 5). In all, 1/19 ‘oncogene-negative’ and 0/3 ‘oncogene-positive cell lines’ had a non-synonymous change in RIT1; NCI-H2110 cells harbored the p.M90I mutation also seen in human primary lung adenocarcinomas (Figure 3a).
Figure 3.
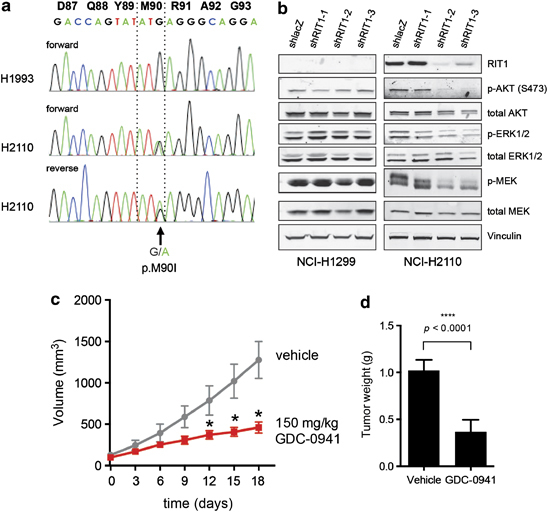
Endogenous mutated RIT1 regulates MEK and PI3K in NCI-H2110 cells. (a) Sanger sequencing of RIT1 RT–PCR products generated from NCI-H1993 or NCI-H2110 cell line cDNA. Numbering in black bold refers to amino-acid positions and colored letters refer to nucleotide sequence. An arrow indicates the position of a heterozygous p.M90I mutation. (b) Western blot of lysates from NCI-H1299 and NCI-H2110 following expression of shRNA hairpins targeting RIT1 (shRIT1-1, -2 and -3) or non-targeting hairpin control (shlacZ). (c) Tumor volume of NCI-H2110 xenografts in nude mice. 2 × 106 cells were injected subcutaneously into the flanks of nude mice. When tumors reached ∼100 mm3, drug treatment was initiated (day 0). Mice were treated daily with 150 mg/kg GDC-0941 or vehicle control by oral gavage. *P<0.05. n=9 tumors per condition. (d) Weight of tumors from NCI-H2110 xenografts shown in b. At day 18, animals were euthanized and tumors excised and weighed.
To determine if RIT1-mediated PI3K and MEK activation are involved in RIT1-mutated human lung adenocarcinoma, we interrogated the role of these pathways in NCI-H2110 cells. Using lentiviral delivery of RIT1-targeting (shRIT1-1, shRIT1-2 and shRIT1-3) and non-targeting (shLacZ) short hairpin RNA, we knocked down RIT1 levels in NCI-H2110 and NCI-H1299 cells, the latter of which express low or no RIT1 (Figure 3b). Knockdown of RIT1 in NCI-H2110 cells resulted in loss of AKT phosphorylation and a reduction in MEK and ERK phosphorylation, whereas expression of the same hairpins in NCI-H1299 cells had no consistent effect. Therefore, RIT1 regulates PI3K and MEK signaling in NCI-H2110 cells.
Given the potent ability of the PI3K/mTOR inhibitor GDC-0941 to inhibit RIT1-induced cellular transformation (Figure 2f), we sought to determine whether GDC-0941 would impair the growth of RIT1-mutated human lung adenocarcinoma cells. NCI-H2110 xenografts were generated via subcutaneous injection into the flanks of nude mice. After tumors were established, mice were dosed daily with 150 mg/kg GDC-0941 as previously described.28 GDC-0941 significantly impaired tumor growth (Figure 3c), resulting in markedly smaller tumor masses in the GDC-0941-treated group at the experimental end point (Figure 3d).
Discussion
RIT1 encodes a RAS-family small GTPase24, 29 with significant domain and sequence homology to KRAS, HRAS and NRAS (Figures 1a and b). Although RIT1 is ubiquitously expressed, expression of its closest homolog, RIT2/RIN, is restricted to the nervous system.29 Like RAS proteins, RIT1 possesses intrinsic GTP hydrolysis activity,24 which can be inactivated by construction of a point mutation, Q79L, at the glutamine homologous to Q61 in RAS.24 Expression of Q79L RIT1 is sufficient to transform NIH3T3 cells30 and induces activation of p38, ERK or AKT signaling pathways depending on cellular context.31, 32 Interestingly, the codon for Q79 also provides the 5′ splice site for intron 4–5 of RIT1; mutation of glutamine 79 to leucine would be predicted to disrupt the splice site, which may make natural Q79L mutation incompatible with RIT1 expression.
In PC6 cells, a derivative of the PC12 pheochromocytoma line commonly used for study of RAS/MAPK signaling and neuronal differentiation,33, 34 RIT1 activates MEK/ERK through direct binding with BRAF.31 Moreover, RIT1 activity is induced by stimulation with EGF or NGF in PC6 cells.31 Recently, studies in Rit knockout mice demonstrated that Rit promotes survival after oxidative stress through activation of a p38-AKT-BAD signaling cascade.35 Rit −/− cells are hypersensitive to apoptosis after exposure to reactive oxygen species (ROS) whereas cells expressing Q79L RIT1 are protected.35
The RIT1 switch II domain mutations identified here in ∼2% of lung adenocarcinomas define a new subset of lung adenocarcinoma. RIT1 switch II domain mutations are mutually exclusive with all other known lung adenocarcinoma oncogenes and rapidly induce transformation in vitro and in vivo. RIT1 mutation induces activation of PI3K and MEK signaling and these pathways are required for RIT1-mediated cellular transformation. These experimental and observational data indicate that RIT1 likely acts in the RTK/MAPK pathway to promote tumorigenesis. In agreement with this notion is the recent identification of germline RIT1 mutations in Noonan syndrome, a developmental disorder caused by mutations in RAS-pathway genes.36 In cancer, targeted inhibition of PI3K and MEK should be explored as a possible therapeutic strategy for patients with RIT1 mutations.
Materials and methods
Identification of mutations
Somatic RIT1 mutations were identified in two independent cohorts of lung adenocarcinoma sequence data from Imielinski et al.1 or The Cancer Genome Atlas (http://cancergenome.nih.gov, manuscript submitted).
Construct generation and virus production
pDONR223-RIT1 was obtained from the Broad Institute’s ORF collection37 and confirmed to be wild-type by Sanger sequencing. Mutations were introduced into pDONR223-RIT1 by site-directed mutagenesis using the Quikchange II kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s protocol. Primer sequences are available upon request. RIT1 mutations were then subcloned from pDONR223 into pBabe-puro for generation of retrovirus or p3xFlag-CMV10 (Sigma-Aldrich, St Louis, MO, USA) for transient transfection experiments. Retrovirus was generated by co-transfection of pBabe-puro and a pCL-Eco packaging vector into 293T cells using Fugene6 (Promega, Madison, WI, USA). Forty-eight hours post-transfection, virus was harvested and filtered through a 0.45 μm syringe filter before addition to exponentially growing NIH3T3 cells. Forty-eight hours post-infection, cells were selected with 2 μg/ml puromycin for 2–3 days before proceeding with downstream soft agar assays or other experiments.
Cell line sequencing
We considered ‘known’ adenocarcinoma oncogenes as previously defined,2 with the addition of the recently identified MET exon 14 skipping variant.16, 38 We curated the genotypes of these genes in 64 non-squamous cell lines (Supplementary Table 5) from previously generated Oncomap data.39, 40 Nineteen cell lines lacking known lung adenocarcinoma oncogenes and three control cell lines were selected for RIT1 cDNA sequencing. Cell line culturing and cDNA preparation was as previously described.41 The switch II domain region of RIT1 was PCR amplified using the following primers: RIT1-c-ex3-F, TCATCAGCCACCGATTCC; RIT1-c-ex5-R, CGTCGGACTCGATAAATAAGC. PCR was performed using the HotStarTaq mastermix (Qiagen, Valencia, CA, USA) with the following thermocycling conditions: 5 min at 95 °C, 38 cycles of 15 s at 94 °C, 1 min at 50 °C, 1 min at 72 °C and an additional extension time of 10 min at 72 °C. PCR products were purified using the Qiaquick PCR purification kit (Qiagen) and sequenced by Sanger sequencing (Genewiz, Cambridge, MA, USA) using the same primers as above. Seqman (DNAStar, Madison, WI, USA) was used to align sequencing traces and identify single nucleotide polymorphisms. The p.M90I mutation identified in NCI-H2110 cells was validated in an independent cDNA sample from an independent aliquot of cells obtained directly from ATCC.
Soft agar assay
NIH3T3 cells were transduced with retrovirus generated with pBabe-puro-RIT1 plasmids or pBabe-puro empty vector. Forty-eight hours post-infection, cells were selected with 2 ug/ml puromycin. After 48 h of selection, cells were split for seeding in a soft agar assay or lysis for verification of protein expression by western blotting. For the soft agar assay, 5 × 103–5 × 104 cells were suspended in 1 ml of 0.33% select agar in DMEM/FBS and plated on a bottom layer of 0.5% select agar in DMEM/FBS in six-well dishes. Each cell line/mutation was analyzed in triplicate. Colonies were photographed after 14–21 days and quantified using CellProfiler.42 An anti-RIT1 antibody (#ab13322, Abcam, Cambridge, UK) and an anti-vinculin (loading control) antibody (Sigma-Aldrich) were used to confirm RIT1 expression. For soft agar inhibitor experiments, AZD6244, erlotinib or GDC-0941 were purchased from Selleckchem, Houston, TX, USA and suspended in the top agar solution at a final concentration of 1 μM. Agar wells were hydrated weekly with media containing 1 μM drug.
Xenograft assays
Xenografts were performed as previously described.43 For transformation assays, RIT1- or empty vector-transduced NIH3T3 cells, generated as above, were harvested by trypsinization, washed in PBS and resuspended at 106 cells/ml in PBS. Two hundred microliters (2 × 105 cells) were injected into each injection site, n=9 sites per cell line, in 4–6-week-old female nu/nu mice (Jackson Laboratory, Bar Harbor, ME, USA). Cells were allowed to engraft for 1 week, then tumors were measured every 3 days using a digital caliper (VWR, Radnor, PA, USA). Tumor volume was calculated with the formula 0.5 × L × W2 where L is the longest diameter and W is the diameter perpendicular to L. NCI-H2110 studies, cells were prepared as above and then resuspended at 107 cells/ml. Two hundred microliters (2 × 106 cells) were injected per site into flanks of nu/nu mice. Tumors were monitored until ∼100 mm3, at which time inhibitor dosing was initiated. GDC-0941 (Selleckchem) was resuspended at 20 mg/ml in 0.5% methylcellulose/0.2% Tween-80 and 150 mg/kg administered daily by oral gavage. All animal experiments were carried out in accordance with the DFCI Institutional Animal Care and Use Committee guidelines.
Antibodies and cell culture
Monolayers of PC6 cells were transfected with 2 μg of plasmid DNA per well of a six-well plate as described previously.31 After a 60 h incubation, whole cell lysates were prepared in kinase lysis buffer, an equal amount of protein from each lysate was resolved by 10% SDS–PAGE and subjected to immunoblot analysis with the indicated antibody as described previously.31 For inhibitor experiments, cells were pre-treated with 10 μM PD98059 or LY294002 or dimethylsulfoxide for 30 min prior to transfection. 60 h post-transfection, cells were serum-starved for 5 h in the presence of the inhibitor or dimethylsulfoxide before lysis in kinase buffer. Commercial antibodies were used to total- and phospho-specific MEK, ERK, AKT (Cell Signaling, Danvers, MA, USA), Flag (Sigma-Aldrich) and RIT1 (Abcam).
Knockdown experiments
Hairpins targeting RIT1 (shRIT1-1, -2 and -3) and a non-targeting hairpin (shLacZ) were obtained from The RNAi Consortium (TRC) in the pLKO lentiviral backbone. Virus was generated by co-transfection of pLKO, VSV-G and Δ8.9 packaging vectors in 293T cells. Lentivirus was harvested in DMEM+30% FBS and filtered or frozen before application to exponentially growing NCI-H1299 or NCI-H2110 cells. Cells were selected in 2 μg/ml puromycin for 3 days and then expanded for 1–2 weeks before analysis. Hairpin TRC clone IDs and target sequences were as follows:
shLacZ,/TRCN0000072223, TGTTCGCATTATCCGAACCAT
shRIT1-1/TRCN0000047888, CGCTACTATATTGATGATGTT
shRIT1-2/CGTCGAAGTTTCCATGAAGTT, CGTCGAAGTTTCCATGAAGTT
shRIT1-3/TRCN0000047890, CGAGAATTCAGCTGTCCCTTT.
Supplementary information
Supplementary Table 4 and 5 Legends (DOC 43 kb)
Acknowledgements
We thank A. Brooks, P. Choi, H. Gannon, H. Greulich, L. De Waal, B. Kaplan, R. Liao, T. Sharifnia, J. Boehm and A. Koehler for critical advice and discussion. AHB is supported by a postdoctoral fellowship from the American Cancer Society. MI is supported by NCI Training Grant T32CA9216. JW is supported by NIH Training Grant T32HG002295. DAA is supported by NIH NS045103, KSCHIRT 12-1A, and a University of Kentucky Research Professorship. This work was supported by grants from the Department of Defense (#W81XWH-12-1-0269) National Cancer Institute (P01CA154303 and R01CA109038), Uniting Against Lung Cancer, the Lung Cancer Research Foundation and the American Lung Association to MM.
Competing interests
MM is a founder and equity holder of Foundation Medicine, a for-profit company that provides next-generation sequencing diagnostic services.
Footnotes
Supplementary Information accompanies this paper on the Oncogene website
Change history
4/13/2022
A Correction to this paper has been published: 10.1038/s41388-022-02300-2
References
- 1.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med. 2012;18:349–351. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 3.Janne PA, Meyerson M. ROS1 rearrangements in lung cancer: a new genomic subset of lung adenocarcinoma. J Clin Oncol. 2012;30:878–879. doi: 10.1200/JCO.2011.39.4197. [DOI] [PubMed] [Google Scholar]
- 4.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 5.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 6.Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012;22:436–445. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 8.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. The Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 9.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 10.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 12.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. 2009;6:201–205. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]
- 14.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–2119. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 18.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawazu M, Ueno T, Kontani K, Ogita Y, Ando M, Fukumura K. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc Natl Acad Sci USA. 2013;110:3029–3034. doi: 10.1073/pnas.1216141110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genomes Project C. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM. An integrated map of genetic variation from 1092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Atlas TCG Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomez-Segui I, Makishima H, Jerez A, Yoshida K, Przychodzen B, Miyano S. Novel recurrent mutations in the RAS-like GTP-binding gene RIT1 in myeloid malignancies. Leukemia. 2013;27:1943–1946. doi: 10.1038/leu.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao H, Kadono-Okuda K, Finlin BS, Andres DA. Biochemical characterization of the Ras-related GTPases Rit and Rin. Arch Biochem Biophys. 1999;371:207–219. doi: 10.1006/abbi.1999.1448. [DOI] [PubMed] [Google Scholar]
- 25.Zenker M, Lehmann K, Schulz AL, Barth H, Hansmann D, Koenig R. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet. 2007;44:131–135. doi: 10.1136/jmg.2006.046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A. Cosmic 2005. Br J Cancer. 2006;94:318–322. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Brien C, Wallin JJ, Sampath D, GuhaThakurta D, Savage H, Punnoose EA. Predictive biomarkers of sensitivity to the phosphatidylinositol 3' kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010;16:3670–3683. doi: 10.1158/1078-0432.CCR-09-2828. [DOI] [PubMed] [Google Scholar]
- 29.Lee CH, Della NG, Chew CE, Zack DJ. Rin, a neuron-specific and calmodulin-binding small G-protein, and Rit define a novel subfamily of ras proteins. J Neurosci. 1996;16:6784–6794. doi: 10.1523/JNEUROSCI.16-21-06784.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rusyn EV, Reynolds ER, Shao H, Grana TM, Chan TO, Andres DA. Rit, a non-lipid-modified Ras-related protein, transforms NIH3T3 cells without activating the ERK, JNK, p38 MAPK or PI3K/Akt pathways. Oncogene. 2000;19:4685–4694. doi: 10.1038/sj.onc.1203836. [DOI] [PubMed] [Google Scholar]
- 31.Shi GX, Andres DA. Rit contributes to nerve growth factor-induced neuronal differentiation via activation of B-Raf-extracellular signal-regulated kinase and p38 mitogen-activated protein kinase cascades. Mol Cell Biol. 2005;25:830–846. doi: 10.1128/MCB.25.2.830-846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi GX, Rehmann H, Andres DA. A novel cyclic AMP-dependent Epac-Rit signaling pathway contributes to PACAP38-mediated neuronal differentiation. Mol Cell Biol. 2006;26:9136–9147. doi: 10.1128/MCB.00332-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pittman RN, Wang S, DiBenedetto AJ, Mills JC. A system for characterizing cellular and molecular events in programmed neuronal cell death. J Neurosci. 1993;13:3669–3680. doi: 10.1523/JNEUROSCI.13-09-03669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bar-Sagi D, Feramisco JR. Microinjection of the ras oncogene protein into PC12 cells induces morphological differentiation. Cell. 1985;42:841–848. doi: 10.1016/0092-8674(85)90280-6. [DOI] [PubMed] [Google Scholar]
- 35.Cai W, Rudolph JL, Harrison SM, Jin L, Frantz AL, Harrison DA. An evolutionarily conserved Rit GTPase-p38 MAPK signaling pathway mediates oxidative stress resistance. Mol Biol Cell. 2011;22:3231–3241. doi: 10.1091/mbc.e11-05-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Human Genet. 2013;93:173–180. doi: 10.1016/j.ajhg.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8:659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. 2009;4:5–11. doi: 10.1097/JTO.0b013e3181913e0e. [DOI] [PubMed] [Google Scholar]
- 39.Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 40.Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119:1727–1740. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–4283. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamprecht MR, Sabatini DM, Carpenter AE. CellProfiler: free, versatile software for automated biological image analysis. BioTechniques. 2007;42:71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
- 43.Clark GJ, Cox AD, Graham SM, Der CJ. Biological assays for Ras transformation. Methods Enzymol. 1995;255:395–412. doi: 10.1016/S0076-6879(95)55042-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 4 and 5 Legends (DOC 43 kb)