

Abstract
Fibroblast growth factor-2 (FGF-2) plays a critical role in endothelial survival, proliferation, and angiogenesis and is localized on the cell membrane by binding to heparan sulfate proteoglycans. Here we established a neutralizing monoclonal antibody, 1B9B9, against FGF-2 using the rat medial iliac lymph node method. 1B9B9 blocked the binding of FGF-2 to its receptor, inhibiting FGF-2-induced proliferation and corresponding downstream signaling in endothelial cells. Treatment of human umbilical vein endothelial cells with 1B9B9 reduced the basal phosphorylation levels of Akt and MAPK. Furthermore, continued treatment with 1B9B9 induced cell death by apoptosis. Compared with FGF-2 knockdown, 1B9B9 significantly reduced cell survival. In addition, the combination of FGF-2 siRNA and 1B9B9 showed a synergistic effect. The data indicate that 1B9B9 established by the rat iliac lymph node method is a fully compatible neutralizing antibody.
Introduction
Fibroblast growth factor-2 (FGF-2), a member of the heparin-binding growth factor family, plays key roles during development and morphogenesis as well as in several physiological and pathological functions such as wound healing, neovascularization, and tumor growth and progression. In particular, the physiological functions of vascular endothelial cells depend on FGF-2, as it promotes endothelial cell proliferation, survival, and migration, which lead to angiogenesis and thereby contribute to the regulation of vascular homeostasis.
The dynamics of FGF-2 in vivo is also well characterized. FGF-2 is a secreted glycoprotein that is readily sequestered to the extracellular matrix as well as to the cell surface by heparan sulfate proteoglycans (HSPGs). The interaction between FGF-2 and HSPG protects FGF-2 against denaturation and proteolysis and may limit its diffusion and release into interstitial spaces.(1–5) It is thought that FGF-2 signaling is initiated by the release of FGF-2 that is stored by association with HSPGs. FGF-2-induced phosphorylation of fibroblast growth factor receptor (FGFR) leads to the activation of multiple signal transduction pathways. Activated FGFR phosphorylates FGFR substrate 2 (FGRS2) at several sites, allowing the recruitment of the adaptor proteins SOS and GRB2 to activate RAS and the downstream Raf and MAPK cascades.(6,7) A major downstream effect of MAPK signaling is the promotion of cell proliferation. Furthermore, FGF-2 signaling has the potential to activate anti-apoptotic pathways through the activation of either PI3K/Akt or STAT signaling.(8)
FGF-2 and vascular endothelial growth factor (VEGF) are the most potent angiogenesis inducers and have a synergistic effect on angiogenesis.(9,10) A significant amount of cross-talk is thought to exist between VEGF and FGF-2 during angiogenesis. VEGF plays an integral role in the development of new blood vessels in tumor formation, and angiogenesis inhibitors targeting the VEGF pathway have been used to develop anti-angiogenic strategies for cancer therapy. Recently, emerging evidence has suggested that upregulation of FGF and FGFR may serve as a mechanism of resistance to anti-VEGF therapy.(11) Clinical evidence in colon cancer also supports the role of FGF-2 in resistance to bevacizumab-containing regimens.(12) Dysfunction of FGF-2 can result in severe immune deficiencies, which can potentially be neutralized by monoclonal antibodies. Unfortunately, most current methods to neutralize the activity of endogenous cytokines are toxic, and the produced effects are not stable in vivo in immunized animals. One effective approach to avoid this problem is to use an animal different from that from which the immunogen was derived. The rat iliac lymph node method is one such alternative.(13) Instead of using lymphocytes from the spleen, the iliac lymph node is employed as a rich source of active lymphocytes, allowing just one immunization to achieve highly efficient hybridoma formation. While promising, the use of the rat iliac lymph node method for establishing neutralizing monoclonal antibodies is unproven.
In the present study, we obtained a new neutralizing monoclonal antibody (MAb) against FGF-2 by the rat iliac lymph node method. Treatment with this FGF-2 antibody prevented basal activities of MAPK and Akt, and induced endothelial cell death. These results indicate that the rat iliac lymph node method has the potential to establish neutralizing antibodies rapidly and effectively.
Materials and Methods
Materials
Antibodies were obtained as follows. Anti-Akt (p-Ser473), ERK1,2 (p-Thr202/p-Tyr204), cleaved-caspase3, and GST antibodies were purchased from Cell Signaling (Danvers, MA); anti-FGF-2 and PARP antibodies from Santa Cruz Biotechonology (Santa Cruz, CA); anti-FGFR (p-Tyr653/p-Tyr654) antibody from R&D Systems (Minneapolis, MN); and anti-β-actin antibody from Sigma (St. Louis, MO). HRP-coupled secondary antibodies were purchased from GE Healthcare (Uppsala, Sweden). IgG from rat serum and recombinant FGF-2 were obtained from Sigma-Aldrich and Invitrogen (Carlsbad, CA), respectively. Alexa Fluor 488-conjugated anti-rat IgG and Alexa Fluor 568-phalloidin were also purchased from Invitrogen. The FGFR1 inhibitor, PD166866, was obtained from Sigma.
Production and purification of recombinant proteins
Expression vectors of glutathione S-transferase (GST) fusion proteins that bind to a 146-amino-acid section of human FGF-2 (C-terminus) were transformed into Escherichia coli BL21 (DE3) (Novagen, Madison, WI).(14) The E. coli cells were grown in MMI medium containing 50 μg/mL carbenicillin (Nacalai Tesque, Kyoto, Japan) at 37°C. Expression was induced by the addition of 0.5 mM isopropyl-1-thio-β-D-galactopyranoside (Nacalai Tesque) followed by incubation for 12 h at 37°C. Bacteria were lysed and fusion proteins purified according to previously described methods.(15–17)
Immunization of rat and production of monoclonal antibodies
Anti-FGF-2 rat MAbs were generated by the rat lymph node method as previously described.(13,16,18) A 10-week-old female lzm rat (Japan SLC, Shizuoka, Japan) was injected via the hind footpads with 500 μL of an emulsion containing 1 mg of recombinant human FGF-2 protein and Freund's complete adjuvant. After 2 weeks, cells from the lymph nodes of this rat were fused with mouse myeloma Sp2/0-Ag14 cells at a ratio of 5:1 in a 50% polyethylene glycol solution (PEG1500; Merck, Darmstadt, Germany). The resulting hybridoma cells were plated onto 96-well plates and cultured in HAT selection medium (hybridoma SFM medium [Invitrogen], 10% fetal bovine serum [FBS], 10% BM-Condimed H1 [Roche, Basel, Switzerland], 100 μM hypoxanthine, 0.4 μM aminopterin, and 1.6 μM thymidine). At 7 days post-fusion, the hybridoma supernatants were screened using an enzyme-linked immunosorbent assay (ELISA) against GST-fused FGF-2. Positive clones were subcloned and rescreened by ELISA and immunoblotting. To prepare hybridoma supernatants containing highly concentrated antibodies, the resulting positive clone 1B9B9 was then cultured at a high density using a miniPERM bioreactor (Vivascience, Hannover, Germany).
ELISA
FGF-2 antigen (5 μg/mL) in TBS-T (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 0.05% Tween-20) was adsorbed on the surface of 96-well flexible microplates (Thermo Scientific, Rockford, IL) and incubated overnight at 4°C. To avoid non-specific binding, the plates were blocked with 1% bovine serum albumin (BSA, Nacalai Tesque) in TBS-T. Hybridoma supernatants were incubated for 1 h at room temperature and then washed with TBS-T three times. The plates were incubated for 30 min at room temperature with alkaline phosphatase-conjugated anti-rat IgG antibody (Sigma) diluted at 1:10,000. After washing with TBS-T three times, immunoreactivity was visualized using a pNPP phosphatase substrate system (KPL, Gaithersburg, MD).(19)
Cell culture
EGM2 and EBM2 culture media were purchased from Lonza (Walkersville, MD). Human umbilical vein endothelial cell (HUVECs, passage 5 or fewer) were obtained from Lonza and grown at 37°C in a humidified atmosphere of 5% CO2 in EGM2 medium. Fetal bovine heart endothelial (FBHE) cells were obtained from the Health Science Research Resources Bank (Osaka, Japan). These cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and 2 ng/mL recombinant FGF-2 (Wako, Osaka, Japan) at 37°C in a 5% CO2 incubator. All procedures were in accordance with institutional guidelines for animal research.
Immunoblotting
Details of the applied immunoblotting method have been described previously.(20) Cells were washed with phosphate-buffered saline (PBS) and lysed in RIPA lysis buffer consisting of 50 mM HEPES (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1% CHAPS, 10% glycerol, 100 mM NaF, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (Nacalai Tesque). Lysates were characterized by centrifugation, the supernatant was recovered, and protein concentrations were assayed using the bicinchoninic acid protein assay reagent (Thermo Scientific). Lysates for immunoblotting (20 μg of protein) were separated on SDS-polyacrylamide gels under reducing conditions, followed by electrophoretic transfer to polyvinylidine difluoride membranes (Immobilon-P; Millipore, Billerica, MA). After blocking, the membranes were probed with the appropriate primary antibodies. Membrane-bound primary antibodies were detected using secondary antibodies conjugated with HRP. Immunoblots were detected with LAS-3000 (Fujifilm, Tokyo, Japan) using the enhanced chemiluminescence technique (Immobilon Western HRP Substrate; Millipore). Immunoblots were quantified using Image Gauge software (Fujifilm).
Immunoprecipitation
Cells were washed with PBS and lysed in RIPA lysis buffer consisting of 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 1% CHAPS, 10% glycerol, 100 mM NaF, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail. For immunoprecipitation, cell lysates (500 μg of protein) clarified with protein G Sepharose beads (Sigma) for 1 h at room temperature were mixed with primary antibodies at room temperature for 2 h. Protein G Sepharose beads were then added, and the mixture was gently rocked at room temperature for 2 h. The immunoprecipitates were washed three times with RIPA buffer, resolved by SDS-PAGE, and immunoblotted.
Immunofluorescence microscopy
HUVECs grown on coverslips were washed twice with PBS and fixed with 4% paraformaldehyde at room temperature for 20 min. After rapid washing with PBS, the cells were quenched with 50 mM NH4Cl in PBS for 10 min, washed twice with PBS, and permeabilized with 0.1% Triton X-100 in PBS for 5 min. After the cells were immersed in blocking buffer (10% goat serum/1% BSA/PBS) for 30 min, they were treated with the primary antibodies in 50% blocking buffer for 1 h at room temperature, followed by Alexa Fluor 488-conjugated goat anti-rat IgG secondary antibodies and Alexa Fluor 568-phalloidin for actin for 40 min at room temperature. Coverslips were mounted with FluorSave™ reagent (Calbiochem, Merck). Stained cells were visualized using a TCA-SP5 confocal laser-scanning microscope (Leica, Wetzlar, Germany) with a 63×oil immersion objective.
Neutralization assay with FBHE cells
FBHE cells (1×104 cells) were seeded in 6-well plates and cultured with DMEM/RPMI 1640 (1:1) supplemented with 2% FBS and 0.1 μg/mL heparin for 24 h. Thereafter, FGF-2 (5 ng/mL) and/or 1B9B9 were added to the cells for 4 days. Cells were then trypsinized and counted using a hemocytometer.
Cell viability assays
HUVECs were seeded in 96-well plates at a density of 1×104 cells per well. At 24 h after the addition of antibodies, cell viability was determined by assaying with Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Absorbance was measured at 450 nm with a microplate reader.
RNA interference
All siRNAs against human FGF-2 (#4286) and Silencer negative control number 1 siRNA (NC) were obtained from Ambion (Austin, TX). Lipofectamine RNAiMAX (Invitrogen) was used to transfect siRNAs into the HUVECs (final concentration, 10 nmol/L) as suggested in the manual. After 48 h of reverse transfection, cells were washed using PBS, and the assay medium was replaced.
Nanoelectrospray LC-MS/MS analysis and protein identification
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses were performed on a DiNa-AI nano LC system (KYA Technology, Tokyo, Japan) coupled to a QSTAR Elite hybrid mass spectrometer (AB Sciex, Concord, Canada) through a NanoSpray ion source (AB Sciex). The details of this analysis are described elsewhere.(21) Data acquisition was performed using Analyst QS Software 2.01 (AB Sciex) in the positive-ion mode. Both sets of data were processed by ProteinPilot using the Paragon™ search algorithm (AB Sciex). MS/MS data were searched against the NCBI database (UniProt, June 2013; ftp.hgc.jp/pub/mirror/ncbi/refseq/) using a H. sapiens taxonomy filter. The minimum threshold for protein identification was set at a protein score of 0.47, which corresponds to a confidence level greater than 66% and 1% false discovery rate.
iTRAQ labeling and quantification of protein expression
Cell lysates were collected and concentrated, followed by a buffer exchange against 100 mM triethyl-ammonium bicarbonate (pH 8.0) with Microcon centrifugal filters (3K nominal molecular weight limit; Millipore). Proteins (100 μg) were reduced, alkylated, digested with trypsin, and labeled with iTRAQ reagent (AB Sciex) according to the manufacturer's instructions. The samples were labeled as follows: 114, IgG treatment for 6 h; 115, 1B9B9 treatment for 6 h; 116, IgG treatment for 24 h; and 117, 1B9B9 treatment for 24 h. Each sample contained 100 μg of protein.
Results
Generation of neutralizing antibodies for FGF-2
To establish a specific antibody against FGF-2, we utilized a 146-amino-acid section of human FGF-2 as the antigen. We then generated a monoclonal antibody (MAb) specific for FGF-2 using GST-FGF-2 by the rat lymph node method. ELISA screening was performed, and 41 positive identical clones were selected. The selected clones were screened for their capacity to dephosphorylate FGFR. Serum-starved HUVECs were stimulated with conditioned medium of each clone, which had been pre-incubated with GST-FGF-2 for 1 h. Finally, the clone with the strongest inhibitory effect, 1B9B9, was selected. This MAb was identified in terms of class and subclass as IgG2a/κ.
To evaluate the reactivity against FGF-2, we performed immunoprecipitation using 1B9B9 against GST-FGF-2. Immunoblot analysis indicated that both FGF-2 polyclonal antibody and GST antibody detected FGF-2 in terms of binding at a band corresponding to estimate the molecular weight of FGF-2 (Fig. 1A). To examine whether 1B9B9 could recognize endogenous FGF-2, GST-FGF-2-treated HUVECs were lysed, and the lysate was immunoprecipitated with 1B9B9. The precipitates were immunoblotted with FGF-2 polyclonal antibody. 1B9B9 precipitated both GST-FGF-2 and endogenous 18-kDa-type FGF-2 (lane 6) as well as input (lane 2) (Fig. 1B). FGF-2 exists in multiple isoforms that range in weight from 18 to 24 kDa and are derived from the same gene locus under unique translational conditions. The 18 kDa isoform is primarily localized in the cytosol, but is also secreted and may form reservoirs of FGF-2 in the extracellular matrix. In contrast, the larger isoforms are predominantly nuclear. To evaluate whether 1B9B9 can be used in immunofluorescence analysis, HUVECs were immunostained by 1B9B9. Immunostaining indicated that FGF-2 is localized in the cytosol and nucleus (Fig. 1C). Furthermore, the staining pattern of FGF-2 partially co-localized with phalloidin, which suggests that FGF-2 also exists on the cell membrane or extracellular matrix. These findings argue that 1B9B9 can detect multiple isoforms ranging from 18 to 24 kDa in weight.
FIG. 1.

1B9B9 recognized both recombinant and endogenous FGF-2. (A) GST-FGF-2 (0.5 μg) or GST (0.5 μg) was immunoprecipitated with 1B9B9 followed by immunoblotting with an anti-FGF2 polyclonal antibody (upper panel) and GST antibody (lower panel). (B) HUVECs were treated with GST-FGF-2 (10 ng/mL) for 1 h before lysis. Cell lysates were immunoprecipitated with rat IgG or 1B9B9 followed by immunoblotting with an anti-FGF-2 polyclonal antibody. (C) HUVECs were immunostained with 1B9B9 and phalloidin. A representative microscopic image of the cells is shown. Scale bar, 40 μm.
1B9B9 suppressed bioactivity of exogenous FGF-2
The survival of FBHE cells has been shown to be dependent on FGF-2. To assess the FGF inhibitory effect of 1B9B9 as a neutralizing antibody, FGF-2 (5 ng/mL) pre-incubated with 1B9B9 (10, 5, and 1 μg/mL) was added to serum-starved FBHE cells. All doses of 1B9B9 fully suppressed FGF-2-induced phosphorylation of FGFR (Fig. 2A). Next, we examined the antibody's ability to inhibit the FGF-2-induced proliferation of FBHE cells in order to evaluate its immunoneutralizing activity. Figure 2B shows the effects of 1B9B9 on the growth of FBHE cells in the presence of exogenous FGF-2. Growth of FBHE cells in medium containing 2% FBS was stimulated by the addition of 5 ng/mL FGF-2. Cell numbers on day 3 were 1.24×105 and 5.3×104 with and without FGF-2 (5 ng/mL), respectively (Fig. 2B). On the other hand, when these cells were inoculated with 10 μg/mL 1B9B9, the cell number decreased to 8.3×104 cells. This suppression even occurred upon inoculation with 1 μg/mL 1B9B9 (9.2×104 cells). As a result, 1B9B9 (10 μg/mL) neutralized 40% of the growth of FBHE cells stimulated with 5 ng/mL FGF-2.
FIG. 2.
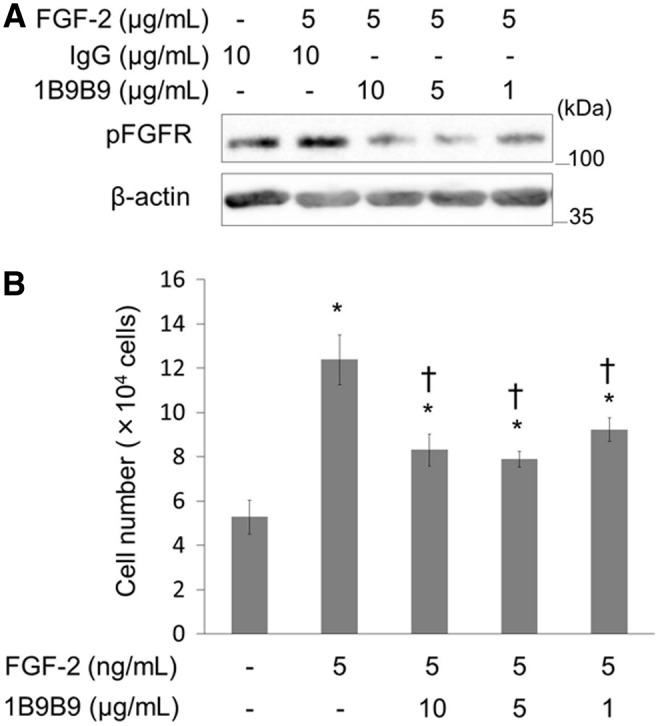
1B9B9 suppressed the bioactivity of FGF-2. (A) Recombinant FGF-2 was incubated with 1B9B9 or IgG for 1 h and then used to treat FBHEs for 30 min. Phosphorylation of FGFR was analyzed by immunoblotting. Data are representative of three independent experiments that yielded similar results. (B) FBHE (2×104) cells were cultured in 2% FBS-DMEM/RPMI 1640 (1:1) for 24 h. The cells were then cultured with IgG (10 μg/mL) as a negative control or 1B9B9 (10 μg/mL, 5 μg/mL, 1 μg/mL) in the presence of FGF-2 (2 ng/mL). After 3 days, the cell number was assessed by Trypan blue staining. *p<0.05 vs. (-), †p<0.05 vs. FGF-2 by one-way ANOVA; values are means±SE (n=4).
The stimulation of FGFR by FGF-2 involves sequential activation of the MAPK and PI3K/Akt pathways in HUVECs. To assess whether 1B9B9 inhibits the activation of ERK1,2 and Akt by FGF-2, serum-starved HUVECs were stimulated with FGF-2 (10 ng/mL), which bound to 1B9B9 for 1 h. FGF-2 bound to IgG induced phosphorylation of Akt at 30 min, whereas binding of FGF-2 to 1B9B9 completely inhibited the FGF-2-induced Akt phosphorylation (Fig. 3A). Likewise, FGF-2-induced phosphorylation of ERK1,2 was suppressed by 1B9B9. Accordingly, we judged that 1B9B9 acts as a neutralizing antibody. Next, we examined whether 1B9B9 suppressed basal activity of ERK1,2 and Akt in endothelial cells. HUVECs were treated with 1B9B9 (10, 5, and 1 μg/mL) for 1 h without exogenous FGF-2, and the basal activities of Akt and ERK1,2 were evaluated. At the basal levels, both Akt and ERK1,2 were phosphorylated to some extent. The Akt basal activity was reduced 50% more by 1B9B9 than by IgG (Fig. 3B). Likewise, 1B9B9 reduced ERK1,2 basal activity 75% more than IgG (Fig. 3B).
FIG. 3.

1B9B9 suppressed the activity of ERK1,2 and Akt in HUVECs. (A) (upper) Serum-starved HUVECs were stimulated for 30 min with FGF-2, which had been incubated with 1B9B9 (1 μg/mL, 5 μg/mL, 10 μg/mL) or IgG (10 μg/mL) for 1 h. Thereafter lysates were immunoblotted with pAkt or pERK1,2 antibodies. (lower) Densitometric quantitation of each protein using β-actin for normalization. *p<0.05 vs. FGF-2+IgG, by one-way ANOVA; values are means±SE (n=5). (B) HUVECs were incubated with IgG (10 μg/mL) or 1B9B9 (1 μg/mL, 5 μg/mL, 10 μg/mL) for 1 h under serum- and growth factor-starved conditions. (upper) Phosphorylation of Akt or ERK1,2 was analyzed by immunoblotting. (lower) Densitometric quantitation of each protein using β-actin for normalization. *p<0.05 vs. IgG by one-way ANOVA; values are means±SE (n=4).
1B9B9 suppressed endothelial cell survival in dose-dependent manner
If 1B9B9 reduced basal activity of ERK1,2 and Akt, 1B9B9 should provoke physiological change. Thus, we initially examined the effect of 1B9B9 on cell morphology. HUVECs were incubated with IgG or 1B9B9 for 24 h, and their cell morphology was observed (Fig. 4A). Since 1B9B9-treated cells detached from the substratum, cell viability was quantified by the MTS assay (Fig. 4B). IgG alone showed no toxic effects on HUVEC growth. In contrast, 1B9B9 treatment remarkably reduced viability in a dose-dependent manner.
FIG. 4.

1B9B9 induced endothelial cell death via apoptosis. (A) HUVECs were treated with 1B9B9 (1 μg/mL, 5 μg/mL, 10 μg/mL) for 24 h under serum- and growth factor-starved conditions. Representative photomicrographs of HE staining are shown. (a) No treatment, (b) IgG (10 μg/mL), (c) 1B9B9 (10 μg/mL), (d) 1B9B9 (5 μg/mL), (e) 1B9B9 (1 μg/mL). Scale bars, 200 μm. (B) HUVECs (1×104 cells) were treated with 1B9B9 (1 μg/mL, 5 μg/mL, 10 μg/mL) for 24 h. Viable cells were evaluated by the MTS assay. *p<0.05 vs. IgG by one-way ANOVA; values are means±SE (n=4). (C) HUVECs were treated with IgG or 1B9B9 for 6 or 24 h. Thereafter lysates were immunoblotted with PARP or cleaved caspase3 antibodies.
To confirm that the mechanism of cell death under these conditions involved apoptosis, apoptosis markers were assessed by immunoblotting (Fig. 4C). The 1B9B9 treatment markedly increased cleaved caspase3 or cleaved PARP. These results suggest that extracellular FGF-2 is critical for endothelial cell survival.
FGF-2 binding on cell membrane is critical for cell survival
To determine the target of 1B9B9, HUVECs were cultured with or without heparin or 1B9B9. Heparin treatment could remove any FGF-2 bound to the cell membrane, as previously described.(22) Heparin significantly reduced cell viability, but substantially less so than the 1B9B9-induced reduction (Fig. 5A). The combination of heparin and 1B9B9 enhanced the reduction of cells further. These results show that 1B9B9 should target not only free FGF-2, but FGF-2 associated on the membranes.
FIG. 5.
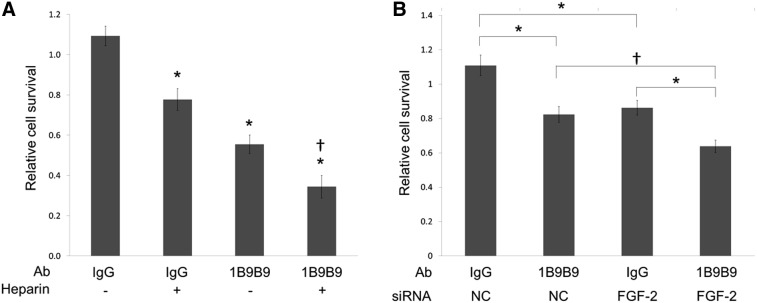
1B9B9 induced cell death in a synergistic manner with heparin or FGF-2 siRNA. (A) HUVECs were cultured with IgG (10 μg/mL) or 1B9B9 (10 μg/mL) with or without heparin (100 μg/mL) under serum- and growth factor-reduced medium conditions. After 24 h, viable cells were evaluated by the MTS assay. *p<0.01, †p<0.01, two-way ANOVA followed by Tukey-Kramer post-hoc test; values are means±SE (n=4). (B) After reverse transfection with FGF-2 siRNA or negative control, HUVECs were cultured with IgG (10 μg/mL) or 1B9B9 (10 μg/mL) under serum- and growth factor-reduced medium conditions. After 24 h, viable cells were evaluated by the MTS assay. *p<0.01, †p<0.05, two-way ANOVA followed by Tukey-Kramer post-hoc test; values are means±SE (n=6).
The relationship between the suppression of extracellular FGF-2 by 1B9B9 and the blocking of endogenous expression of the FGF-2 gene is unknown. As such, FGF-2-knockdown HUVECs were cultured with or without 1B9B9 for 24 h (Fig. 5B). The decrease of 1B9B9 in surviving cells occurred to a similar extent as that upon FGF-2 knockdown. The effect of knockdown was significantly enhanced by 1B9B9 treatment.
Quantitative differential proteomics in 1B9B9-treated cells
To investigate the mechanism of 1B9B9-induced cell death, we performed quantitative proteomics analysis of 1B9B9-treated cells based on the iTRAQ technique. iTRAQ-labeled proteins extracted from the 1B9B9-treated cells for 6 or 24 h were analyzed and compared with the proteome. Twenty-eight proteins that had a change of expression of <0.8-fold were considered to be downregulated at 6 h (Table 1). Of these, seven proteins were significantly reduced even at 24 h (bold), with Ral-B,(23) Rho GDP-dissociation inhibitor 2,(24) and calmodulin,(25) being related to signal transduction, and endothelial cell-specific chemotaxis regulator (ECSM2)(26) to cell differentiation, chemotaxis, and angiogenesis. These results suggest that 1B9B9-inhibited ERK1,2 and Akt activity decreased the expression of these molecules; as a result, endothelial cell death may have been induced. Taken together, these findings indicate that FGF-2 binding on the plasma membrane may have a constitutive function in maintaining cell survival.
Table 1.
Identification of Downregulated Proteins in 1B9B9 Treatment
| Ratio: 1B9B9/IgG | ||||
|---|---|---|---|---|
| Accession no. | Protein name | Coverage (%) | 6 h | 24 h |
| P11234 | Ras-related protein Ral-B | 64.1 | 0.485 | 0.664† |
| P52566 | Rho GDP-dissociation inhibitor 2 | 64.7 | 0.524 | 0.709* |
| P61081 | NEDD8-conjugating enzyme Ubc12 | 29.5 | 0.532 | 0.767 |
| P30050 | 60S ribosomal protein L12 | 29.1 | 0.653 | 0.897 |
| Q1349 | Phosphatidylinositol-binding clathrin assembly protein | 24.8 | 0.683 | 1.120 |
| P14735 | Insulin-degrading enzyme | 25.1 | 0.691 | 1.268 |
| O15067 | Phosphoribosylformylglycinamidine synthase | 27.4 | 0.698 | 1.053 |
| Q19T08 | Endothelial cell-specific chemotaxis regulator | 31.7 | 0.699 | 0.735 |
| P26583 | High mobility group protein B2 | 45.0 | 0.706 | 0.827 |
| P49189 | 4-trimethylaminobutyraldehyde dehydrogenase | 31.0 | 0.716* | 0.834 |
| P21291 | Cysteine and glycine-rich protein 1 | 50.3 | 0.716† | 0.934 |
| P62158 | Calmodulin | 44.3 | 0.717† | 0.750† |
| P09429 | High mobility group protein B1 | 44.2 | 0.725† | 0.920 |
| O15144 | Actin-related protein 2/3 complex subunit 2 | 27.7 | 0.739 | 1.106 |
| P23381 | Tryptophan-tRNA ligase, cytoplasmic | 40.6 | 0.757† | 0.973 |
| Q9H7C9 | Mth938 domain-containing protein | 41.0 | 0.759 | 0.972 |
| Q9H993 | UPF0364 protein C6orf211 | 23.6 | 0.761 | 0.859 |
| P24844 | Myosin regulatory light polypeptide 9 | 18.0 | 0.763* | 1.094 |
| P62633 | Cellular nucleic acid-binding protein | 46.9 | 0.764 | 1.300 |
| Q9NUV9 | GTPase IMAP family member 4 | 56.2 | 0.772* | 0.597* |
| Q9NWY4 | UPF0609 protein C4orf27 | 26.9 | 0.773 | 0.788 |
| P04181 | Ornithine aminotransferase, mitochondrial | 32.3 | 0.775 | 0.821 |
| Q9Y6C9 | Mitochondrial carrier homolog 2 | 28.7 | 0.784 | 0.996 |
| Q13162 | Peroxiredoxin-4 | 39.9 | 0.785 | 1.004 |
| P17980 | 26S protease regulatory subunit 6A | 38.5 | 0.793 | 1.553 |
| Q13630 | GDP-L-fucose synthase | 36.4 | 0.793 | 1.393 |
| P27824 | Calnexin | 58.4 | 0.794* | 0.854* |
| Q9NYL4 | Peptidyl-prolyl cis-trans isomerase FKBP11 | 16.9 | 0.799 | 1.162 |
p<0.01, †p<0.05.
Discussion
Here we report on a neutralizing MAb against FGF-2, 1B9B9, that was made using the rat iliac lymph node method and a GST fusion protein that binds to a 146-amino-acid section of human FGF-2 (C-terminus) as the antigen. 1B9B9 blocked FGF-2-induced endothelial cell proliferation and/or survival via the FGFR signaling cascade. We also demonstrated that pre-incubation of 1B9B9 with FGF-2 suppressed FGF-2-induced ERK1,2 and Akt phosphorylation. Accordingly, we conclude that 1B9B9 is a functional blocking antibody. Furthermore, it can be applied to immunoblotting, immunoprecipitation, and immunofluorescence. FGF-2 exists in multiple isoforms that range in weight from 18 to 24 kDa and are produced by different processes of translation of a single mRNA species. This isoform is primarily cytosolic, but is also secreted and stored in the extracellular matrix. In contrast, the larger isoforms are predominantly nuclear. 1B9B9 could detect FGF-2 in the nucleus, cytosol, and extracellular matrix, demonstrating that it recognizes all isoforms of FGF-2. Additionally, the pattern of immunofluorescence staining was in good agreement with the findings obtained using commercial antibodies.
In this report, we show that 1B9B9 effectively suppresses the physiological function of FGF-2. Several labs have reported on MAbs against FGF-2, which blocked its biological activities(27–29) FGF-2-neutralizing antibody, hIgG1-1A2, inhibited the proliferation, migration, and tube formation of HUVECs.(29) Likewise, MAbF7 also inhibited the tube formation of HUVECs.(30) However, both hIgG1-1A2 and MAbF7 did not lead to apoptosis in HUVECs. On the other hand, bFM-1 induced endothelial cell death in serum- and FGF-2-deprived HUVECs.(31) These results are in agreement with our findings for 1B9B9, which we established from rat. 1B9B9 suppressed cell viability less than heparin treatment did, while the combination of the two exerted a synergistic effect. Disruption of FGF-2 signaling both by an FGF-2-neutralizing MAb and by FGF-2 suppression by siRNA inhibited cell survival. In addition, the combination of FGF-2 siRNA and 1B9B9 showed a synergistic effect. Endogenous FGF-2 produced by endothelial cells may also play important autocrine and paracrine roles, not in angiogenesis but in survival. Intracellular and extracellular FGF-2, a component of the FGF-2 autocrine loop, is suggested to be a positive regulator of cell survival. 1B9B9 may suppress the autocrine loop of FGF-2 in HUVECs. Moreover, treatment of HUVECs with 1B9B9 for 1 h significantly reduced both Akt and ERK1,2 basal phosphorylation (Fig. 3B). Possible mechanisms of survival suppression by FGF-2 MAb were suggested by the reduced phosphorylation of MAPK and Akt.
The clinical evidence indicates that FGF-2 may play a role in tumor relapse in patients treated with anti-VEGF agents.(32) FGF-2 elicits its angiogenic effect via VEGF-dependent as well as VEGF-independent pathways.(33–35) Thus, the effect of anti-FGF-2 and anti-VEGF combinatory treatment might be superior to that of anti-VEGF treatment alone.(36) Accordingly, targeting FGF-2, in addition to VEGF, might result in synergistic effects in the treatment of angiogenesis-related diseases, including cancer.
In conclusion, we show that the rat iliac lymph node method can be used to produce neutralizing antibodies that react to FGF-2. Various approaches based on the inhibition of FGF-2 have been reported extensively.(37) This anti-FGF-2 antibody might be useful for resistance to anti-VEGF agents in cancer therapy. Further studies will be required, however, to test the universality of this approach.
Acknowledgments
The authors thank Ms. K. Onishi and Ms. H. Koga for providing technical support. We also wish to thank members of the Central Laboratory of Osaka City University Graduate School of Medicine for providing technical support. This work was supported by MEXT KAKENHI (grant no. 19790389 to M.S.; grant nos. 23310134 and 23114718 to Y.O).
Author Disclosure Statement
The authors confirm that there is no conflict of interest.
References
- 1.Flaumenhaft R, Moscatelli D, and Rifkin DB: Heparin and heparan sulfate increase the radius of diffusion and action of basic fibroblast growth factor. J Cell Biol 1990;111:1651–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flaumenhaft R, and Rifkin DB: The extracellular regulation of growth factor action. Mol Biol Cell 1992;3:1057–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forsten KE, Courant NA, and Nugent MA: Endothelial proteoglycans inhibit bFGF binding and mitogenesis. J Cell Physiol 1997;172:209–220 [DOI] [PubMed] [Google Scholar]
- 4.Ornitz DM, and Marie PJ: FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 2002;16:1446–1465 [DOI] [PubMed] [Google Scholar]
- 5.Duchesne L, Octeau V, Bearon RN, Beckett A, Prior IA, Lounis B, and Fernig DG: Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol 2012;10:e1001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eswarakumar VP, Lax I, and Schlessinger J: Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 2005;16:139–149 [DOI] [PubMed] [Google Scholar]
- 7.Turner N, and Grose R: Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116–129 [DOI] [PubMed] [Google Scholar]
- 8.Altomare DA, and Testa JR: Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005;24:7455–7464 [DOI] [PubMed] [Google Scholar]
- 9.Pepper MS, Ferrara N, Orci L, and Montesano R: Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun 1992;189:824–831 [DOI] [PubMed] [Google Scholar]
- 10.Asahara T, Bauters C, Zheng LP, Takeshita S, Bunting S, Ferrara N, Symes JF, and Isner JM: Synergistic effect of vascular endothelial growth factor and basic fibroblast growth factor on angiogenesis in vivo. Circulation 1995;92:II365–371 [DOI] [PubMed] [Google Scholar]
- 11.Casanovas O, Hicklin DJ, Bergers G, and Hanahan D: Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005;8:299–309 [DOI] [PubMed] [Google Scholar]
- 12.Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, Adinin R, Overman MJ, Valero V, Wen S, Lieu C, Yan S, Tran HT, Ellis LM, Abbruzzese JL, and Heymach JV: Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol 2010;28:453–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sado Y, Kagawa M, Kishiro Y, Sugihara K, Naito I, Seyer JM, Sugimoto M, Oohashi T, and Ninomiya Y: Establishment by the rat lymph node method of epitope-defined monoclonal antibodies recognizing the six different alpha chains of human type IV collagen. Histochem Cell Biol 1995;104:267–275 [DOI] [PubMed] [Google Scholar]
- 14.Muchardt C, Reyes JC, Bourachot B, Leguoy E, and Yaniv M: The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J 1996;15:3394–3402 [PMC free article] [PubMed] [Google Scholar]
- 15.de La Serna IL, Carlson KA, Hill DA, Guidi CJ, Stephenson RO, Sif S, Kingston RE, and Imbalzano AN: Mammalian SWI-SNF complexes contribute to activation of the hsp70 gene. Mol Cell Biol 2000;20:2839–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harada A, Okada S, Saiwai H, Aoki M, Nakamura M, and Ohkawa Y: Generation of a rat monoclonal antibody specific for Pax7. Hybridoma (Larchmt) 2009;28:451–453 [DOI] [PubMed] [Google Scholar]
- 17.Shiota M, Saiwai H, Mun S, Harada A, Okada S, Odawara J, Tanaka M, Iwao H, and Ohkawa Y: Generation of a rat monoclonal antibody specific for heat shock cognate protein 70. Hybridoma (Larchmt) 2010;29:453–456 [DOI] [PubMed] [Google Scholar]
- 18.Kishiro Y, Kagawa M, Naito I, and Sado Y: A novel method of preparing rat-monoclonal antibody-producing hybridomas by using rat medial iliac lymph node cells. Cell Struct Funct 1995;20:151–156 [DOI] [PubMed] [Google Scholar]
- 19.Ohkawa Y, Harada A, Nakamura M, Yoshimura S, and Tachibana T: Production of a rat monoclonal antibody against Brg1. Hybridoma (Larchmt) 2009;28:463–466 [DOI] [PubMed] [Google Scholar]
- 20.Shiota M, Kusakabe H, Izumi Y, Hikita Y, Nakao T, Funae Y, Miura K, and Iwao H: Heat shock cognate protein 70 is essential for Akt signaling in endothelial function. Arterioscler Thromb Vasc Biol 2010;30:491–497 [DOI] [PubMed] [Google Scholar]
- 21.Kakehashi A, Ishii N, Shibata T, Wei M, Okazaki E, Tachibana T, Fukushima S, and Wanibuchi H: Mitochondrial prohibitins and septin 9 are implicated in the onset of rat hepatocarcinogenesis. Toxicol Sci 2011;119:61–72 [DOI] [PubMed] [Google Scholar]
- 22.Rhoads DN, Eskin SG, and McIntire LV: Fluid flow releases fibroblast growth factor-2 from human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol 2000;20:416–421 [DOI] [PubMed] [Google Scholar]
- 23.Lim IK, Won Hong K, Kwak IH, Yoon G, and Park SC: Cytoplasmic retention of p-Erk1/2 and nuclear accumulation of actin proteins during cellular senescence in human diploid fibroblasts. Mech Age Dev 2000;119:113–130 [DOI] [PubMed] [Google Scholar]
- 24.Li X, Wang J, Zhang X, Zeng Y, Liang L, and Ding Y: Overexpression of RhoGDI2 correlates with tumor progression and poor prognosis in colorectal carcinoma. Ann Surg Oncol 2012;19:145–153 [DOI] [PubMed] [Google Scholar]
- 25.D'Angelo G, Struman I, Martial J, and Weiner RI: Activation of mitogen-activated protein kinases by vascular endothelial growth factor and basic fibroblast growth factor in capillary endothelial cells is inhibited by the antiangiogenic factor 16-kDa N-terminal fragment of prolactin. Proc Natl Acad Sci USA 1995;92:6374–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi C, Lu J, Wu W, Ma F, Georges J, Huang H, Balducci J, Chang Y, and Huang Y: Endothelial cell-specific molecule 2 (ECSM2) localizes to cell-cell junctions and modulates bFGF-directed cell migration via the ERK-FAK pathway. PloS One 2011;6:e21482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuzaki K, Yoshitake Y, Matuo Y, Sasaki H, and Nishikawa K: Monoclonal antibodies against heparin-binding growth factor II/basic fibroblast growth factor that block its biological activity: invalidity of the antibodies for tumor angiogenesis. Proc Natl Acad Sci USA 1989;86:9911–9915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hori A, Sasada R, Matsutani E, Naito K, Sakura Y, Fujita T, and Kozai Y: Suppression of solid tumor growth by immunoneutralizing monoclonal antibody against human basic fibroblast growth factor. Cancer Res 1991;51:6180–6184 [PubMed] [Google Scholar]
- 29.Tao J, Xiang JJ, Li D, Deng N, Wang H, and Gong YP: Selection and characterization of a human neutralizing antibody to human fibroblast growth factor-2. Biochem Biophys Res Commun 2010;394:767–773 [DOI] [PubMed] [Google Scholar]
- 30.Li D, Wang H, Xiang JJ, Deng N, Wang PP, Kang YL, Tao J, and Xu M: Monoclonal antibodies targeting basic fibroblast growth factor inhibit the growth of B16 melanoma in vivo and in vitro. Oncol Rep 2010;24:457–463 [DOI] [PubMed] [Google Scholar]
- 31.Bolitho C, Xu W, and Zoellner H: Negative feedback for endothelial apoptosis: a potential physiological role for fibroblast growth factor. J Vasc Res 2008;45:193–204 [DOI] [PubMed] [Google Scholar]
- 32.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, Drappatz J, Louis DN, Ivy P, Scadden DT, Benner T, Loeffler JS, Wen PY, and Jain RK: AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007;11:83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stavri GT, Zachary IC, Baskerville PA, Martin JF, and Erusalimsky JD: Basic fibroblast growth factor upregulates the expression of vascular endothelial growth factor in vascular smooth muscle cells. Synergistic interaction with hypoxia. Circulation 1995;92:11–14 [DOI] [PubMed] [Google Scholar]
- 34.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, and Mignatti P: Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol 1998;141:1659–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zubilewicz A, Hecquet C, Jeanny JC, Soubrane G, Courtois Y, and Mascarelli F: Two distinct signalling pathways are involved in FGF2-stimulated proliferation of choriocapillary endothelial cells: a comparative study with VEGF. Oncogene 2001;20:1403–1413 [DOI] [PubMed] [Google Scholar]
- 36.Stahl A, Paschek L, Martin G, Feltgen N, Hansen LL, and Agostini HT: Combinatory inhibition of VEGF and FGF2 is superior to solitary VEGF inhibition in an in vitro model of RPE-induced angiogenesis. Graefe Arch Clin Exp Ophthalmol 2009;247:767–773 [DOI] [PubMed] [Google Scholar]
- 37.Rusnati M, and Presta M: Fibroblast growth factors/fibroblast growth factor receptors as targets for the development of anti-angiogenesis strategies. Curr Pharm Des 2007;13:2025–2044 [DOI] [PubMed] [Google Scholar]