

Abstract
Some metabolic pathway enzymes are known to organize into multi-enzyme complexes for reasons of catalytic efficiency, metabolite channeling, and other advantages of compartmentalization. It has long been an appealing prospect that de novo purine biosynthesis enzymes form such a complex, termed the “purinosome.” Early work characterizing these enzymes garnered scarce but encouraging evidence for its existence. Recent investigations led to the discovery in human cell lines of purinosome bodies—cytoplasmic puncta containing transfected purine biosynthesis enzymes, which were argued to correspond to purinosomes. New discoveries challenge both the functional and physiological relevance of these bodies in favor of protein aggregation.
Introduction
Multi-enzyme complexes often engage in various forms of substrate channeling, in which sequential pathway enzymes “hand off” intermediate metabolic products amongst each other rather than release them into bulk solution. The advantages of such complexes include improved efficiency and optimized usage of short-lived intermediates.1-5
One such possible complex involves the enzymes for de novo purine biosynthesis. Termed the “purinosome,” the complex's existence has been long suggested by circumstantial evidence. The search for direct in vivo evidence of the purinosome culminated in the discovery of intracellular punctate bodies formed in part by purine biosynthesis enzymes.6 It was argued, based in part upon evidence for purine dependency, that these were functional complexes.6 However, new discoveries support a model where these bodies behave in a manner unlike that expected for functional purinosomes and instead have numerous features one might expect of simple protein aggregates or stress bodies.7 Here we review the literature evidence characterizing purinosome bodies, and discuss the models that involve metabolically active associations as opposed to general protein aggregation.
Purine biosynthesis
Purines are ubiquitous and essential components of DNA and RNA, and their derivatives participate in numerous biological processes. Adenine and guanine nucleotides are derived from the compound inosine monophosphate (IMP), which is synthesized de novo from phosphoribosyl pyrophosphate (PRPP) through a highly conserved multi-step de novo purine biosynthesis pathway. In higher eukaryotes (such as humans), the pathway consists of six enzymes catalyzing ten sequential reactions converting PRPP to IMP (Table 1). De novo purine biosynthesis activity is up-regulated when the cellular demand for purines exceeds that supplied by the purine salvage pathway, a single-step conversion of hypoxanthine to IMP catalyzed by hypoxanthine phosphoribosyltransferase (HPRT). Conversely, de novo biosynthesis is down-regulated when exogenous purine, i.e. hypoxanthine, is available.
Table 1.
Six human enzymes catalyze the ten-step conversion of phosphoribosyl pyrophosphate to inosine monophosphate.
| Step(s) | Gene symbol | Description |
|---|---|---|
| 1 | PPAT | Phosphoribosylpyrophosphate amidotransferase |
| 2,3,5 | GART | Trifunctional phosphoribosylglycinamide formyltransferase / phosphoribosylglycinamide synthetase / phosphoribosylaminoimidazole synthetase |
| 4 | FGAMS | Phosphoribosylformylglycinamidine synthase |
| 6,7 | PAICS | Bifunctional phosphoribosylaminoimidazole carboxylase / phosphoribosylaminoimidazole succinocarboxamide synthetase |
| 8 | ADSL | Adenylosuccinate lyase |
| 9,10 | ATIC | Bifunctional 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase / IMP cyclohydrolase |
The search for a complex
The hypothesis that the de novo purine biosynthesis enzymes organize into a multi-enzyme complex has long been attractive based at least in part on the chemical instability of 5-phosphoribosylamine, the first intermediate substrate in the pathway, which suggests an essential direct transfer between PPAT and GART.8 Additionally, the consolidation of several individual enzymatic functions onto single bifunctional or trifunctional polypeptide chains has been observed in many organisms,9, 10 which suggests stable physical interactions between these enzymes may exist even in organisms which do not consolidate these enzymes on a single polypeptide chain.11 The joining of the non-sequential steps 2, 3, and 5 into a single trifunctional enzyme in humans also suggests that this polypeptide may be further, non-covalently juxtaposed with the enzyme for step 4.
However, historic experiments to isolate an intact purinosome have been largely unsuccessful. Kinetic studies revealed evidence for substrate channeling between PPAT and GART, but attempts to detect physical protein-protein interactions failed.12 Later studies found that pairs of purine biosynthesis pathway members (including PPAT and GART, and ATIC and GART) could be enriched by co-fractionation under some conditions,13-15 and purine-dependent protein-protein interactions have been observed using a luciferase reporter system.16 Even so, transfected recombinant GART was not found to be co-localized to any cellular architecture that might serve as a structural scaffold to assemble a multi-enzyme complex,17 and no biophysical support was found for a larger multi-enzyme complex or a fully intact purinosome.13-15 While a complex representing the purinosome may physically exist, it may form only transiently or in a condition-specific manner.
The discovery of purinosome bodies
In 2008, the human purinosome was thought to have finally been identified by the discovery that fluorescent protein-tagged recombinant purine biosynthesis enzymes form intracellular punctate bodies when transiently expressed in human cell culture.6 Bodies formed by FGAMS-GFP were shown to co-localize with bodies formed by the five other enzymes of the pathway, suggesting that the bodies contained all direct participants of de novo purine biosynthesis pathway. These so-called “purinosome bodies” apparently formed in purine-depleted medium and disassembled in purine-rich medium, a potentially strong indication of function. Moreover, microtubule inhibitors that disfavored purinosome body formation also led to decreased purine synthesis.18 Finally, it has been shown that purinosome body assembly is under casein kinase II (CK2) and GPCR control, and could be modulated by heat shock chaperones,7, 19-21 and in parallel, modulation of CK2 and GPCR activities have been shown to correlate with changes in the overall spatial distribution of bulk intracellular protein.20
Conflicting evidence
While there is evidence that supports the hypothesis that purinosome bodies form in a purine-dependent and reversible manner, it is possible that these effects are not solely due to the presence or absence of purines. For example, while it has been reported that purinosome bodies form when cells are cultured in purine-depleted medium, the specific growth medium employed for these experiments actually significantly differed from the purine-rich control medium in ways that went beyond the mere presence or absence of purines. The purine-depleted medium was generated by serum dialysis with a 25,000 Da pore size, which would have removed a variety of compounds other than purines (which are only ~100-500 Da); additionally, the serum supplementation was doubled for the “purine-rich” medium.6 These non-conservative changes confound interpretation of the purine-dependency of the observations. Notably, in other experiments, altering only purine levels did not affect purinosome body formation: purinosome bodies were unaffected after specifically adding an exogenous purine source (hypoxanthine) back to “purine-depleted” medium,6 or after specifically adding a purine antagonist (azaserine).6 These contrary observations suggest that the bodies may not form in response to purine levels, but are instead dependent on other factors (we will suggest aggregation, below). To further complicate matters, independent laboratories have reported both induction and no induction of purinosome body formation by similar bulk nutrient-depletion experiments,6, 7, 22, 23 further suggesting that unknown factors unrelated to purine concentration might account for the observed phenomena.
As structures for enhancing purine synthesis, purinosomes should exhibit abundant metabolic flux. It has been shown that flux through the de novo purine biosynthesis pathway is suppressed when purinosome body assembly is disrupted by nocodazole. While this correlation supports a role for microtubules in establishing purinosome bodies in live cells18 these observations may also arise from an alternative explanation, namely that given that nocodazole is a cell cycle arresting agent, the combination of nutrient-poor medium and arrested cell cycle might also be expected to greatly impede metabolic flux. Such experiments illustrate the intrinsic difficulties in distinguishing flux contributed by the bodies and flux contributed by the free, un-localized pool of the same enzymes. Experiments have so far monitored 14C-glycine incorporation into the pool of cellular purines and measured the complete cellular complement of purine biosynthetic enzymes, not explicitly flux through the bodies; to date, no experiments have demonstrated that the bodies themselves provide any metabolic flux. Thus, further kinetic experiments are required before purinosome bodies meet the standard of proof associated with other well-characterized metabolic enhancing structures like acetyl-CoA carboxylase polymers24-27 or quaternary structures verified for substrate channeling, such as for tryptophan synthase.4, 28,29
Additionally, with one exception,22 all literature evidence for purinosome bodies has thus far relied on transiently expressed recombinant fluorescent protein fusion constructs. This may be a consequence of reported difficulties and possible artifacts surrounding native immunofluorescent labeling of purine biosynthesis enzymes.6, 7 For example, it was observed that while endogenous GART behaved similarly to the GART fusion construct in “purine-depleted” medium, its endogenous behavior did not correspond to that observed for its fusion construct in “purine-rich” medium. 6 The sole reliance on microscopy with fusion proteins, and the absence of other physical demonstrations of the purinosome as a whole, argues for caution in interpretation.
What are purinosome bodies?
Somewhat problematically, published purinosome bodies have varied widely in their morphologies, ranging from pinpoint foci to oil droplet-like, and methods have not been developed yet either to classify purinosome bodies or to distinguish them from aggregates based on morphology. In our own studies, we have observed a dynamic spectrum of morphologies, as well as both increases and decreases in the numbers of bodies per cell even over the course of unperturbed growth.7 Future work clearly remains to address and characterize the different morphologies and kinetics of purinosome bodies, and to discover cellular markers that can securely distinguish bona fide purinosomes, should they exist.30
Another issue that confounds interpretation is that it is unclear whether the purine enzyme fusion constructs function similarly to endogenous enzymes. Others have observed that fluorescent protein fusions are subject to aggregation.31, 32 Even if we assume that some fluorescent protein fusions function similarly to the native proteins, there is large heterogeneity observed in purinosome body formation. The penetrance of body formation following transient transfection varies widely across enzymes, with individual enzyme body formation rates ranging from 5% to 77% of the transfected cells.6, 7 This heterogeneity, persisting even in cell populations treated with pharmacophores that promote purinosome bodies (15 to 95% penetrance of assembly),19 is inconsistent with the hypothesis that each body contains all members of the pathway, and at a minimum, such bodies are unlikely to represent complete purinosomes.
The constituents of purinosome bodies as measured by microscopy are also sometimes in disagreement with biochemically-captured purine enzyme protein-protein associations. As previously shown through partial co-purification, two folate metabolism enzymes (serine hydroxymethyltransferase (SHMT1) and C1THF synthase) associate with the folate-utilizing purine biosynthesis enzymes GART and ATIC.15, 33 GART's catalytic activity actually requires interaction with C1THF synthase or its analog.33, 34 However, these two folate metabolism enzymes were found to be excluded from purinosome bodies.6, 21 Meanwhile, an additional folate enzyme MTHFS (which catalyzes the conversion of a product from SHMT1 into substrate for C1THF synthase) was independently discovered to co-localize to purinosome bodies.23 Beyond C1THF synthase, a multitude of additional proteins not previously implicated in purine biosynthesis have been localized to purinosome bodies, including Hsp70, Hsp90, ubiquitin, Bag2, Bag5, Stip1/Hop, p23, DnaJ-C7 (Hsp40), and DnaJ-A1 (Hsp40).7, 21 Many of these proteins found associated with the purinosome body are commonly associated with deleterious protein aggregates.35-44 The relatively high rate of co-localization between an individual purine biosynthesis enzyme and Hsp70 compared to that between pairs of purine biosynthesis enzymes gives rise to the hypothesis that the purinosome bodies are non-specific aggregates rather than specific functional metabolic complexes.7
Growing evidence for protein aggregation
Many enzymes can aggregate under conditions of cellular stress or recombinatorial expression, and as we discussed above, the medium used to observe inducibility of purinosome bodies was broadly depleted of nutrients.6 Unsurprisingly, a switch to this nutrient-depleted medium might be stressful for some cells (e.g., HTB-125 cells cannot survive in the depleted medium).6 It is therefore possible that this stress contributed to the visible aggregation of recombinant purine enzyme constructs as stress bodies, which would then have been reversed by providing the cell with nutrients necessary to dissociate or clear aggregated protein bodies. Our study in 2013 supports this simpler explanation that purinosome bodies may be aggregated proteins that commonly result from fusion protein expression.7 Many characteristics of purinosome bodies were found to be shared between those of canonical protein aggregates, including body morphology, inducibility by diverse stress, co-localization with ubiquitin and chaperones, and correlation with transfected DNA quantity, protein aggregation potential, and increased cell death.7 In this regard, purinosome bodies exhibit overlapping co-localization with a recombinant fusion protein shown to mark aggregates and aggresomes21, 31 (Fig. 1), and many literature figures of purinosome bodies closely resemble the spectrum of morphologies formed by disease-associated protein aggregates, including both huntingtin and α-synuclein (Fig. 2). In accordance with the notion that aggregated proteins are inherently cytotoxic,45 cells exhibiting purinosome bodies were also found to be associated with early cell death,7 although it is unclear whether the bodies were a cause of that stress or rather an indicator of stressed cells.
Figure 1.

Partial co-localization of [A] gp-250 (also known as GFP-250) with [B] FGAMS-OFP, co-transfected in the same cell, as reported by French et al.21 The overlay in [C] shows the merge of panel A in green and panel B in red (Pearson's coefficient of 0.4 and overlap coefficient of 0.41)21 , with regions of co-localized expression extracted and plotted in [D] for clarity. Notably, over-expression of the GFP-chimera, gp-250, has been reported to form insoluble aggregates that are delivered to aggresomes.31, 63 Reprinted with permission from the Proceedings of the National Academy of Sciences.
Figure 2.
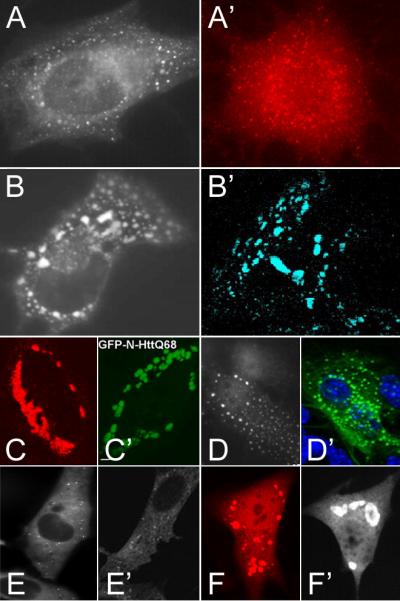
Intracellular purinosome bodies display a spectrum of morphologies similar to bodies formed by canonical protein aggregates. [A-F] are purinosome bodies formed in HeLa cells. [A’-F’] are various aggregated disease-associated protein inclusions formed in various mammalian cell lines. We can roughly classify these representative (but not exhaustive) morphologies into arbitrary categories: Pinpoint foci include [A] TrifGART-GFP from An et al. 20086 and [A’] α-synuclein aggregates in oligodendroglial cells, from Riedel et al.66 Droplet-like clusters are evident in [B] FGAMS-GFP from An et al. 20086 and [B’] a mutated form of glial fibrillary acidic protein (GFAP) attached to CFP transiently transfected into modified astrocytes, from Mignot et al.67 Bulk clumps are shown in [C] FGAMS-OFP transiently expressed in HeLa cells from Field et al.23 and [C’] GFP-tagged Huntingtin fragment with polyglutamine expansion transiently expressed in HeLa cells, from Bjørkøy et al.68 Spherical puncta are shown in [D] FGAMS-OFP from An et al. JBC 201019 and [D’] GFAP-GFP transiently transfected into astrocytes, from Mignot et al.67 Sparse pinpoint foci are formed by [E] FGAMS-GFP from An et al. PNAS 2010,18 and [E’] a fragment of p62 protein, a common component of disease-associated protein aggregates,69-72 tagged to GFP and transiently expressed in NIH3T3 fibroblasts (although the authors Bjørkøy et al.68 classify this subtler morphology as diffuse). Large oil-droplet-like bodies are formed by [F] hTrifGART-OFP transfected in HeLa cells from Field et al.23 and [F’] α-synuclein-EGFP transfected into H4 neuroglioma cells, from McLean et al.73 All images are reproduced with permission from the respective publishers.
Follow-up studies characterizing purinosome bodies as functional assemblies have also uncovered a surprising number of additional features that parallel features of disease-associated aggregates. For example, as pointed out earlier, inhibition of microtubule polymerization with nocodazole blocks formation of purinosome bodies and reduces cellular flux of de novo purine biosynthesis.18 Likewise, nocodazole also blocks formation of inclusion bodies and aggresomes (bodies containing protein aggregates)46-48 and places cells in G2/M arrest. In a similar fashion, partial inhibition of casein kinase 2 (CK2) by small molecule inhibitors was found to induce purinosome body formation.19 Inhibition of CK2 is also known to disrupt hundreds of cellular processes,49 among them being protein homeostasis which regulates protein aggregation.50, 51 Notably, CK2 inhibitors are highly non-specific,52 while some induce oxidative stress53, 54 (a promoter of purinosome body formation)7 and apoptosis.55 Additionally, the observation that CK2 inhibitors affect FGAMS, GART, and PPAT but do not affect PAICS, ADSL, or ATIC unless a member from the latter set is co-transfected with one in the former19 suggests either that individual members of the pathway are differentially and complexly regulated to assemble bodies or that these observations may be transfection artifacts. As well, the claim that the formation of purinosome bodies can be controlled by the addition of Gαi agonists or antagonists20 may again be more simply explained by aggregation: downstream Gαi targets (e.g., the PI3K/Akt pathway) include regulators of various stress-related cellular responses, such as cell survival and protein synthesis, which will also influence protein homeostasis and aggregation. Notably, G protein signaling has been implicated in regulating autophagy (a mechanism known to clear aggregated proteins)56, 57 through Gαi-control of autophagic sequestration58, 59 and through autophagic vacuole formation.60
While it appears that purinosome bodies may be the result of stress-induced aggregation, it is still not entirely clear what they are. Purinosome bodies are distinct from stress granules21 (messenger ribonucleoprotein bodies composed of translation initiation factors and mRNAs),61, 62 but additional work is necessary to differentiate them from other forms of stress-inducible bodies (Table 2). While some data suggest chaperones promote purinosome body assembly, observations supporting the contrary also exist. Experiments perturbing the activity of heat shock chaperones (e.g., Hsp70 or Hsp90) by pharmacophores can promote purinosome body assembly or disassembly depending on the drug concentration and treatment duration,7, 21 and further study is necessary to clarify the disparity.
Table 2.
Some of the protein-rich bodies identified in the eukaryote cytosol.
| Term | Description | Example markers |
|---|---|---|
| Aggresomes | Large pericentriolar core of aggregated, ubiquitinated proteins encaged by vimentin; emerges from microtubule-dependent collection of small aggregates distributed throughout the cell typically in response to inhibited or overwhelmed proteasome. | CTFR46, GFP-25031, 63 |
| Inclusion bodies | Typically used to describe insoluble, dense protein particles in bacteria and virus infected cells. This term is sometimes applied to describe human disease-associated aggregates. | Huntingtin64 |
| Liquid droplets | Polymeric assemblies of multivalent proteins that produce a liquid-liquid phase separation in vitro and manifest in cells as dynamic puncta. | SH3+PRM65 |
| P-bodies | Stress-inducible cytoplasmic RNA/protein granules involved in mRNA decay; can physically associate with stress granules to receive sequestered mRNAs shuttled for degradation. | DCP1a62 |
| Stress bodies | General term for proteinaceous assemblies of unknown nature correlated with stress; frequently, but not exclusively, refers to nuclear stress bodies. | FGAMS-GFP7 |
| Stress granules | Stress-inducible cytoplasmic granules composed of stalled translation pre-initiation complexes, postulated to protect untranslated mRNAs from potentially harmful conditions. | G3BP62 |
Speculations and conclusions
The idea of a purinosome has many compelling features. Assorted evidence ranging from metabolic flux and channeling considerations to pairwise kinetic or physical interactions observed between particular biosynthetic enzymes support the idea of some form of physical association between purine biosynthetic enzymes. Thus, there is a substantial body of literature suggesting the existence of the purinosome. Here, we address confounding issues surrounding the interpretation of punctate bodies containing purine biosynthetic enzymes (purinosome bodies). Although current observations do not negate the possible formation of functional purinosomes, it also does not suggest that purinosome bodies must be the purinosome. A far simpler explanation is plausible: that the purine biosynthetic enzymes can aggregate under conditions of cellular stress or recombinant expression. Indeed, it is well known that many other intracellular enzymes form aggregates under similar conditions, and purine biosynthetic enzymes are not known to be special in this regard. This discrepancy between purinosome body interpretations highlights the need for caution regarding reliable methodologies for observing punctate body formation and determining their function and physiological relevance.
Acknowledgements
The authors acknowledge support by grants from the National Institutes of Health, National Science Foundation, Cancer Prevention and Research in Texas, and Welch Foundation to EMM (F-1515) and to ADE (F-1654). MT acknowledges funding from the University of Cambridge. This review had its inception in discussions of the purinosome on Wikipedia and in PLoS One readers’ comments.30
References
- 1.Spivey HO, Ovadi J. Methods. 1999;19:306–321. doi: 10.1006/meth.1999.0858. [DOI] [PubMed] [Google Scholar]
- 2.Miles EW, Rhee S, Davies DR. J. Biol. Chem. 1999;274:12193–12196. doi: 10.1074/jbc.274.18.12193. [DOI] [PubMed] [Google Scholar]
- 3.Holden HM, Thoden JB, Raushel FM. Curr. Opin. Struct. Biol. 1998;8:679–685. doi: 10.1016/s0959-440x(98)80086-9. [DOI] [PubMed] [Google Scholar]
- 4.Pan P, Woehl E, Dunn MF. Trends Biochem. Sci. 1997;22:22–27. doi: 10.1016/s0968-0004(96)10066-9. [DOI] [PubMed] [Google Scholar]
- 5.Ovadi J. J. Theor. Biol. 1991;152:1–22. [PubMed] [Google Scholar]
- 6.An S, Kumar R, Sheets ED, Benkovic SJ. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- 7.Zhao A, Tsechansky M, Swaminathan J, Cook L, Ellington AD, Marcotte EM. PLoS One. 2013;8:e56203. doi: 10.1371/journal.pone.0056203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schendel FJ, Cheng YS, Otvos JD, Wehrli S, Stubbe J. Biochemistry. 1988;27:2614–2623. doi: 10.1021/bi00407a052. [DOI] [PubMed] [Google Scholar]
- 9.Henikoff S, Keene MA, Sloan JS, Bleskan J, Hards R, Patterson D. Proc. Natl. Acad. Sci. USA. 1986;83:720–724. doi: 10.1073/pnas.83.3.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aimi J, Qiu H, Williams J, Zalkin H, Dixon JE. Nucleic Acids Res. 1990;18:6665–6672. doi: 10.1093/nar/18.22.6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 12.Rudolph J, Stubbe J. Biochemistry. 1995;34:2241–2250. doi: 10.1021/bi00007a019. [DOI] [PubMed] [Google Scholar]
- 13.McCairns E, Fahey D, Sauer D, Rowe PB. J. Biol. Chem. 1983;258:1851–1856. [PubMed] [Google Scholar]
- 14.Rowe PB, McCairns E, Madsen G, Sauer D, Elliott H. J. Biol. Chem. 1978;253:7711–7721. [PubMed] [Google Scholar]
- 15.Caperelli CA, Benkovic PA, Chettur G, Benkovic SJ. J. Biol. Chem. 1980;255:1885–1890. [PubMed] [Google Scholar]
- 16.Deng Y, Gam J, French JB, Zhao H, An S, Benkovic SJ. J. Biol. Chem. 2012;287:36201–36207. doi: 10.1074/jbc.M112.407056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gooljarsingh LT, Ramcharan J, Gilroy S, Benkovic SJ. Proc. Natl. Acad. Sci. USA. 2001;98:6565–6570. doi: 10.1073/pnas.121182998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.An S, Deng Y, Tomsho JW, Kyoung M, Benkovic SJ. Proc. Natl. Acad. Sci. USA. 2010;107:12872–12876. doi: 10.1073/pnas.1008451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.An S, Kyoung M, Allen JJ, Shokat KM, Benkovic SJ. J. Biol. Chem. 2010;285:11093–11099. doi: 10.1074/jbc.M110.101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verrier F, An S, Ferrie AM, Sun H, Kyoung M, Deng H, Fang Y, Benkovic SJ. Nat. Chem. Biol. 2011;7:909–915. doi: 10.1038/nchembio.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.French JB, Zhao H, An S, Niessen S, Deng Y, Cravatt BF, Benkovic SJ. Proc. Natl. Acad. Sci. USA. 2013;110:2528–2533. doi: 10.1073/pnas.1300173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baresova V, Skopova V, Sikora J, Patterson D, Sovova J, Zikanova M, Kmoch S. Hum. Mol. Genet. 2012;21:1534–1543. doi: 10.1093/hmg/ddr591. [DOI] [PubMed] [Google Scholar]
- 23.Field MS, Anderson DD, Stover PJ. Front. Genet. 2011;2:36. doi: 10.3389/fgene.2011.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beaty NB, Lane MD. J. Biol. Chem. 1983;258:13051–13055. [PubMed] [Google Scholar]
- 25.Beaty NB, Lane MD. J. Biol. Chem. 1983;258:13043–13050. [PubMed] [Google Scholar]
- 26.Meredith MJ, Lane MD. J. Biol. Chem. 1978;253:3381–3383. [PubMed] [Google Scholar]
- 27.Clarke BA, Clarke SD. Arch. Biochem. Biophys. 1982;218:92–100. doi: 10.1016/0003-9861(82)90324-1. [DOI] [PubMed] [Google Scholar]
- 28.Hyde CC, Miles EW. Biotechnology (N Y) 1990;8:27–32. doi: 10.1038/nbt0190-27. [DOI] [PubMed] [Google Scholar]
- 29.Dunn MF. Arch. Biochem. Biophys. 2012;519:154–166. doi: 10.1016/j.abb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.PLoS One Reader's Comments, Transiently Transfected Purine Biosynthetic Enzymes Form Stress Bodies. doi: 10.1371/journal.pone.0056203. http://www.plosone.org/article/comments/info%3Adoi%2F10.1371%2Fjournal.pone.0056203. [DOI] [PMC free article] [PubMed]
- 31.Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. J. Cell Biol. 1999;146:1239–1254. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. Nat. Methods. 2012;9:480–482. doi: 10.1038/nmeth.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith GK, Mueller WT, Wasserman GF, Taylor WD, Benkovic SJ. Biochemistry. 1980;19:4313–4321. doi: 10.1021/bi00559a026. [DOI] [PubMed] [Google Scholar]
- 34.Smith GK, Mueller WT, Benkovic PA, Benkovic SJ. Biochemistry. 1981;20:1241–1245. doi: 10.1021/bi00508a029. [DOI] [PubMed] [Google Scholar]
- 35.Broadley SA, Hartl FU. FEBS Lett. 2009;583:2647–2653. doi: 10.1016/j.febslet.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 36.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Nat. Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chai Y, Koppenhafer SL, Bonini NM, Paulson HL. J. Neurosci. 1999;19:10338–10347. doi: 10.1523/JNEUROSCI.19-23-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uryu K, Richter-Landsberg C, Welch W, Sun E, Goldbaum O, Norris EH, Pham CT, Yazawa I, Hilburger K, Micsenyi M, Giasson BI, Bonini NM, Lee VM, Trojanowski JQ. Am. J. Pathol. 2006;168:947–961. doi: 10.2353/ajpath.2006.050770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jana NR, Tanaka M, Wang G, Nukina N. Hum. Mol. Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt T, Lindenberg KS, Krebs A, Schols L, Laccone F, Herms J, Rechsteiner M, Riess O, Landwehrmeyer GB. Ann. Neurol. 2002;51:302–310. doi: 10.1002/ana.10101. [DOI] [PubMed] [Google Scholar]
- 41.McLean PJ, Kawamata H, Shariff S, Hewett J, Sharma N, Ueda K, Breakefield XO, Hyman BT. J. Neurochem. 2002;83:846–854. doi: 10.1046/j.1471-4159.2002.01190.x. [DOI] [PubMed] [Google Scholar]
- 42.Kalia LV, Kalia SK, Chau H, Lozano AM, Hyman BT, McLean PJ. PLoS One. 2011;6:e14695. doi: 10.1371/journal.pone.0014695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shinder GA, Lacourse MC, Minotti S, Durham HD. J. Biol. Chem. 2001;276:12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- 44.Muchowski PJ, Wacker JL. Nat. Rev. Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 45.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 46.Johnston JA, Ward CL, Kopito RR. J. Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaminosono S, Saito T, Oyama F, Ohshima T, Asada A, Nagai Y, Nukina N, Hisanaga S. J. Neurosci. 2008;28:8747–8755. doi: 10.1523/JNEUROSCI.0973-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muchowski PJ, Ning K, D'Souza-Schorey C, Fields S. Proc. Natl. Acad. Sci. USA. 2002;99:727–732. doi: 10.1073/pnas.022628699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meggio F, Pinna LA. FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 50.Watabe M, Nakaki T. J. Cell Sci. 2011;124:1519–1532. doi: 10.1242/jcs.081778. [DOI] [PubMed] [Google Scholar]
- 51.Watabe M, Nakaki T. Commun. Integr. Biol. 2012;5:278–280. doi: 10.4161/cib.19473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pagano MA, Bain J, Kazimierczuk Z, Sarno S, Ruzzene M, Di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F, Pinna LA. Biochem. J. 2008;415:353–365. doi: 10.1042/BJ20080309. [DOI] [PubMed] [Google Scholar]
- 53.Schneider CC, Hessenauer A, Gotz C, Montenarh M. Oncol. Rep. 2009;21:1593–1597. doi: 10.3892/or_00000392. [DOI] [PubMed] [Google Scholar]
- 54.Ahmad KA, Wang G, Ahmed K. Mol. Cancer Res. 2006;4:331–338. doi: 10.1158/1541-7786.MCR-06-0073. [DOI] [PubMed] [Google Scholar]
- 55.Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K. Adv. Enzyme Regul. 2008;48:179–187. doi: 10.1016/j.advenzreg.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tyedmers J, Mogk A, Bukau B. Nat. Rev. Mol. Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 57.Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Curr. Top. Dev. Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 58.Ogier-Denis E, Couvineau A, Maoret JJ, Houri JJ, Bauvy C, De Stefanis D, Isidoro C, Laburthe M, Codogno P. J. Biol. Chem. 1995;270:13–16. doi: 10.1074/jbc.270.1.13. [DOI] [PubMed] [Google Scholar]
- 59.Ogier-Denis E, Houri JJ, Bauvy C, Codogno P. J. Biol. Chem. 1996;271:28593–28600. doi: 10.1074/jbc.271.45.28593. [DOI] [PubMed] [Google Scholar]
- 60.Kadowaki M, Venerando R, Miotto G, Mortimore GE. J. Biol. Chem. 1994;269:3703–3710. [PubMed] [Google Scholar]
- 61.Anderson P, Kedersha N. J. Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kedersha N, Anderson P. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 63.Garcia-Mata R, Gao YS, Sztul E. Traffic. 2002;3:388–396. doi: 10.1034/j.1600-0854.2002.30602.x. [DOI] [PubMed] [Google Scholar]
- 64.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Molecular biology of the cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, Russo PS, Jiang QX, Nixon BT, Rosen MK. Nature. 2012;483:336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Riedel M, Goldbaum O, Schwarz L, Schmitt S, Richter-Landsberg C. PLoS One. 2010;5:e8753. doi: 10.1371/journal.pone.0008753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mignot C, Delarasse C, Escaich S, Della Gaspera B, Noe E, Colucci-Guyon E, Babinet C, Pekny M, Vicart P, Boespflug-Tanguy O, Dautigny A, Rodriguez D, Pham-Dinh D. Exp. Cell Res. 2007;313:2766–2779. doi: 10.1016/j.yexcr.2007.04.035. [DOI] [PubMed] [Google Scholar]
- 68.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. J. Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuusisto E, Salminen A, Alafuzoff I. Neuropathol. Appl. Neurobiol. 2002;28:228–237. doi: 10.1046/j.1365-2990.2002.00394.x. [DOI] [PubMed] [Google Scholar]
- 70.Kuusisto E, Salminen A, Alafuzoff I. Neuroreport. 2001;12:2085–2090. doi: 10.1097/00001756-200107200-00009. [DOI] [PubMed] [Google Scholar]
- 71.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H. Am. J. Pathol. 2002;160:255–263. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nagaoka U, Kim K, Jana NR, Doi H, Maruyama M, Mitsui K, Oyama F, Nukina N. J. Neurochem. 2004;91:57–68. doi: 10.1111/j.1471-4159.2004.02692.x. [DOI] [PubMed] [Google Scholar]
- 73.McLean PJ, Kawamata H, Hyman BT. Neuroscience. 2001;104:901–912. doi: 10.1016/s0306-4522(01)00113-0. [DOI] [PubMed] [Google Scholar]