

Abstract
Prior to the 1990s, genetic analyses indicated that many autoimmune diseases are driven by T cell responses; however, the identity of the pathogenic T cell populations responsible for dysfunctional autoimmune responses remained unclear. Some 20 years ago, the discovery of numerous chemokines and their receptors along with the development of specific mAbs to these provided a distinct advance. These new tools revealed a remarkable dichotomy and disclosed that some chemokine receptors guided the constitutive migration of T cells through lymphoid tissues, whereas others, such as CCR5 and CXCR3, guided effector and memory T cell migration to inflammatory lesions. These T cell markers enabled a new understanding of immune responses and the types of T cells involved in different inflammatory reactions.
Chemokines and their receptors: leukocyte traffic control
Chemokines were all the rage in the 1990s and seemed to explain so many things, notably the specificity of a leukocyte to enter a given tissue. Furthermore, the discovery of chemokines indicated that it wasn’t just cell adhesion molecules, such as selectins and integrins, that were important for directing leukocyte trafficking. As originally proposed by Springer and Butcher (1, 2), chemokines participate in the multistep process of leukocyte extravasation and contribute to the combinatorial code that determines selectivity. In fact, the ensuing discovery of so many different chemokines and chemokine receptors — many more than the subtypes of selectins or integrins — suggested that the chemokine system dictated much of the fine specificity of leukocyte traffic. It was proposed and shown that chemokines and their receptors determine gut homing, skin homing, and lymphoid tissue homing as well as Th1- and Th2- dependent immune responses (3). In addition, chemokines and their receptors appeared to associate with particular types of immune responses, allowing for collective recruitment and interaction of several leukocyte types. A good example is the eotaxin receptor CCR3, which is expressed by multiple parasite/allergy-associated leukocytes, including eosinophils, basophils, mast cells, and Th2 cells. Likewise CXCR5 is expressed by T follicular helper (Tfh) cells and B cells and facilitates germinal center reactions. The identification and characterization of chemokines and their receptors explained, to a large degree, how the immune system was organized and how immune cells were recruited to and interacted within specific tissues.
T cells that enter nonlymphoid tissues provide front-line protection against pathogens; however, in autoimmune disease and allergy, their role is more sinister. It was therefore important to understand the nature of the T cells that actually enter inflammatory sites, as this might be informative of the type of immune responses that develop in response to invasion and in association with autoimmune disease (Th1, Th2, etc.). Prior to 1990, immunologists had assumed that most lymphocytes randomly recirculated throughout the body. Studies then emerged demonstrating that naive T cells migrated preferentially through lymph nodes via high endothelial venules (HEV), whereas memory/effector (CD45RO+) T cells migrated to peripheral tissues, such as skin (4, 5). At this time, the precise nature of the T cells that traffic to inflammatory lesions received little attention.
In the 1990s, the discovery of numerous chemokine receptors provided new T cell markers, including CCR5 and CXCR3, which bound the so-called “inflammatory” chemokines, RANTES (CCL5) and IP-10 (CXCL10), respectively. At LeukoSite Inc., Boston, we pioneered methods for developing mAbs to chemokine receptors, and these mAbs proved valuable for characterizing chemokine receptor biology. For instance, use of these antibodies revealed that CCR5 and CXCR3 preferentially mark Th1 cells rather than Th2 cells (3). The results of these and other studies led to the notion that certain chemokines and their receptors were intimately associated with certain types of inflammatory responses, whereas others were associated with constitutive homing by naive as well as a subset of memory T cells through lymph nodes. In support of this concept, CXCR4 and CCR7 proved to be receptors for lymph node homing by T cells, whereas CCR5, CXCR3, and later CCR6 were determined to mark subsets of effector/memory T cells (6).
Chemokine receptors define T cell subsets
The above information provided a backdrop to the relatively simple question: what is the actual phenotype of T cells associated with different inflammatory reactions, particularly autoimmune lesions? The answer to this may enable development of novel inhibitors to chemokine receptors with potential to limit inflammation in autoimmune and allergic diseases, such as rheumatoid arthritis (RA), asthma, inflammatory bowel disease (IBD), multiple sclerosis (MS), and others. One of the striking results from our 1998 study published in the JCI (7) was the revelation that CXCR3 and CCR5 marked a relatively small subset of T cells in blood, but were present on the vast majority of T cells in synovial fluid or synovium from patients with RA. This striking discrepancy indicated that there was a strong and highly selective recruitment of T cells from blood to the sites of this inflammatory reaction. Furthermore, our results implied that these CXCR3+CCR5+ T cells actually contribute to disease pathogenesis and suggested that relevant chemokines, CCR5 ligands, or CXCR3 ligands might be attracting these T cells, thereby promoting disease. It now appears that this CXCR3+ and/or CCR5+ T cell subset associates with many other inflammatory lesions in human autoimmune diseases, notably the colonic mucosa in ulcerative colitis and myelinated neural tissue in MS (8). CCR5+CXCR3+ T cells are an effector population associated with IFN-γ production (3), although CXCR3 can also be expressed by Th17 T cells. This suggests that IFN-γ or IL-17 is associated with the abundance of CCR5+CXCR3+ T cells in the arthritic joint and so contributes to disease pathogenesis. Interestingly, polymorphisms within the gene encoding IFN-γ have been linked to human RA (9).
Targeting CCR5 and CXCR3 in inflammatory disease
The results of our 1998 study (7) suggested that chemokine or receptor inhibition could be an effective treatment for inflammatory diseases such as RA. In animal models of inflammatory arthritis and the experimental autoimmune encephalomyelitis (EAE) model of MS, chemokine and receptor inhibitors ameliorate disease (reviewed in ref. 10); however, use of these inhibitors in human clinical trials has so far been frustrating. Maraviroc, a small molecule CCR5 inhibitor, failed to demonstrate efficacy in a trial of patients with RA; therefore, at least in humans, CCR5 appears not to be the critical molecule for recruitment of disease-causing cells. CXCR3 inhibitors have not yet entered clinical trials, although several companies have performed clinical trials with mAb inhibitors of one CXCR3 ligand, IP-10 (CXCL10), with promising results; however, it still remains to be determined whether CCR5+CXCR3+ T cells are driving pathogenesis in human RA, IBD, and other inflammatory diseases. There is evidence that other pathways may underlie these diseases. For example, in KxB/N, SKG, and collagen-induced murine models of inflammatory arthritis, inhibition of C5aR, a chemoattractant receptor for complement component C5a, results in a striking disease inhibition (11), and trials are currently under way to evaluate the efficacy of an anti-C5aR drug in human RA. These RA models suggest that development of rheumatoid factor or citrullinated peptide antibodies might activate the complement cascade that then attracts tissue-damaging C5aR+ leukocytes, such as neutrophils and activated macrophages, to the joints. Activated macrophages produce TNF or IL-6 (12), which in turn mediate synovial inflammation (Figure 1). The chemokine ligands of CCR5 or CXCR3 are then induced by inflammatory cytokines such TNF and IFN-γ. Perhaps T cells in the joint then exacerbate disease through production of inflammatory cytokines, particularly IL-17 and IFN-γ. Of course, the genetic element that implicates T cell involvement in human RA could relate entirely to another arm of the T cell response, such as T cell help for B cells that produce pathogenic autoantibodies (Figure 1). Regarding other inflammatory diseases, effector T cells actually do appear to drive pathogenesis. In psoriasis, agents that block the Th17 cytokine IL-17 have been highly promising in human clinical trials. Th17 cells are marked by the chemokine receptor CCR6, which likely facilitates Th17 cell migration to the skin, although CXCR3 probably also plays a role.
Figure 1. Likely phases of RA development, emphasizing CXCR3+ effector T cells.
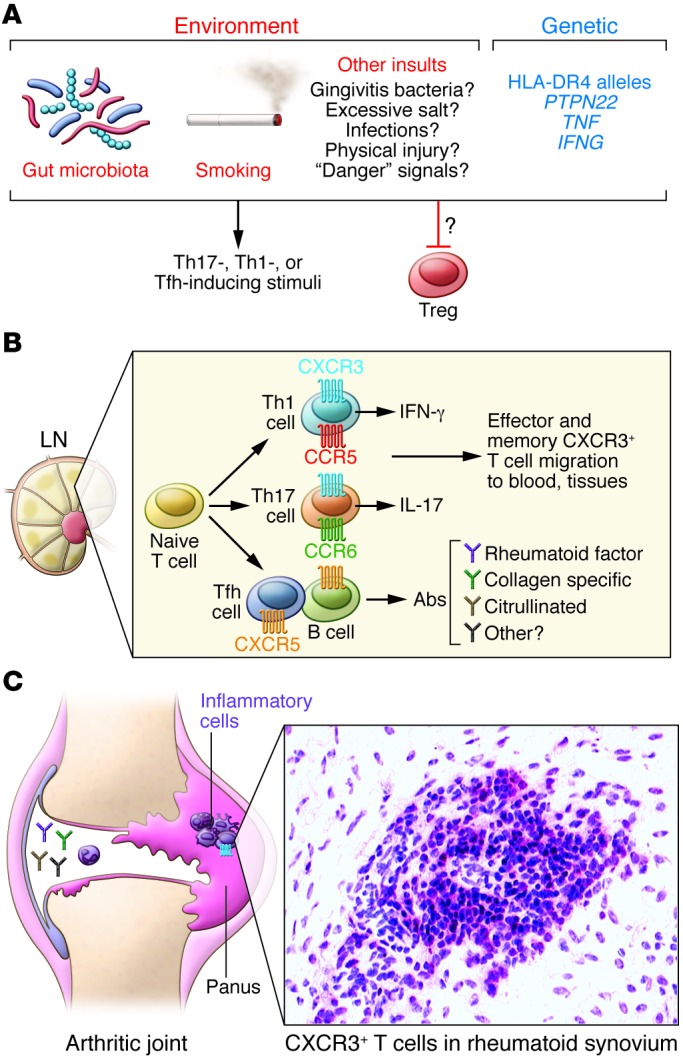
(A) Environmental and genetic triggers, including gut microbiota, smoking, infection, and PTPN22, TNF, and IFNG polymorphisms may promote Th17 and Th1 differentiation, and limit Treg development. Numerous environmental stimuli likely trigger disease through poorly understood processes. RA-associated genetic predispositions, including HLA-DR4 alleles, indicate that T cells drive RA; however, the pathogenic T cell subset is unknown. Dysfunctional MHCII and/or T cell responses could also alter gut microbiota, subsequently influencing disease. (B) During the lymphoid phase, naive T cells differentiate into disease-associated effector and memory T cells. IFN-γ–producing Th1 cells generally express CCR5 and CXCR3, whereas Th17 cells express CCR6 and CXCR3 to some extent. Tfh cells, which facilitate B cell antibody production, express CXCR5. Following generation in the lymph nodes (LN), CXCR3+ effector/memory T cells enter the blood and are a minority of T cells. During this stage, development and distribution of autoreactive effector cells and autoantibody-producing cells promote loss of immune tolerance. (C) The articular phase involves numerous cell types and inflammatory mediators, although the sequence of events remains uncertain. CXCR3+ effector/memory T cells are enriched in RA synovium, where their cytokine products, such as IFN-γ or IL-17, may drive inflammation and leukocyte recruitment. Additionally, deposition of immune complexes and complement activation in joints may initiate inflammation and leukocyte recruitment. Leukocyte products may damage joint tissue or promote synovial fibroblast proliferation. It is not clear whether CXCR3+ Th1 or Th17 cells underlie rheumatoid joint pathogenesis. See ref. 12 for more detailed information.
Conclusions
Our 1998 JCI report (7) reinforced the concept that the migration of T cells to inflammatory lesions is very different than their migration to lymphoid tissue. Accordingly, chemokine receptors, such as CCR5, CXCR3, and CCR6, remain excellent markers for functional T cell subsets, and these molecules likely facilitate the highly directed migration of effector T cells to inflammatory lesions. However, the jury is still out on which T cells pull the trigger to initiate disease.
Acknowledgments
Charles Mackay is supported by an Australian National Health and Medical Research Council Australia Fellowship.
Footnotes
Conflict of interest: Charles Mackay has part ownership of a company that has licensed anti-C5aR antibodies and related technology to Novo Nordisk. Charles Mackay has patented anti-CXCR3 antibodies for use in inflammatory diseases. These are the subject of a license agreement with a pharmaceutical company.
Reference information:J Clin Invest. 2014;124(9):3682–3684. doi:10.1172/JCI77837.
References
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76(2):301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67(6):1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 3.Sallusto F, Lanzavecchia A, Mackay CR. Chemokines and chemokine receptors in T-cell priming and Th1/Th2- mediated responses. Immunol Today. 1998;19(12):568–574. doi: 10.1016/S0167-5699(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 4.Mackay CR, Marston WL, Dudler L. Naive and memory T cells show distinct pathways of lymphocyte recirculation. J Exp Med. 1990;171(3):801–817. doi: 10.1084/jem.171.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343(14):1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 6.Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci U S A. 1997;94(5):1925–1930. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin S, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101(4):746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci U S A. 1999;96(12):6873–6878. doi: 10.1073/pnas.96.12.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khani-Hanjani A, et al. Association between dinucleotide repeat in non-coding region of interferon-γ gene and susceptibility to, and severity of, rheumatoid arthritis. Lancet. 2000;356(9232):820–825. doi: 10.1016/S0140-6736(00)02657-X. [DOI] [PubMed] [Google Scholar]
- 10.Mackay CR. Moving targets:cell migration inhibitors as new anti-inflammatory therapies. Nat Immunol. 2008;9(9):988–998. doi: 10.1038/ni.f.210. [DOI] [PubMed] [Google Scholar]
- 11.Lee H, et al. Human C5aR knock-in mice facilitate the production and assessment of anti-inflammatory monoclonal antibodies. Nat Biotechnol. 2006;24(10):1279–1284. doi: 10.1038/nbt1248. [DOI] [PubMed] [Google Scholar]
- 12.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7(6):429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]