

Abstract
Despite advancement in breast cancer treatment, 30% of patients with early breast cancers experience relapse with distant metastasis. It is a challenge to identify patients at risk for relapse; therefore, the identification of markers and therapeutic targets for metastatic breast cancers is imperative. Here, we identified DP103 as a biomarker and metastasis-driving oncogene in human breast cancers and determined that DP103 elevates matrix metallopeptidase 9 (MMP9) levels, which are associated with metastasis and invasion through activation of NF-κB. In turn, NF-κB signaling positively activated DP103 expression. Furthermore, DP103 enhanced TGF-β–activated kinase-1 (TAK1) phosphorylation of NF-κB–activating IκB kinase 2 (IKK2), leading to increased NF-κB activity. Reduction of DP103 expression in invasive breast cancer cells reduced phosphorylation of IKK2, abrogated NF-κB–mediated MMP9 expression, and impeded metastasis in a murine xenograft model. In breast cancer patient tissues, elevated levels of DP103 correlated with enhanced MMP9, reduced overall survival, and reduced survival after relapse. Together, these data indicate that a positive DP103/NF-κB feedback loop promotes constitutive NF-κB activation in invasive breast cancers and activation of this pathway is linked to cancer progression and the acquisition of chemotherapy resistance. Furthermore, our results suggest that DP103 has potential as a therapeutic target for breast cancer treatment.
Introduction
Breast carcinoma is one of the most common malignancies in women around the world. Mortality from breast cancer is almost entirely the result of invasion and metastasis of neoplastic cells from the primary tumors to distant organ sites (1–8); therefore, identifying genes involved in breast cancer metastasis is important. Since it is not possible to accurately predict the risk of metastasis in individual patients, at the present time, more than 80% of them receive adjuvant chemotherapy, but approximately 40% of the patients suffer relapse and ultimately die of metastatic disease. Cancer metastasis is a multistage process by which cancer spreads from the place at which it first arose as a primary tumor to distant locations in the body. These stages are interconnected through a series of adhesive interactions and invasive processes, including tumor angiogenesis, invasion, and colonization (9–15). Due to its heterogeneity and nature, mechanisms involving metastasis remain poorly understood. Degradation of the extracellular matrix (ECM) by matrix metalloproteinases (MMPs) (16, 17) is essential for almost every step in metastasis (18, 19). It is now accepted that the strong and direct causal association of MMPs in breast cancers makes the inhibition of these enzymes a worthwhile strategy for impeding tumor metastasis. However, due to the multiplicity of MMPs and the widespread effects of their actions, inhibitors of MMPs have not been successful as anticancer agents. Indeed, there is a significant unmet need for identifying novel mechanisms to inhibit selective MMP functions in invading tumor cells.
DP103 belongs to the family of DExD/H-box proteins, named after the signature Asp-Glu-Ala-Asp/His motif within the helicase domain (20–26). The 824-aa DEAD-box protein, DP103 (Gemin3, DDX20), was originally cloned and characterized as a component of the splicing machinery in concert with SMN, Sm, and other Gemin proteins (27–30). DP103 is a transcriptional repressor for Egr2 in hind brain development (31) and forms a repressor complex with METS–PE-1 to silence transcription of Ets target genes involved in Ras-dependent macrophage proliferation and differentiation (26). We have previously shown that DP103 transcriptionally represses the nuclear receptor SF-1 in a SUMO-dependent manner (23). SUMOylation of SF-1 is mediated by PIASy, an E3 SUMO ligase, and this reaction is catalyzed in the presence of DP103 as a cofactor. However, the role of DP103 as a cofactor in SUMOylation or other covalent modifications of cellular substrates and its involvement in cancer initiation or metastasis have never been explored.
The NF-κB family of transcription factors comprises 5 structurally related proteins that form homo- and heterodimers through their highly conserved DNA binding/dimerization Rel homology domain (32). Binding of NF-κB to IκB proteins maintains NF-κB in an inactive state (33). Activation of NF-κB in normal cells is inducible and is a tightly controlled event. Upon stimulation, IκBs are phosphorylated by the IκB kinase (IKK) complex (consisting of IKK1, IKK2, and NEMO proteins) (34–37). IκB phosphorylation leads to its rapid proteolysis, thereby allowing NF-κB to function as a transcription factor. Temporally, IκBα is one of the first target genes that NF-κB activates very rapidly, and hence, this forms a feedback loop that shuts off the activity of NF-κB under normal circumstances. In many malignancies, including that of the breast, however, NF-κB is found to be constitutively activated (38–49). Yet the molecular mechanism for such constitutive activation of NF-κB signaling in solid tumors is not clear. TGF-β–activated kinase-1 (TAK1), a member of the MAPK family, is a key regulator of signal transduction cascades, leading to the activation of the transcription factor NF-κB. Stimulation of cells with cytokines and microbial pathogens results in the activation of TAK1, which subsequently activates the IKK complex and MAPKs, culminating in the activation of NF-κB and AP-1, respectively (39, 40, 50–54). TAK1 is recruited to the TNF-α receptor complex in an RIP-dependent manner following the stimulation of TNF-α receptor 1 and serves as a pivotal intermediate for IKK activation (39, 40, 44). Apart from its critical role in immunity (55), wherein TAK1 is mostly activated by membrane or cytoplasmic cues, TAK1 is also essential for NF-κB activation in response to DNA damage, which is initiated from the nucleus (46, 56). Although hypothesized, overexpression of molecules such as IKK2 and TAK1 as likely causes of constitutive NF-κB apparent in most malignancies has not been documented, despite sequencing of several cancer genomes.
In a search for markers of metastasis, we uncovered a molecule whose levels are upregulated in metastatic human breast cancers. Depletion of endogenous DP103 in invasive breast cancer cells led to decreased expression of matrix metallopeptidase 9 (MMP9) and impeded cell migration and invasion. Conversely, forced expression of DP103 in mammary carcinoma and nonmalignant cells both in vitro and in vivo increased oncogenicity and the cells’ capacity to invade. Mechanistic studies showed that DP103 is a critical cofactor for TAK1-mediated activation of IKK2, the key NF-κB–activating kinase in its activation loop. Hence, the observed effects of DP103 are mostly due its ability to activate NF-κB, an oncogenic transcription factor. Since a body of research indicates that TAK1-dependent NF-κB activation has a direct role in various aspects of cancer (39, 56), our identification of DP103 as the rate-limiting factor that mediates TAK1 signaling adds an important previously unidentified regulator that could be used as a drug target, since its levels specifically increase in cancer cells. Interestingly, NF-κB signaling also positively activates DP103 expression. Since levels of DP103 are limiting in normal cells, the existence of a tonic DP103–NF-κB–positive feedback loop in cancer cells could also explain how constitutive NF-κB activation is documented in most human cancers despite the absence of amplification of TAK1 or IKK2.
Results
Expression of DP103 is significantly upregulated in basal subtype human breast cancers.
Recently, a study showed increased expression of DP103 in a protein microarray done on tissues from mantle-cell lymphoma patients (57). This prompted us to explore the role of DP103 in breast cancer. Tissue microarray (TMA) slides consisting of invasive ductal carcinoma (IDC) cases from 399 patients and normal nonmalignant ductal tissues from 61 women were obtained from the Department of Pathology, Singapore General Hospital. Clinicopathological features are shown in Supplemental Table 1A (supplemental material available online with this article; doi:10.1172/JCI73451DS1). Typical DP103 staining in normal versus tumor samples is shown in Figure 1, A and B, respectively. Based on DP103 staining, the cut-off value for low or high expression of DP103 was scored as 0 and 1+ versus 2+ and 3+. As a result of tissue loss during immunohistochemical processing, 330 cases of IDC and 38 cases of normal nonmalignant ductal tissues could be evaluated. As shown in Supplemental Table 1B, all 38 normal tissues showed low DP103 expression, while a significantly higher proportion (267 out of 330 patients) of specimens showed high DP103 expression in the tumor cores (P < 0.001). These findings were further extended using another cohort from the Montreal University Health Centre (Supplemental Figure 1, A and B, and Supplemental Table 1C). Clinicopathological features are shown in Supplemental Table 1D. Taken together, these data sets reveal that expression of DP103 is significantly higher in tumors (across ethnic groups and regardless of the source of patient material).
Figure 1. DP103 levels correlate with invasiveness and malignancy.

DP103 staining of (A) normal ductal tissue and (B) an IDC. (C) Gene expression value of DP103 (y axis) plotted for each breast cancer subtype, namely basal, claudin-low, luminal-A, luminal-B, ERBB2 (HER2+), and normal-like. (D) Kaplan-Meier curves showing DP103 expression in relation to patients’ OS. Cases that have not experienced a positive event are censored at the date of last follow-up (small vertical lines on the line plots). (E) Kaplan-Meier curves showing DP103 expression in relation to SAR. Cases that have not experienced a positive event are censored at the date of last follow-up (small vertical lines on the line plots). (F) Breast cancer progression model showing isogenic cell lines with increasing invasive potential. (G) Western blotting with DP103 antibody in lysates from the isogenic cell lines (F). (H) qPCR with DP103 mRNA expression in RNA from the isogenic cell lines (F). (I) Gene expression of DP103 correlates with breast metastasis activity by Spearman correlation (64). Red dotted line is curve fitted by linear regression. (J) Primary breast tissues from patients with benign disease, no lymph node metastases (Non-Met), and lymph node metastases (Met) collected and analyzed for DP103 mRNA expression **P < 0.01; ***P < 0.001. (K) RNA from breast cell lines and qPCR performed with DP103 primers. (L) Protein from breast cell lines extracted and levels of DP103 protein evaluated. Fold difference in protein expression indicated in G and L.
To further delineate the expression profile of DP103 in various subtypes of breast cancer, 11 cohorts containing 1,325 breast tumors were collected and compiled from the NCBI’s Gene Expression Omnibus (GEO) (see Methods). These 1,325 tumors were then classified using Single-Sample Gene Set Enrichment Analysis (ssGSEA) (58) and breast cancer subtype signature from Prat et al. (59). As shown in Figure 1C, DP103 expression is significantly higher in basal subtype (Mann-Whitney test, P = 4.88 × 10–11). No significant difference in DP103 expression in claudin-low and luminal B were seen, while luminal A, ERBB2, and normal-like subtypes showed significantly lower expression (Mann-Whitney test, P = 4.5 × 10–5; P = 0.0048; P = 0.0281, respectively). Consistent findings were seen on a validation data set (GEO GSE3494) not included in the 11 meta-analysis cohorts, where basal subtype had significantly higher DP103 expression when compared against other subtypes (Mann-Whitney test, P = 1.09 × 10–4; see Supplemental Figure 1C). Since protein expression levels provide a more reliable quantification for function compared with mRNA quantification, as shown in Figure 1C and Supplemental Figure 1C, we then assessed protein expression of DP103 in the same 2 cohorts by immunohistochemistry (IHC). In agreement with our microarray results, the highest protein expression of DP103 correlated with the basal subtypes (Supplemental Figure 1, D–H; Supplemental Table 2A, Singapore cohort; Supplemental Figure 1, I–M; and Supplemental Table 2B, Canada cohort).
DP103 expression levels correlate with malignancy and with patient survival.
We then analyzed DP103 expression in breast tumors using a multi-institutional “microarray meta-analysis cohort” with a sample size of 669 primary breast cancer cases and found levels of DP103 to be significantly elevated in poorly differentiated grade 3 tumors compared with those in well-differentiated grade 1 or 2 tumors (n = 669, P = 0.008) (Supplemental Figure 1N). We also validated the microarray data in Supplemental Figure 1N by analyzing DP103 protein expression by IHC. Corroborating our microarray results was the finding that DP103 protein is significantly higher in high-grade IDC compared with low-grade IDC (Supplemental Figure 2, A–D; Supplemental Table 2C, Singapore cohort; Supplemental Figure 2, E–G; and Supplemental Table 2D, Canada cohort).
Kaplan-Meier analysis using the cohort consisting of 399 patients (Supplemental Table 1A) revealed that high DP103 protein levels correlated with reduced survival. Patients with high DP103 expression (mean overall survival [OS], 132 months; n = 61) had significantly shorter survival (P = 0.010) compared with those with low DP103 expression (mean OS, 149 months) (Figure 1D). In addition, analysis of survival after relapse (SAR) revealed that patients with high DP103 expression (mean SAR, 63 months; n = 61) had significantly shorter survival (P = 0.009) compared with those with low DP103 expression (mean, 116 months) (Figure 1E). To investigate whether a meaningful functional correlation between DP103 expression levels and increasing tumor malignancy does exist, we determined basal DP103 expression in an isogenic cell xenograft–derived MCF10 breast cancer progression model system (60–62) that included the immortalized breast epithelial cell line MCF10A (10A1), the premalignant MCF10AT (AT1k), low-grade MCF10CA1h (CA1h), and high–metastatic grade MCF10CA1aCl.1 (CA1a) cell lines (Figure 1F). We found that DP103 protein (Figure 1G) and mRNA (Figure 1H) levels directly correlated with increasing invasive potential of these cell lines, with the highest levels of DP103 expression observed in the most metastatic CA1a cells (Figure 1, G and H).
DP103 expression correlates with metastasis gene signatures and breast cancer metastasis.
A recent US patent application on the use of compounds that block cancer metastasis showed DP103 (DDX20) as the only DEAD-box helicase member listed in the microarray data (publication number: US 2010/0004190 A1; ref. 63), thereby encouraging us to determine whether expression of DP103 correlates to the metastatic potential of tumor cells. Using breast cancer metastasis signature from Zucchi et al. (64), we employed ssGSEA to estimate the degree of cancer metastasis for breast tumors. Figure 1I shows a strong positive correlation in the meta-analysis cohort for breast cancer metastasis with DP103 expression (Spearman ρ = 0.21, P = 7 × 10–15), suggesting that DP103 may have a role in breast cancer metastasis. From this metastasis gene signature list (64), the association of the 2 most positively correlated metastasis genes, HMGB1 and H2AFZ, to expression of DP103 was then validated by quantitative PCR (qPCR) in a cohort of 63 breast cancer and 22 benign breast tissue samples derived from 85 patients who underwent surgery at the First Affiliated Hospital of Anhui Medical University, Hefei, China (Supplemental Figure 2, H and I). Clinicopathological status of this cohort is provided in Supplemental Table 3A. In this cohort, we then assessed whether expression of DP103 alone is predictive for lymph node metastasis. This analysis mirrored our gene expression analysis from above in that DP103 expression was lower in primary breast tissues that did not develop lymph node metastasis, while increased DP103 levels were observed in primary breast cancer tissues of patients who developed lymph node metastasis (Figure 1J). Benign breast tissue samples that were used as a reference demonstrated the least DP103 expression levels (Figure 1J). The role of DP103 in metastasis gained further credence by its listing in microarray data done on colorectal cancer (CRC) patients’ tissues (n = 100), comparing gene expression profile in primary tumors with and without distant metastasis. When retrieved (ArrayExpress E-GEOD-18105), DP103 was found to be upregulated in CRC patients that developed distant metastasis (n = 100; P < 1 × 10–10). These results then encouraged us to screen a panel of breast cancer cell lines for DP103 expression. Interestingly, cell lines that are highly metastatic, such as MDA-MB-231 and BT549, display high levels of DP103 mRNA (Figure 1K) and protein (Figure 1L). On the other hand, noninvasive SKBr3, MCF7, and BT474 cells have much lower DP103 expression, while normal breast cells MCF10A and 184A1 have the lowest DP103 expression.
Suppression of DP103 decreases migratory ability of breast cancer cells.
To evaluate the functional consequence of DP103 expression in breast cancer metastasis, we assessed cells’ migratory behavior following RNAi-mediated knockdown of DP103 in metastatic MDA-MB-231 cells. To exclude off-target effects, we performed these assays using 2 independent siRNAs against DP103 (Supplemental Figure 3A) either singly or in combination. DP103 depletion led to decreased migration (by 40%) when siDP103#1 and siDP103#2 were used alone or (by 80%) when they were used together (siDP103 #1+2) (Figure 2, A and B) in a classical wound-healing assay. These observations were further confirmed by using a 2D nonwound migration assay (Supplemental Figure 3B). The 2D track plots of the cells are depicted in Supplemental Figure 3C, which shows reduction in cell motility, with the left panels showing cells transfected with control siRNA and the right panels showing cells transfected with siRNA against DP103. The top panels show the entire tracks made by the 50 cells, while the bottom panels show the ending points of each cell track (Supplemental Figure 3C). The data of individual cell tracks from this assay were then analyzed for various cell migration parameters, as shown in Supplemental Figure 3D. Accumulated distance is an indicator of motility of individual cells, whereas Euclidean distance gives an idea about the actual dispersion of the cells with respect to their starting positions. Confinement ratio is an indicator of directionality of the cells over time. In summary, we concur that suppression of DP103 affects the intracellular motility mechanisms in MDA-MB-231 cells but not the directionality of motion (Supplemental Figure 3D).
Figure 2. DP103 regulates invasive abilities of cancer cells.

(A) MDA-MB-231 cells transfected with control siRNA (ctsi) and 2 different siRNAs against DP103. Scratch wound healing assay is shown. (B) MDA-MB-231 cells represented in percentage of siDP103 or ctsi cell movement after scratching. *P < 0.05. (C) MDA-MB-231 and BT549 cells transfected with control siRNA (ctsi) and 2 different siRNAs against DP103 in Transwell assays. Inset shows representative photograph. (D) Quantification of MDA-MB-231 cells invaded through Transwell invasion chambers from C. *P < 0.05. (E) Quantification of BT549 cells invaded through the Transwell invasion chambers from C. *P < 0.05. (F) MCF10A cells transfected with control pcDNA3 vector (EV) and pcDNA3-FLAG-DP103 (DP103). Cell extracts immunoblotted with anti-DP103 and anti–β-actin antibodies. (G) MCF10A cells transfected as in F and analyzed in an invasion assay as in C. (H) MCF10A cells that invaded through the chambers quantified and represented. *P < 0.05. (I) Pulmonary and liver metastases from primary tumors evaluated by bioluminescence imaging. Color scale depicts photon flux (p/s) emitted from the organs. (J) Left: DP103 expression in primary tumor injected with empty vector–transfected MDA-MB-231 cells (MDA-MB-231-EV). Right: DP103 expression in mouse mammary fat pad injected with DP103-transfected cells (MDA-MB-231-DP103). (K) Distal metastasis to lung tissues quantified by mRNA levels of human GAPDH. *P = 0.05. (L) Metastasis to liver tissues quantified by mRNA levels of human GAPDH. *P = 0.05. Fold difference in protein expression indicated in F. Original magnification, ×10; ×40 (insets).
Suppression of DP103 decreases invasive ability of breast cancer cells.
Given that DP103 is upregulated in invasive breast cancer cells, we next explored effects of decreasing DP103 expression on tumor cell invasion by the use of Matrigel-coated Transwell chambers. In highly invasive MDA-MB-231 and BT549 cells, transient knockdown of DP103 resulted in about 50% less invasion through the Matrigel-coated inserts (Figure 2, C–E). The combined use of siDP103 resulted in about an 80% decrease in invading cells (Figure 2, C–E). To mimic in vivo conditions as closely as possible, we then monitored luciferase-expressing MDA-MB-231 cells with and without suppression of DP103 expression using a 3D collagen gel assay. siDP103 reduced cells’ invasiveness in 3D collagen hydrogel; notably, the spread of green tracks (ctsi) surpassed that of red tracks (siDP103) (Supplemental Figure 4A). Cell speed of the knockdown cells was also averaged over each track and plotted into a histogram (Supplemental Figure 4B). As an additional indicator of invasive potential, MDA-MB-231 cells characteristically induce a branching morphogenesis. Interestingly, we observed a loss of this branching morphogenesis and reduction in cell spreading in cells depleted of DP103 expression (Supplemental Figure 4C).
Ectopic expression of DP103 enhances cell invasion.
Induction of tumor cell invasion is an important step in tumor metastasis and transition to a more malignant form of cancer at distant sites (65). To further substantiate our findings, we then evaluated whether ectopic expression of DP103 would result in altered invasion by noncancer cells. Ectopic expression of DP103 alone had a dramatic effect on the phenotypic conversion of the MCF10A normal human breast epithelial cell line, increasing its invasion potential by 4-fold (Figure 2, F–H). In addition, forced overexpression of DP103 in MDA-MB-231 cells (Supplemental Figure 4, D–F) significantly promoted cells’ invasive capacity. To validate these findings in vivo, we injected luciferase-expressing MDA-MB-231 cells stably transfected with either empty vector (MDA-MB-231-EV) or DP103 plasmid (MDA-MB-231-DP103) in the mammary fat pads of nude mice. Supplemental Table 3B compares the incidence of pulmonary and liver metastases at necropsy from primary tumors formed by the 2 breast cancer cell lines. Of 12 mice inoculated with MDA-MB-231-EV, only one was found to have lung and liver metastases by bioluminescence imaging. In contrast, 4 out of 11 mice injected with MDA-MB-231-DP103 cells developed distant metastasis to lung and liver (Supplemental Table 3B). Figure 2I shows the ex vivo images of the lung and liver lobes from all mice that developed distant metastasis. We validated differences in protein expression levels of DP103 in primary tumors of MDA-MB-231-EV cells versus MDA-MB-231-DP103 cells by IHC (Figure 2J). In addition, we also quantified distal metastasis in lung and liver tissues by qPCR (human GAPDH gene expression normalized to mouse Gapdh gene expression). Our results show a 2-fold increase in distal metastasis to lung (Figure 2K) and liver (Figure 2L) in the MDA-MB-231-DP103 mouse group compared with the MDA-MB-231-EV mouse group. Taken together, mice injected with MDA-MB-231-DP103 had higher incidence of lung and liver metastases than mice injected with MDA-MB-231-EV.
MMP9 levels predict survival and strongly correlate with DP103 levels in breast cancer.
Cell invasion requires the complex coregulation of cytoskeletal reorganization and cell motility as well as proteolysis and interaction with the ECM. Therefore, we measured expressions of several such genes in a “metastasis qPCR array” (Table 1). While ectopic expression of DP103 did not change the expression of genes such as NME1, PLAU, SERPINB5, or MTA1, it led to upregulation of several other genes, such as MMP1, TIMP1, and TIMP3. Notably, MMP9 mRNA levels were increased significantly (Table 1). Since high MMP9 expression is closely associated with poor prognosis in other cancer types (66–68), we stained tissues from the same cohorts used in Supplemental Table 1, A and D, for MMP9 protein expression. Similarly to expression of DP103, we observed high expression of MMP9 in breast tumor tissues compared with normal breast tissues (Figure 3, A and B, and Supplemental Table 4A, Singapore cohort; Supplemental Figure 5, A and B, and Supplemental Table 4B, Canada cohort). Kaplan-Meier analysis showed that high MMP9 levels were correlated with higher tumor recurrence risk and reduced survival. Similarly to DP103, MMP9 status correlated with OS, with high expression (mean OS, 132 months; n = 69) showing significantly shorter survival duration (P = 0.002) compared with low MMP9 expression (mean OS, 150 months) (Figure 3C). Furthermore, cases with high MMP9 expression have shorter SAR (mean, 66 months; n = 69) and significantly shorter survival (P = 0.028) compared with those with low MMP9 expression (mean, 118 months) (Figure 3D). Having observed similar clinical significance for DP103 and MMP9, we then analyzed to determine whether there is a positive correlation between these 2 markers. Supplemental Table 4, C and D, indeed showed a strong positive clinical correlation between expression of DP103 and MMP9 (P < 0.001 in both cohorts). To further substantiate this finding, we next investigated whether a similar correlation between DP103 and MMP9 expression existed in the cohort from Supplemental Table 3A. Importantly, much like DP103, the highest levels of MMP9 were found in primary breast tumors that showed metastasis compared with nonmetastatic or benign tumors (Figure 3E). Importantly, the correlation between DP103 and MMP9 expression was statistically significant (P < 0.001) (Supplemental Figure 5C) a trend that was also observed (Supplemental Figure 5D) in data obtained from the same microarray consisting of the 11 meta-analysis cohort used above (Figure 1C).
Table 1.
Metastatic qPCR array
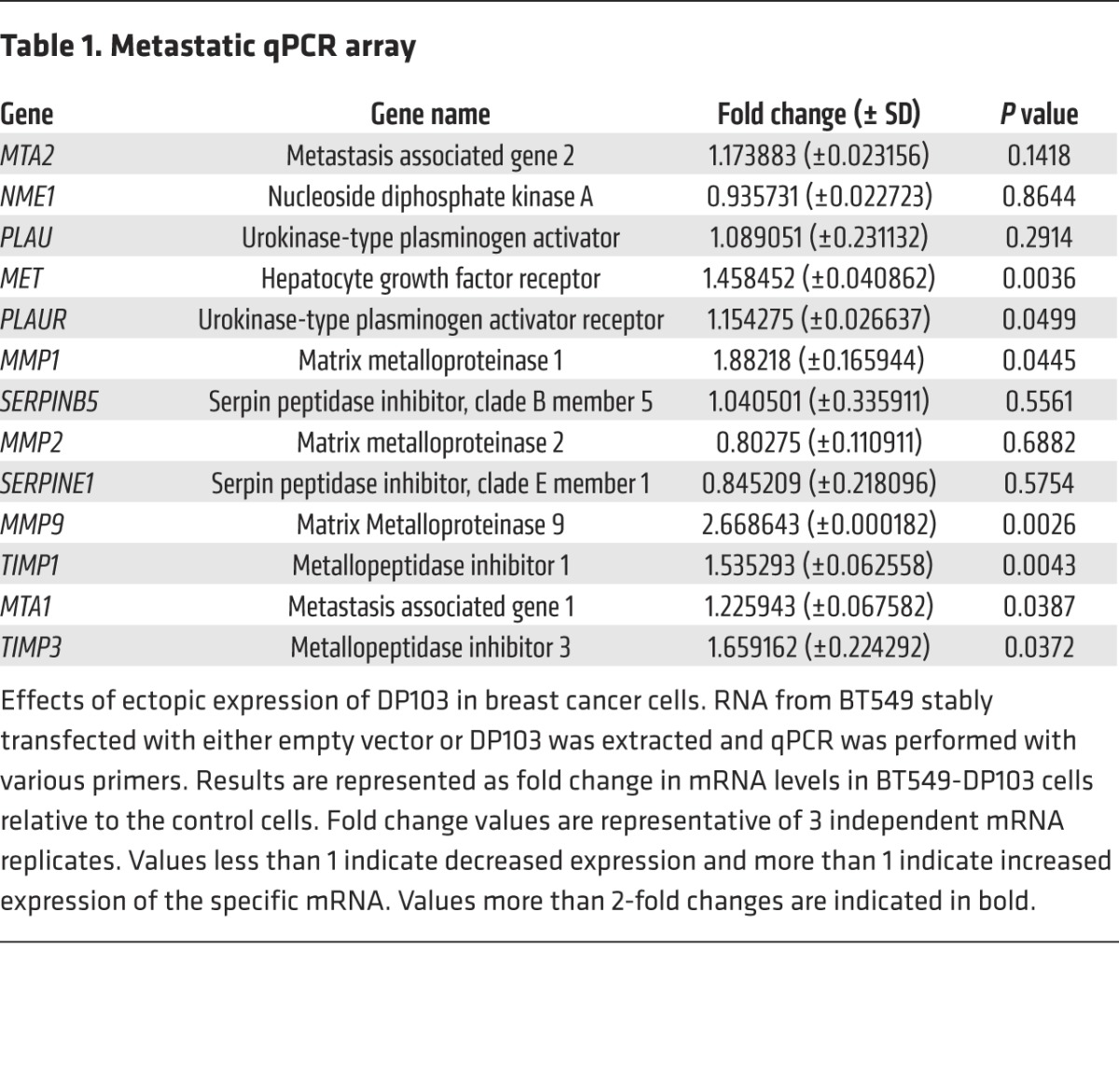
Figure 3. DP103 regulates MMP9 expression and function.

MMP9 staining of (A) normal ductal tissue and (B) IDC. (C) Kaplan-Meier curves of MMP9 expression in relation to OS. Cases that have not experienced a positive event are censored at the date of last follow-up (small vertical lines on the line plots). (D) Kaplan-Meier curves of MMP9 expression in relation to SAR. Cases that have not experienced a positive event are censored at the date of last follow-up (small vertical lines on the line plots). (E) Primary breast tissues from patients with benign diseases, no metastases, and displaying metastases analyzed for MMP9 mRNA expression. **P < 0.01; ***P < 0.001. (F) Total RNA of different breast cell lines extracted and qPCR performed with MMP9 primers. (G) MDA-MB-231 and BT549 cells were transfected with control siRNA (ctsi) and siRNA against DP103. Total RNA analyzed for mRNA expression of DP103 and MMP9. (H) Expression of DP103 and MMP9 in cells used in G, evaluated using DP103 and MMP9 antibodies. (I) Suppression of DP103 in BT549 cells decreases MMP9 activity. Enzymatic activity of MMP-9 was determined by gelatin zymography.
Suppression of DP103 expression decreases MMP9 gene expression.
We then screened for MMP9 expression in the panel of breast cell lines used in Figure 1K. We found that, mirroring the expression of DP103, metastatic breast cancer cell lines MDA-MB-231 and BT549 displayed the highest levels of endogenous MMP9 mRNA expressions, while the nonmetastatic and normal breast cell lines displayed relatively low levels (Figure 3F). In addition, we observed that MMP9 mRNA expression positively correlated with DP103 mRNA expression across a panel of cell lines (Pearson coefficient = 0.913) (Supplemental Figure 5E). Given the strong correlation observed between DP103 and MMP9 expression levels, we probed for a causal link between these enzymes. Transient knockdown of DP103 resulted in a significant decrease in MMP9 mRNA levels at 24 hours and 48 hours compared with cells transfected with a scrambled siRNA (ctsi) in MDA-MB-231, BT549 (Figure 3G), and Hs578t cells (Supplemental Figure 5F). Decrease in MMP9 mRNA corroborated well with an observed drop in MMP9 protein levels (Figure 3H and Supplemental Figure 5G) and enzyme activities (Figure 3I) without affecting cell viability (Supplemental Figure 5, H and I). Conversely, ectopic expression of DP103, much like what is seen in cancer tissues, resulted in increased MMP9 expression in both malignant MDA-MD-231 and nonmalignant MCF10A cells (Supplemental Figure 6A).
MMP9 mediates effects of DP103 in invasiveness.
In order to ascertain whether MMP9 is the primary molecular effector of the observed effects mediated by DP103, we evaluated invasion of MDA-MB-231 in a Transwell assay using a pan-MMP inhibitor (GM6001), a MMP2/9 inhibitor (SB-3CT), and a selective inhibitor of MMP9 (MMP9 inhibitor I). We found that inhibition by either GM6001 or SB-3CT prevented invasion of MDA-MB-231 cells through the Matrigel matrix by 40% (Supplemental Figure 6, B and C). More importantly, we found that specific inhibition of MMP9 activity alone was sufficient to significantly (~50%) impede the invasion of MDA-MB-231 cells either with basal or ectopic expression of DP103 (Supplemental Figure 6D), which suggests MMP9 is the key molecular effector mediating effects of DP103 seen in our experiments and in clinical settings. MMPs are known to play roles not only in metastatic progression, but also in tumor initiation and formation (69), thereby suggesting that DP103-mediated regulation of these enzymes may have widespread implication in cancer initiation and progression. Indeed, although there was no difference in tumor size between control and DP103-overexpressing cells (Figure 2J and Supplemental Table 3B), there were more tumors formed in DP103-expressing mice compared with control and more mice developed tumors in this group (data not shown).
DP103 regulates NF-κB–dependent gene expression.
Since an increase in MMP9 mRNA levels correlated with an increase in DP103 levels, it was likely that DP103 could be controlling the transcription of MMP9. Transcriptional regulation of MMP9 involves a relatively large repertoire of transcriptional factors that includes AP-1, AP-2, Ets, NF-κB, and SP-1, out of which AP-1 (70–72) and NF-κB (73, 74) are considered the major regulators. Given that depletion of DP103 expression in 3 invasive cell lines decreased MMP9 mRNA, we questioned whether this downregulation is due to inhibition of NF-κB or AP-1 activity. Suppression of DP103 led to downregulation of basal NF-κB activity, but had no effect on AP-1 activity (Figure 4A). Interestingly, DP103 levels were found to change in response to ecteinascidin 743 (ET-743), a genotoxic stress inducer (75). Given that DP103 regulated MMP9 expression via activation of NF-κB, whose transcriptional activity is also regulated by genotoxic chemotherapeutic agents (46, 76–78), we then sought to investigate effects of DP103 in the presence of known chemotherapeutic agents that induce NF-κB, namely doxorubicin, etoposide (VP16), and camptothecin. Indeed, luciferase assays with NF-κB– and AP-1–dependent reporters showed that suppression of DP103 reduced drug-induced NF-κB reporter activity in both MDA-MB-231 and BT549 cells (Figure 4, B–D, and Supplemental Figure 7, A–C, respectively). Significantly, drug-induced AP-1 activity was not affected by DP103 levels (Supplemental Figure 7, D–F). EMSA analysis showed that siRNA-mediated reduction of DP103 reduced both basal and drug-induced NF-κB DNA–binding activity seen in both MDA-MB-231 and BT549 breast cancer cells (Figure 4E). AP-1 DNA–binding in both MDA-MB-231 and BT549 cells remained unaffected by DP103 levels and served as control (Figure 4E). Conversely, ectopic expression of DP103, much like what is seen in tumors, increased NF-κB DNA–binding ability (Figure 4F). siRNA to DP103 also reduced nuclear accumulation of p65 while causing it to accumulate in the cytoplasm (Supplemental Figure 7G). We next evaluated whether DP103 could regulate other clinically relevant NF-κB target genes in response to stress. Indeed, siRNA to DP103 significantly reduced the levels of NF-κB target genes, namely ICAM-1, MMP9, and CXCR4 (Figure 4G).
Figure 4. DP103 is an essential regulator of NF-κB signaling.

(A) Control siRNA–treated (ctsi) and siDP103-treated MDA-MB-231 cells transfected with Renilla and luciferase reporter plasmid containing NF-κB or AP-1. Luciferase activity normalized and quantified. Results are average of 3 separate experiments. *P < 0.05. Control siRNA–treated (ctsi) and siDP103-treated MDA-MB-231 cells transfected with Renilla and luciferase reporter plasmid driven by NF-κB–binding sites. Cells were stimulated with (B) 10 μM VP16, (C) 25 μM doxorubicin, (D) or 10 μM CPT for 0, 6, and 12 hours and harvested for luciferase assays. Results are the average of 3 separate experiments. *P < 0.05. (E) MDA-MB-231 and BT549 cells transfected with either control siRNA (ctsi) or siDP103. Cells were left either untreated (–) or treated with CPT (10 μM), doxorubicin (10 μM), or VP16 (10 μM) for 2 hours. Protein extracts were analyzed by EMSA (top panel) and Western blotting using DP103 and GAPDH antibodies (bottom panel). (F) MDA-MB-231 cells infected either with lentiviral empty vector (lanes 1–3) or pBOBI-DP103 (lanes 4–6) were either left untreated or treated with CPT (10 μM) for indicated times. Protein extracts analyzed by EMSA and Western blotting using anti-DP103 and anti–β-actin antibodies (top panel). (G) MDA-MB-231 cells transfected with either control siRNA (ctsi) or siRNA against DP103 and subjected to CPT (10 μM) stimulation at the indicated times. Cells were harvested and lysates evaluated by Western blotting for the indicated proteins. Fold difference in protein DNA binding indicated in E and F for EMSA and for protein expression changes indicated in G for Western blot.
Activation of NF-κB is known to protect cells from cell death induced by genotoxic agents. To evaluate whether reduction of NF-κB DNA binding (Figure 4, E and F, and Supplemental Figure 7G) and transcriptional activity (Figure 4, A–D, and Supplemental Figure 7, A–C) has functional significance, we treated MDA-MB-231 cells with doxorubicin or CPT and measured cell viability. Indeed, cells depleted of DP103 and hence with defective NF-κB activation were more sensitive to cell death induced by these genotoxic agents (Supplemental Figure 7, H and I). In addition, we found that silencing of DP103 led to decreased NF-κB binding to the DP103 promoter (Supplemental Figure 7J), suggesting that the DP103–NF-κB–positive feedback loop likely contributes to oncogenesis.
Activated NF-κB (p-p65-276) levels correlate with expression of DP103 and patient survival.
Since our results indicated that DP103, a key biomarker of cancer metastasis, functions via activation of NF-κB, we next explored whether there is a clinical value for NF-κB activation per se by evaluating expression levels of phospho-p65 in patient tissues. IHC performed on clinical specimens from the cohort shown in the Supplemental Table 1A revealed that, similar to expression of DP103, expression of phospho-p65 (Ser276), which represents the activated form of NF-κB, the competent form for chromatin remodeling, is significantly higher in breast tumor tissues compared with normal breast tissues (P < 0.001) (Figure 5, A and B, and Supplemental Table 5A). Analysis of phospho-p65 protein expression mirrored that of DP103, with the highest expression observed in basal subtypes compared with other subtypes (P = 0.0141) (Figure 5, C–G, and Supplemental Table 5B) and closely associated with high-grade tumors (P < 0.001) (Figure 5, H–K, and Supplemental Table 5C). Kaplan-Meier analysis on the same cohort revealed that high phospho-p65 (Ser276) expression (mean, 132 months; n = 62) correlated with reduced OS, (P = 0.002) compared with low phospho-p65 expression (mean, 151 months) (Figure 5L). Furthermore high phospho-p65 (Ser276) expression (mean, 66 months; n = 62) predicted shorter SAR (P = 0.011) compared with low phospho-p65 expression (mean, 120 months) (Figure 5M). Most importantly, high expression of phospho-p65 correlated positively with high expression of DP103 in clinical specimens (P < 0.001) (Supplemental Table 5D).
Figure 5. Expression of active p65 (phospho-p65, Ser276) has a prognostic value.

Staining of phospho-p65 (Ser276) in (A) normal ductal tissue and (B) IDC. Representative staining of phospho-p65 (Ser276) in (C) normal ductal tissue, (D) luminal A subtype, (E) luminal B subtype, (F) HER2 subtype, and (G) basal subtype. Phospho-p65 (Ser276) staining in (H) normal nonmalignant ductal tissue, (I) low-grade IDC, (J) IDC grade 2, and (K) IDC grade 3. Cell nuclei were counterstained with hematoxylin. (L) Kaplan-Meier curves showing phospho-p65 (Ser276) expression in relation to OS. Cases that have not experienced a positive event are censored at the date of last follow-up (small vertical lines on the line plots). (M) Kaplan-Meier curves showing phospho-p65 (Ser276) expression in relation to SAR. Cases that have not experienced a positive event are censored at the date of last follow-up (small vertical lines on the line plots).
DP103 regulates NF-κB in response to multiple stimuli.
NF-κB can regulate gene expression in response to multiple stimuli (39, 40, 79). To test whether DP103 is an essential component of NF-κB signaling, we determined whether DP103 can also regulate NF-κB in response to a diverse array of well-known NF-κB–activating agents such as TNF-α, IL-1, LPS, CPT, and Dox. These agents initiate NF-κB activation via very distinct cellular-signaling players (40). However all the distinct signaling to NF-κB initiated by these agents converges on the IKK complex. DP103 depletion impeded NF-κB DNA binding in response to all the tested stimuli (Figure 6A). DP103 also affected kinetics of NF-κB binding in response to membrane (TNF-α) and nuclear (CPT) activators of NF-κB (Figure 6, B and C). Furthermore, reduction of DP103 caused significant reduction in TNF-α–induced NF-κB–dependent transcription (Figure 6D). Based on the finding that DP103 promoter contains κB sites (data not shown), we then evaluated whether DP103 itself is a target of NF-κB–activating stimuli. De novo DP103 protein was induced in response to both TNF-α and LPS (Figure 6E) and DP103 induction by LPS can be strongly and specifically inhibited by IKK2 inhibitor IV among other kinase inhibitors tested (Figure 6F), providing evidence that DP103 is an NF-κB target that could form part of a positive feedback loop. Interestingly, we identified 3 putative NF-κB– binding sites within the promoter of the DP103 gene (Figure 6G). By EMSA, we showed NF-κB binds to all 3 sites (data not shown). The specificity and identity of the NF-κB complexes could be ascertained using cold oligonucleotides and supershift with anti-p65 or anti-p50 antibodies, respectively (Figure 6H). Furthermore, EMSA with NF-κB consensus cold oligonucleotides abrogated NF-κB binding within the promoter of the DP103 gene (Figure 6I). In addition, we also showed a feedback loop following silencing of DP103 leading to decreased NF-κB binding to the DP103 promoter (Supplemental Figure 7G), suggesting that the DP103–NF-κB–positive feedback loop likely contributes to this oncogenic signaling arm in cancers. Members of the helicase families, of which DP103 is a member, share several conserved motifs, including the Walker A and B motifs, which are involved in the binding of nucleoside triphosphates required for its helicase activity (20). Changing the conserved GKT to GNT has been shown to reduce ATP binding in RNA helicases by 98% (80). Additionally, the helicase-dead mutant (GNT) also retained its ability to induce invasion in MDA-MB-231 cells (Supplemental Figure 8, A–C), demonstrating that the helicase activity of DP103 is not required for its role in the metastasis of breast cancer cells. The helicase dead mutant of DP103 also interacts with TAK1 (Supplemental Figure 8D).
Figure 6. DP103 regulates NF-κB in response to multiple stimuli.

(A) MDA-MB-231 cells transfected with either control siRNA or siDP103. Cells untreated (UT) or treated as indicated. Extracts analyzed with EMSA (top) and Western blotting (bottom). White separating lines between different stimuli. (B and C) MDA-MB-231 cells transfected with control siRNA or siDP103. Lysates of cells untreated or treated with TNF-α (B) or CPT (C), tested by EMSA. (D) BT549 cells transfected as in C and stimulated with TNF-α. RNA analyzed for mRNA expression of NF-κB target genes. *P < 0.05; **P < 0.01; ***P < 0.001. (E) MCF10A cells treated with TNF-α or LPS. (F) MCF10A cells either untreated or treated with inhibitors for IKK2, MEK1/2, JNK, p38, or PI3K, then stimulated with LPS. (G) White arrowheads indicate putative NF-κB–binding sites on DP103 promoter. (H) Nuclear proteins from MDA-MB-231 cells either untreated or treated with CPT tested by EMSA. Unlabeled DP103 probe as cold or IgG, p65, or p50 was used. (I) Nuclear proteins from MDA-MB-231 cells untreated or treated with TNF-α tested by EMSA using NF-κB consensus (lanes 1–3) or DP103 probe (lanes 4–6). Unlabeled NF-κB consensus cold probe added to lanes 3 and 6. (J) Cells transfected either with siRNA control or siDP103. Lysates immunoprecipitated with NEMO antibody and kinase assay performed as previously described (36). (K) BT549 cells transfected either with control siRNA or siDP103 as indicated and immunoblotted. Fold difference in protein DNA binding indicated in EMSA for A–C and for protein expression changes in E, F, J, and K.
Given that a helicase dead DP103 functions in activating invasion and that DP103 can regulate signaling to NF-κB in response to a wide array of stimuli that converge only on the key TAK1-IKK complex, these results suggest that DP103 likely regulates a central complex in NF-κB activation such as the TAK1 or IKK complex independently of its role as a helicase. We tested kinase activity of the IKK complex on phosphorylation of IκBα with or without DP103 depletion in response to TNF-α as stimuli (Figure 6J). Phosphorylation of IκBα by the IKK complex was significantly impeded upon DP103 depletion. Furthermore, kinetics of endogenous IKK activation, as judged by phosphorylation of IKK1/2 in the activation loop at Ser180/Ser181 and its functional consequence, namely phosphorylation of IκBα (Ser32/Ser36), was markedly reduced when DP103 was depleted (Figure 6K), suggesting that DP103 is a key regulator of activating phosphorylations on the activation loop of IKKs and hence of IKK function in the physiological context.
DP103 enhances Tak1-mediated IKK2 phosphorylation and hence NF-κB activation.
Several mitogen-activated protein kinases, such as MEKK1, MEKK3, and TAK1, are suggested to regulate IKK activation. However, TAK1 is considered the immediate upstream activator of IKK and an essential component of both nuclear and receptor-mediated NF-κB activation, which works by phosphorylating the Ser180/Ser181 in the activation loop of IKKs (39, 40, 44). We first investigated whether endogenous DP103 and TAK1 interact. Indeed, DP103 and TAK1 interacted in vivo, and this interaction was specific to the cytoplasm, since TAK1 is cytoplasmic (Figure 7A). Covalent modification of adaptor proteins by ubiquitin chains (termed as polyubiquitination) has been shown to be critical for activation of IKKs downstream of many NF-κB–activating stimuli. While there is much ongoing debate on the exact nature of the ubiquitin chains, such as in the K-63, linear, or K11 fashion, which is key for IKK activation, the ubiquitin-binding proteins TAB2/3 and NEMO are essential regulatory subunits of TAK1 and IKK complexes, respectively (81–83). Indeed, deubiquitination enzymes A20 and CYLD, which remove these chains from adaptor proteins in NF-κB signaling, inhibit IKK and hence NF-κB activation. They are also known to be lost in inflammatory conditions and cancers, which explains the cause of hyperactivation of the pathway that characterizes these conditions (84). Indeed, we could see TAK1 interacting proteins TAB2/3 interacting with DP103 under endogenous conditions (Supplemental Figure 9, A and B). To further verify whether this is a direct interaction, we purified GST-TAK1 and His-DP103 recombinant proteins (Supplemental Figure 9C) and tested their interaction in vitro (Figure 7B). Pull-down using Ni-NTA beads showed physical interaction between the 2 purified molecules (Figure 7B), suggesting that they indeed directly interact. To test the functionality of this interaction, we evaluated whether DP103 regulates the ability of TAK1 to phosphorylate IKK2 on its activation loop (40, 44). We purified the TAK1 phosphorylation site (residues 152–204) on IKK2 (GST-IKK2-WT) and a protein with mutations in the TAK1 phosphorylation domain (GST-IKK2-Mut) (Supplemental Figure 9D) and included these in a kinase assay with purified TAK1 and an increasing amount of DP103 (Figure 7C). Increasing DP103 concentration caused an increase in TAK1-mediated phosphorylation (hence activation) specifically of IKK2-WT but not that of IKK2-Mut activation loops (Figure 7C). Importantly overexpression of DP103, much like what is seen in cancers, caused increased recruitment of IKK2 to TAK1 upon stimulation (Supplemental Figure 9, E and F). These results further explain the mechanism of constitutive IKK activation in cancers. Furthermore, given that the results in Figure 7C are derived using purified proteins, they suggest that DP103 can directly bind to TAK1 and function as a cofactor to enhance its kinase activity toward IKK2. This reaction is specific, since TAK1 activity toward the GST-IKK2-Mut protein remains unaltered. Indeed, if DP103 levels are limiting in normal cells and overexpression of DP103 as seen in cancers is the mechanistic basis of increased TAK1-mediated IKK and hence NF-κB activation apparent in cancers, this hypothesis could be tested. Next, we included the GST-IKK2-WT in a kinase assay where endogenous TAK1 was immunoprecipitated (Figure 7D). DP103 depletion showed reduced phosphorylation on Ser181 of GST-IKK2 by TAK1 (Figure 7D). Using MDA-MB-231 cells, we next evaluated whether DP103 levels influence the activation of the IKK kinase complex and hence NF-κB activation. Mere ectopic expression of DP103 increased activity of the IKK complex, as seen by phosphorylation of IκBα (Figure 7E). Conversely, depletion of endogenous DP103 expression led to reduced phosphorylation of IκBα (Figure 7E), reiterating that endogenous DP103 levels regulate TAK1 activity toward IKK2. Indeed, TAK1 depletion with siRNAs also showed downregulation of NF-κB target gene transcription (Figure 7F). Furthermore, much like siDP103 (Figure 3I), either TAK1 kinase activity inhibition or IKK2 inhibition or depletion with siRNAs against TAK1 abrogated TNF-α–induced MMP9 activation in MDA-MB-231 cels (Figure 7, G and H). In contrast, MMP2 was not affected, which is in line with the existing literature showing that MMP9 but not MMP2 is the bona fide NF-κB target. Finally, we tested functionality of these observations in MDA-MB-231 cells. Compared with control siRNA–treated cells, invasion of TAK1-depleted cells was greatly decreased in MDA-MB-231 cells (Figure 7I). We conclude that DP103-mediated regulation of TAK1’s activity toward the major NF-κB kinase IKK2 regulates MMP9 expression and invasion.
Figure 7. DP103 is a positive cofactor of TAK1-mediated IKK2 activation in MDA-MB-231 cells.

(A) Nuclear and cytoplasmic fractions immunoprecipitated with TAK1 and IgG control antibodies and immunoprecipitate material and lysates analyzed by immunoblotting with the indicated antibodies. (B) GST-TAK1 and His-DP103 incubated either separately or together. Immunoprecipitates analyzed by immunoblotting with the indicated antibodies. (C) Kinase assay performed using GST-TAK1 either with GST-IKK2-WT or GST-IKK2-Mut substrates with increasing input of His-DP103 protein. (D) Cells transfected either with control siRNA or siDP103 and stimulated with TNF-α. Lysates were immunoprecipitated using anti-TAK1 antibody, and kinase assay was performed using GST–IKK2 (amino acid residues 152–204) as substrate (top panel). (E) Cells transfected with DP103, siDP103, and respective control vectors. IKK complex immunoprecipitated with anti-NEMO antibody. IKK activity was determined using phosphorylation of IκBα. (F) Cells transfected either with control siRNA or siTAK1 and stimulated with TNF-α. Total RNA analyzed for mRNA expression of NF-κB target genes. *P < 0.05; ***P < 0.001. (G) Cells treated with IKK2 inhibitor IV or TAK1 inhibitor (5Z)-7-oxozeaenol and stimulated with TNF-α. MMP9 activity evaluated with zymography. (H) Cells treated with control siRNA (ctsi) or siRNAs against TAK1 (siTAK1) and stimulated with TNF-α. MMP9 activity evaluated with zymography. (I) Cells transfected either with control siRNA or siRNAs against TAK1. Invaded cells through Matrigel detached and lysed in assay buffer were presented as relative fluorescence units (RFU). ***P < 0.001. Fold differences in protein expression are indicated in C–E.
Discussion
Breast carcinoma is the most common malignancy in women, and its treatment is possible with early diagnosis. Despite high survival rates for early stage breast cancer, most such tumors often go unnoticed without regular mammograms. While effective targeted therapeutic modalities exist for women with hormone receptor–positive and HER2-positive disease, chemotherapy is the only systemic therapy available for women with triple-negative breast cancer. Increased heterogeneity of tumors in a larger group of patients poses a huge challenge in determining whether these drugs will show good efficacy in the OS and disease-free survival as well as the extent of the severity of side effects that these drugs will deliver. Strikingly, the number of patients who develop resistance to these therapies as well as patients who show relapse and metastasis is on the rise (85–87). Metastasis is a multifactorial process requiring the concerted effort of many players. As such, it is a highly complex process and therefore unraveling key gene(s) whose expression levels could predict metastasis or tumorigenesis is of immense therapeutic value (12–14).
In this study, we uncovered DP103 as a prognostic marker and a therapeutic target for breast cancer metastasis. While members of the DEAD-box family, p68/p72 and Ddx3, have been linked to tumorigenesis, the potential role of DP103, a relatively new member (28), in cancer, if any, is yet to be uncovered. Using 2 independent cohorts, we found that a significant number of breast cancer patients display high levels of DP103 in their invasive breast tumor tissue, especially the basal subtype, compared with normal breast tissue. The prognostic and clinical significance of DP103 expression is highlighted by the observation that patients with high expression of DP103 have significantly shorter OS and SAR. Furthermore, DP103 levels were found to be upregulated in a statistically significant proportion of grade 3 breast cancers according to a multi-institutional mRNA microarray meta-analysis cohort of primary breast cancer; this was validated by IHC for DP103 protein expression in 2 independent cohorts. In vitro and in vivo analyses revealed that expression of DP103 strongly and positively correlates with the ability of breast cancer cells to metastasize. Remarkably, heightened expression of DP103 (as seen in cancers) in the normal epithelial breast cell line, MCF10A, which normally displays very low levels of DP103 and is unable to invade, was sufficient to confer a 4-fold increased ability to these cells to invade. Overall, our study established that DP103 is not only a prognostic marker for breast cancer tumorigenesis and progression, but also a new predictive biomarker for cancer metastasis.
Since MMP9 expression has a clinical expression profile similar to that of DP103 and its expression is highly correlative to expression of DP103, both in clinical specimens and cell lines, we homed on the expression of the MMP family of proteins as a putative mechanism for observed effects of DP103. Our analysis further suggested that the ability of DP103 to regulate NF-κB but not AP1 activity may be crucial for its regulation of MMP9. Indeed, clinical expression of phospho-p65 (active NF-κB) mirrors the expression of DP103, both in terms of expression profile and survival outcomes, and most significantly, expression of phospho-p65 very significantly associates with expression of DP103. These results are significant because this study is the first, to our knowledge, to establish a role for DP103–NF-κB–MMP9 axis in breast metastasis.
How does DP103 regulate NF-κB–dependent transcription? Interestingly, DP103’s helicase activity was not involved in the regulation of invasion. The only known helicase-independent activity of DP103 is its ability to affect PIASy-mediated SUMOylation of SF1 to act as a corepressor (23, 24). However, the underlying molecular mechanism in the above 2 studies remains unknown. The known function of PIASy in the NF-κB–signaling system is limited to its ability to induce SUMOylation of NEMO in the DNA damage–signaling pathway (76, 88). Indeed, DP103 could also affect PIASy SUMOylation, but this did not have a functional role in its regulation of MMP9 transcription (data not shown). Our results convincingly show that DP103-induced NF-κB and hence MMP9 activation is dependent on its ability to regulate TAK1-mediated activation of IKK via the phosphorylation of IKK activation loop. Knockdown and specific kinase inhibitors further prove that DP103-TAK1 complex is a potent activator of IKK2, the key NF-κB–activating kinase, and this induces MMP9 and hence invasion. Thus, identification of DP103 as a new limiting factor in TAK1-dependent NF-κB activation provides a significant mechanistic insight into the mechanism of constitutive NF-κB activation seen in tumors. It explains why, despite lack of overexpression of the TAK1 and IKK kinases, hyperactivation of NF-κB is apparent in the vast majority of cancers. It is due to the fact that DP103, levels of which increase in invasive or high-grade cancer, provides a tonic activation of TAK1-mediated IKK2 and hence NF-κB activity. Since levels of DP103 are transcriptionally controlled by NF-κB itself in breast cancer cells, this feedback loop maintains constitutive activation of NF-κB and hence chronic inflammation, another hallmark of human cancers (89). A model based on our studies is depicted in Figure 8. Since it is well accepted that constitutive NF-κB activity also imparts cancer cells the chemoresistant phenotype (48, 49), DP103 could also be a therapeutic target in these cancers. Indeed, our data showed that reduction of NF-κB signaling by downregulating DP103 sensitized breast cancer cells to chemotherapy-induced cell death. DP103 on its own may not be a very attractive therapeutic target based on conventional targeting methods. Although small molecules and staple peptides have been designed to target “nondruggable” targets and even transcription factors such as BCL6, our finding that NF-κB itself can positively regulate DP103 expression provides us with an opportunity to break the positive feed-forward loop, the Achilles’ heel of metastatic breast cancer, by blocking NF-κB activation. However, while blocking NF-κB by blocking IKK activity is successful in laboratory settings, this approach hasn’t been successful in the clinic due to toxicity.
Figure 8. Schematic model based on our study showing a role for the RNA helicase DP103 in its ability to bind and stabilize TAK1 and thus activate NF-κB signaling in cancers.

In summary, we describe a function of an RNA helicase DP103 that depends on its ability to bind and stabilize TAK1 and thus activate NF-κB signaling. Although constitutive activation of NF-κB is a well-documented phenomenon in cancer, increased levels of enzymes such as TAK1 that could maintain IKK2 and hence NF-κB in a constitutively active state are not seen in cancer cells. Instead, our current study uncovered that it is the increase in levels of DP103 that marks the switch from a nonmetastatic to a metastatic state in breast cancer cells and possibly other cancer cell types. In addition, we also elucidated a plausible DP103–NF-κB–positive feed-forward loop that could be involved in the maintenance of this oncogenic signaling arm in cancers. Thus, we suggest that DP103 is a novel biomarker as well as a worthwhile therapeutic drug target.
Methods
Cell lines and reagents.
MCF-7, MDA-MB-231, and BT549 human breast cancer cells were grown in RPMI with 10% FBS, 2 mM l-glutamine, and 0.05 mg/ml gentamicin. HEK293, BT474, and SKBr3 cells were grown in DMEM, DMEM/F12, and McCoy’s 5A, respectively, with the same supplements as above. MCF10A and 184A1 cells were grown in MEGM with 10% FBS, 2 mM l-glutamine, bovine pituitary extract, and 0.05 mg/ml gentamicin. Xenograft-derived breast cancer cell lines (MCF10A1, MCF10AT1KCl.2, MCF10CA1h, and MCF10CA1aCl.1) were maintained in DMEM/F12 with 5% horse serum, 100 U/ml penicillin/streptomycin, 10 μg/ml insulin, 20 ng/ml EGF (Millipore), 0.5 μg/ml hydrocortisone, and 100 ng/ml cholera toxin. All cell lines except xenograft-derived were purchased from ATCC; media and supplements were from Hyclone unless otherwise indicated. Doxorubicin, VP16 (Etoposide), and camptothecin were purchased from Calbiochem. All other chemicals and reagents were from Sigma-Aldrich unless otherwise indicated.
Other assays.
Virus preparation and infection, transfections for siRNA and plasmids, Western blot analysis, immunoprecipitation, real-time PCR, GST-IκBα kinase, EMSA, cell viability, and luciferase assays were performed as described previously (36) and are described in detail in Supplemental Methods. Gel zymography (90) is described in detail in Supplemental Methods. Primer sequences are available in Supplemental Methods.
Metastatic qPCR array and microarray.
Total RNA was extracted, and relative expressions of various genes were then analyzed using qPCR (ABI PRISM 7900; Applied Biosystems). Multiple gene markers distributed around the genome and 3 housekeeping genes were used for real-time PCR analysis using the SYBR GreenER qPCR SuperMix for ABI PRISM (Invitrogen), done in triplicate. Sequences of primers used were described previously (91, 92). DP103 expression levels were analyzed in breast tumors using a multi-institutional microarray meta-analysis cohort totaling 759 primary breast cancer cases. The normalized microarray data set and associated clinical annotations were described previously (93). Briefly, the microarray meta-analysis cohort comprised data sets from 6 breast tumor profiling studies. The data sets are accessible through MIAME-compliant public databases. Three data sets are housed in GEO (94) (GSE1456, ref. 95; GSE6532, ref. 96; and GSE9195, ref. 97); 1 data set is housed in the NCI’s caArray database (mille-00271; ref; 98). All tumor samples were analyzed from frozen tissue collected at surgery and profiled on an Affymetrix U133 series microarray according to standard Affymetrix protocols (93). Raw data (CEL files) were normalized by Robust Multichip Average (RMA) using the Bioconductor Affy Package (R) as previously described (99). Batch effects between cohorts were corrected using the Partek Genomics Suite Batch Remover program. Two Affymetrix probe sets (223331_s_at and 224315_at) were designed to detect DP103 expression. The log2 signal intensities of these 2 probe sets were well correlated in the breast tumor meta-analysis cohort (r = 0.65, Pearson correlation).
Data preprocessing of Affymetrix microarray gene expression.
Breast cancer data sets were downloaded from GEO (94). Microarray data on a U133Plus2 platform were utilized for analysis under the following GEO accession numbers: GSE12276 (n = 204), GSE19615 (n = 115), GSE21653 (n = 266), GSE23177 (n = 116), GSE23593 (n = 50), GSE26639 (n = 226), GSE3744 (n = 47), GSE5460 (n = 127), GSE5764 (n = 10), GSE6532 (n = 87), and GSE9195 (n = 77). For this study, we included all publicly available data set in the U133Plus2 platform at the time that the analysis was initiated in October 2011. RMA normalization was performed on each data set. The normalized data were compiled and subsequently standardized using ComBat (100) to remove batch effect. The standardized data yielded a data set of 1,325 breast cancer tumors and 20 normal breast tissue samples. The expression values of the DP103 gene were log averaged from DP103 probes in U133Plus2 platform: 223331_s_at and 224315_at. Apart from the 1,345 samples we compiled, we downloaded GEO GSE3494 (n = 252), data on both U133A and U133B platforms, and used it as validation data. GEO GSE3493 data were processed and normalized using the same method as described above.
ssGSEA.
ssGSEA was originally described in a previous study (58).
Identification of breast cancer subtypes.
Breast cancer subtype signature was obtained from Prat et al. (59). Subsequently, ssGSEA was computed based on the breast cancer subtype signature for each sample. Each sample was then assigned to be the subtype with the maximum ssGSEA score (59).
2D migrational assay, in vitro and 3D invasion assay, wound healing assay.
2D migrational assay, in vitro and 3D invasion assay, and wound healing assay were performed as per standard protocols and are described in Supplemental Methods.
Statistics.
Statistical analyses were performed using the SPSS package (version 15.0 for Windows; SPSS Inc.), with significance set at the 5% level. Statistical significance evaluation by Mann-Whitney test and Spearman correlation test were computed using MATLAB. Dot plot was done using Graphpad Prism. Two-tailed Student’s t tests were performed using the software Origin Pro, and results are represented as mean ± SD. Associations among DP103, MMP9, and phospho-p65 immunohistochemical staining data and clinicopathological parameters were determined using Fisher’s exact and Kendall tau tests. A P value below 0.05 was considered statistically significant. Survival curves were plotted using the Kaplan-Meier method and compared using the log-rank test.
Study approval.
All animal studies were reviewed and approved by the SingHealth Institutional Animal Care and Use Committee, SingHealth Office of Research, Singapore, and are described in Supplemental Methods. Ethics approval for the study using clinical material from Singapore was obtained from the Institutional Review Board, Singapore General Hospital. Protocol for the use of clinical material from China was approved by the Institutional Review Board of Anhui Medical University, while the use of clinical material from Canada was approved by the Institutional Review Board, Montreal University Health Centre, in 2005, with a renewal in 2010. Details on clinical materials and immunohistochemical procedures are described in Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
This work was supported by grants from the Singapore Ministry of Education Tier 2 (MOE2012-T2-2-139), the Academic Research Fund Tier 1 (R-184-000-228-112), and the Cancer Science Institute of Singapore, Experimental Therapeutics I Program (grant R-713-001-011-271) (to A.P. Kumar). A.P. Kumar is also supported by the John Nott Cancer Fellowship from Cancer Council, Western Australia. This work was also supported by the Singapore Ministry of Health’s National Medical Research Council under its Individual Research Grant funding scheme (to G. Sethi); the National Key Scientific Program of China (2010CB912804, 2007CB914801, 2011CBA01103), and the CAS Visiting Professorship for Senior International Scientists (2010T2S03) (to T. Zhu). Core funding was received from A*STAR (to V. Tergaonkar) and NIH R01 CA77474 and GM083681 (to S. Miyamoto).
Footnotes
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Conflict of interest: Alan Prem Kumar is the principal inventor in a patent application based on invention disclosure entitled “Clinical Utility of DEAD-Box Helicase DP103 (DDX20) As a Prognostic and Predictive Biomarker for Drug Response in Cancer” [NUS ILO ref: 14317N-UK/PRV; symbiosis ref: 4439P/GB].
Reference information:J Clin Invest. 2014;124(9):3807–3824. doi:10.1172/JCI73451.
References
- 1.Vernon AE, Bakewell SJ, Chodosh LA. Deciphering the molecular basis of breast cancer metastasis with mouse models. Rev Endocr Metab Disord. 2007;8(3):199–213. doi: 10.1007/s11154-007-9041-5. [DOI] [PubMed] [Google Scholar]
- 2.Guo BH, et al. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol Cancer. 2011;10(1):10. doi: 10.1186/1476-4598-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tavazoie SF, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451(7175):147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao ES, et al. Increased β1 integrin is associated with decreased survival in invasive breast cancer. Cancer Res. 2007;67(2):659–664. doi: 10.1158/0008-5472.CAN-06-2768. [DOI] [PubMed] [Google Scholar]
- 5.Weigelt B, Peterse JL, van ‘t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5(8):591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 6.Zani S, Clary BM. A role for hepatic metastasectomy in stage IV melanoma and breast cancer: reestablishing the surgical modality. Oncology (Williston Park). 2011;25(12):1158–1164. [PubMed] [Google Scholar]
- 7.DeSantis C, Siegel R, Bandi P, Jemal A. Breast cancer statistics, 2011. CA Cancer J Clin. 2011;61(6):409–418. doi: 10.3322/caac.20134. [DOI] [PubMed] [Google Scholar]
- 8.Forouzanfar MH, et al. Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet. 2011;378(9801):1461–1484. doi: 10.1016/S0140-6736(11)61351-2. [DOI] [PubMed] [Google Scholar]
- 9.Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3(1):55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- 10.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 11.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Mego M, Mani SA, Cristofanilli M. Molecular mechanisms of metastasis in breast cancer — clinical applications. Nat Rev Clin Oncol. 2010;7(12):693–701. doi: 10.1038/nrclinonc.2010.171. [DOI] [PubMed] [Google Scholar]
- 13.Onishi T, Hayashi N, Theriault RL, Hortobagyi GN, Ueno NT. Future directions of bone-targeted therapy for metastatic breast cancer. Nat Rev Clin Oncol. 2010;7(11):641–651. doi: 10.1038/nrclinonc.2010.134. [DOI] [PubMed] [Google Scholar]
- 14.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8(4):210–221. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraljevic Pavelic S, Sedic M, Bosnjak H, Spaventi S, Pavelic K. Metastasis: new perspectives on an old problem. Mol Cancer. 2011;10:22. doi: 10.1186/1476-4598-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curran S, Murray GI. Matrix metalloproteinases: molecular aspects of their roles in tumour invasion and metastasis. Eur J Cancer. 2000;36(13 spec no):1621–1630. doi: 10.1016/s0959-8049(00)00156-8. [DOI] [PubMed] [Google Scholar]
- 17.Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011;41(2):271–290. doi: 10.1007/s00726-010-0689-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 19.Meyer T, Hart IR. Mechanisms of tumour metastasis. Eur J Cancer. 1998;34(2):214–221. doi: 10.1016/S0959-8049(97)10129-0. [DOI] [PubMed] [Google Scholar]
- 20.Linder P. Dead-box proteins: a family affair — active and passive players in RNP-remodeling. Nucleic Acids Res. 2006;34(15):4168–4180. doi: 10.1093/nar/gkl468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mouillet JF, et al. DEAD-box protein-103 (DP103, Ddx20) is essential for early embryonic development and modulates ovarian morphology and function. Endocrinology. 2008;149(5):2168–2175. doi: 10.1210/en.2007-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuller-Pace FV. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 2006;34(15):4206–4215. doi: 10.1093/nar/gkl460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee MB, Lebedeva LA, Suzawa M, Wadekar SA, Desclozeaux M, Ingraham HA. The DEAD-box protein DP103 (Ddx20 or Gemin-3) represses orphan nuclear receptor activity via SUMO modification. Mol Cell Biol. 2005;25(5):1879–1890. doi: 10.1128/MCB.25.5.1879-1890.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ou Q, Mouillet JF, Yan X, Dorn C, Crawford PA, Sadovsky Y. The DEAD box protein DP103 is a regulator of steroidogenic factor-1. Mol Endocrinol. 2001;15(1):69–79. doi: 10.1210/mend.15.1.0580. [DOI] [PubMed] [Google Scholar]
- 25.Yan X, Mouillet JF, Ou Q, Sadovsky Y. A novel domain within the DEAD-box protein DP103 is essential for transcriptional repression and helicase activity. Mol Cell Biol. 2003;23(1):414–423. doi: 10.1128/MCB.23.1.414-423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klappacher GW, et al. An induced Ets repressor complex regulates growth arrest during terminal macrophage differentiation. Cell. 2002;109(2):169–180. doi: 10.1016/S0092-8674(02)00714-6. [DOI] [PubMed] [Google Scholar]
- 27.Charroux B, Pellizzoni L, Perkinson RA, Shevchenko A, Mann M, Dreyfuss G. Gemin3: A novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of gems. J Cell Biol. 1999;147(6):1181–1194. doi: 10.1083/jcb.147.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grundhoff AT, et al. Characterization of DP103, a novel DEAD box protein that binds to the Epstein-Barr virus nuclear proteins EBNA2 and EBNA3C. J Biol Chem. 1999;274(27):19136–19144. doi: 10.1074/jbc.274.27.19136. [DOI] [PubMed] [Google Scholar]
- 29.Meister G, Buhler D, Laggerbauer B, Zobawa M, Lottspeich F, Fischer U. Characterization of a nuclear 20S complex containing the survival of motor neurons (SMN) protein and a specific subset of spliceosomal Sm proteins. Hum Mol Genet. 2000;9(13):1977–1986. doi: 10.1093/hmg/9.13.1977. [DOI] [PubMed] [Google Scholar]
- 30.Shpargel KB, Matera AG. Gemin proteins are required for efficient assembly of Sm-class ribonucleoproteins. Proc Natl Acad Sci U S A. 2005;102(48):17372–17377. doi: 10.1073/pnas.0508947102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillian AL, Svaren J. The Ddx20/DP103 dead box protein represses transcriptional activation by Egr2/Krox-20. J Biol Chem. 2004;279(10):9056–9063. doi: 10.1074/jbc.M309308200. [DOI] [PubMed] [Google Scholar]
- 32.Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18(18):2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 33.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev. 1995;9(22):2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 34.Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 35.Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458(7237):430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 36.Teo H, et al. Telomere-independent Rap1 is an IKK adaptor and regulates NF-κB-dependent gene expression. Nat Cell Biol. 2010;12(8):758–767. doi: 10.1038/ncb2080. [DOI] [PubMed] [Google Scholar]
- 37.Wu ZH, et al. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell. 2010;40(1):75–86. doi: 10.1016/j.molcel.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Staudt LM. Oncogenic activation of NF-κB. Cold Spring Harb Perspect Biol. 2010;2(6):a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011;12(8):715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 40.Ghosh S, Karin M. Missing pieces in the NF-κB puzzle. Cell. 2002;109(suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 41.Bhat-Nakshatri P, Sweeney CJ, Nakshatri H. Identification of signal transduction pathways involved in constitutive NF-κB activation in breast cancer cells. Oncogene. 2002;21(13):2066–2078. doi: 10.1038/sj.onc.1205243. [DOI] [PubMed] [Google Scholar]
- 42.Nakshatri H, Goulet RJ, Goulet RJ., Jr NF-κB and breast cancer. Curr Probl Cancer. 2002;26(5):282–309. doi: 10.1067/mcn.2002.129977. [DOI] [PubMed] [Google Scholar]
- 43.Romieu-Mourez R, et al. Mouse mammary tumor virus c-rel transgenic mice develop mammary tumors. Mol Cell Biol. 2003;23(16):5738–5754. doi: 10.1128/MCB.23.16.5738-5754.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 2012;12(2):121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 45.Sovak MA, et al. Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100(12):2952–2960. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyamoto S. Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 2011;21(1):116–130. doi: 10.1038/cr.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-κB addiction and its role in cancer: ‘one size does not fit all’. Oncogene. 2011;30(14):1615–1630. doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basseres DS, Baldwin AS. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25(51):6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 49.Kim HJ, Hawke N, Baldwin AS. NF-κB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13(5):738–747. doi: 10.1038/sj.cdd.4401877. [DOI] [PubMed] [Google Scholar]
- 50.Biswas SK, Tergaonkar V. Myeloid differentiation factor 88-independent Toll-like receptor pathway: sustaining inflammation or promoting tolerance? Int J Biochem Cell Biol. 2007;39(9):1582–1592. doi: 10.1016/j.biocel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 51.Rajendran P, et al. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev Res (Phila). 2012;5(4):631–643. doi: 10.1158/1940-6207.CAPR-11-0420. [DOI] [PubMed] [Google Scholar]
- 52.Dey A, Wong E, Kua N, Teo HL, Tergaonkar V, Lane D. Hexamethylene bisacetamide (HMBA) simultaneously targets AKT and MAPK pathway and represses NFκB activity: implications for cancer therapy. Cell Cycle. 2008;7(23):3759–3767. doi: 10.4161/cc.7.23.7213. [DOI] [PubMed] [Google Scholar]
- 53.Irelan JT, et al. A role for IκB kinase 2 in bipolar spindle assembly. Proc Natl Acad Sci U S A. 2007;104(43):16940–16945. doi: 10.1073/pnas.0706493104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ang HL, Tergaonkar V. Notch and NFκB signaling pathways: do they collaborate in normal vertebrate brain development and function? Bioessays. 2007;29(10):1039–1047. doi: 10.1002/bies.20647. [DOI] [PubMed] [Google Scholar]
- 55.Cildir G, Akincilar SC, Tergaonkar V. Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol Med. 2013;19(8):487–500. doi: 10.1016/j.molmed.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 56.Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441(7092):431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 57.Ghobrial IM, et al. Proteomic analysis of mantle-cell lymphoma by protein microarray. Blood. 2005;105(9):3722–3730. doi: 10.1182/blood-2004-10-3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prat A, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol. 1996;148(1):313–319. [PMC free article] [PubMed] [Google Scholar]
- 61.Choong LY, Lim S, Chong PK, Wong CY, Shah N, Lim YP. Proteome-wide profiling of the MCF10AT breast cancer progression model. PLoS One. 2010;5(6):e11030. doi: 10.1371/journal.pone.0011030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choong LY, et al. Elevated NRD1 metalloprotease expression plays a role in breast cancer growth and proliferation. Genes Chromosomes Cancer. 2011;50(10):837–847. doi: 10.1002/gcc.20905. [DOI] [PubMed] [Google Scholar]
- 63. Chan PK, Mak MS, inventors; Pacific Arrow Limited, assignee. Blocking the migration or metastasis of cancer cells by affecting adhesion proteins the uses of new compounds thereof. US patent application 20,100,004,190. January 7, 2010. [Google Scholar]
- 64.Zucchi I, et al. Gene expression profiles of epithelial cells microscopically isolated from a breast-invasive ductal carcinoma and a nodal metastasis. Proc Natl Acad Sci U S A. 2004;101(52):18147–18152. doi: 10.1073/pnas.0408260101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 66.Liu Z, et al. Increased expression of MMP9 is correlated with poor prognosis of nasopharyngeal carcinoma. BMC Cancer. 2010;10:270. doi: 10.1186/1471-2407-10-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mitra RS, et al. Rap1GAP promotes invasion via induction of matrix metalloproteinase 9 secretion, which is associated with poor survival in low N-stage squamous cell carcinoma. Cancer Res. 2008;68(10):3959–3969. doi: 10.1158/0008-5472.CAN-07-2755. [DOI] [PubMed] [Google Scholar]
- 68.Sakata K, et al. Expression of matrix metalloproteinase 9 is a prognostic factor in patients with non-Hodgkin lymphoma. Cancer. 2004;100(2):356–365. doi: 10.1002/cncr.11905. [DOI] [PubMed] [Google Scholar]
- 69.Beliveau A, et al. Raf-induced MMP9 disrupts tissue architecture of human breast cells in three-dimensional culture and is necessary for tumor growth in vivo. Genes Dev. 2010;24(24):2800–2811. doi: 10.1101/gad.1990410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nair RR, et al. A novel high-throughput screening system identifies a small molecule repressive for matrix metalloproteinase-9 expression. Mol Pharmacol. 2008;73(3):919–929. doi: 10.1124/mol.107.042606. [DOI] [PubMed] [Google Scholar]
- 71.Milde-Langosch K, et al. The role of the AP-1 transcription factors c-Fos, FosB, Fra-1 and Fra-2 in the invasion process of mammary carcinomas. Breast Cancer Res Treat. 2004;86(2):139–152. doi: 10.1023/B:BREA.0000032982.49024.71. [DOI] [PubMed] [Google Scholar]
- 72.Adamson R, Logan M, Kinnaird J, Langsley G, Hall R. Loss of matrix metalloproteinase 9 activity in Theileria annulata-attenuated cells is at the transcriptional level and is associated with differentially expressed AP-1 species. Mol Biochem Parasitol. 2000;106(1):51–61. doi: 10.1016/S0166-6851(99)00213-3. [DOI] [PubMed] [Google Scholar]
- 73.Chou YC, et al. Nuclear-targeted inhibition of NF-κB on MMP-9 production by N-2-(4-bromophenyl) ethyl caffeamide in human monocytic cells. Chem Biol Interact. 2010;184(3):403–412. doi: 10.1016/j.cbi.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 74.Ricca A, Biroccio A, Del Bufalo D, Mackay AR, Santoni A, Cippitelli M. bcl-2 over-expression enhances NF-κB activity and induces mmp-9 transcription in human MCF7(ADR) breast-cancer cells. Int J Cancer. 2000;86(2):188–196. doi: 10.1002/(sici)1097-0215(20000415)86:2<188::aid-ijc7>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 75.Martinez N, et al. Transcriptional signature of Ecteinascidin 743 (Yondelis, Trabectedin) in human sarcoma cells explanted from chemo-naive patients. Mol Cancer Ther. 2005;4(5):814–823. doi: 10.1158/1535-7163.MCT-04-0316. [DOI] [PubMed] [Google Scholar]
- 76.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell. 2003;115(5):565–576. doi: 10.1016/S0092-8674(03)00895-X. [DOI] [PubMed] [Google Scholar]
- 77.Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat Cell Biol. 2006;8(9):986–993. doi: 10.1038/ncb1458. [DOI] [PubMed] [Google Scholar]
- 78.Lee MH, Mabb AM, Gill GB, Yeh ET, Miyamoto S. NF-κB induction of the SUMO protease SENP2: a negative feedback loop to attenuate cell survival response to genotoxic stress. Mol Cell. 2011;43(2):180–191. doi: 10.1016/j.molcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ghosh A, et al. Telomerase directly regulates NF-κB-dependent transcription. Nat Cell Biol. 2012;14(12):1270–1281. doi: 10.1038/ncb2621. [DOI] [PubMed] [Google Scholar]
- 80.Richter L, Bone JR, Kuroda MI. RNA-dependent association of the Drosophila maleless protein with the male X chromosome. Genes Cells. 1996;1(3):325–336. doi: 10.1046/j.1365-2443.1996.26027.x. [DOI] [PubMed] [Google Scholar]
- 81.Haas TL, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36(5):831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 82.Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer. 2006;6(10):776–788. doi: 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- 83.Chen ZJ. Ubiquitin signalling in the NF-κB pathway. Nat Cell Biol. 2005;7(8):758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. 2012;246(1):95–106. doi: 10.1111/j.1600-065X.2012.01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004;11(4):643–658. doi: 10.1677/erc.1.00776. [DOI] [PubMed] [Google Scholar]
- 86.Chabalier C, Lamare C, Racca C, Privat M, Valette A, Larminat F. BRCA1 downregulation leads to premature inactivation of spindle checkpoint and confers paclitaxel resistance. Cell Cycle. 2006;5(9):1001–1007. doi: 10.4161/cc.5.9.2726. [DOI] [PubMed] [Google Scholar]
- 87.Tanaka K, Kawaguchi H, Nakamura Y, Taguchi K, Nishiyama K, Ohno S. Effect of HER2 status on risk of recurrence in women with small, node-negative breast tumours. Br J Surg. 2011;98(11):1561–1565. doi: 10.1002/bjs.7639. [DOI] [PubMed] [Google Scholar]
- 88.Wu ZH, Miyamoto S. Induction of a pro-apoptotic ATM-NF-κB pathway and its repression by ATR in response to replication stress. EMBO J. 2008;27(14):1963–1973. doi: 10.1038/emboj.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Low KC, Tergaonkar V. Telomerase: central regulator of all of the hallmarks of cancer. Trends Biochem Sci. 2013;38(9):426–434. doi: 10.1016/j.tibs.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 90.Karpova TS, Stetler-Stevenson WG, Roberts AB. Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNF-α and TGF-β. J Cell Sci. 2005;118(pt 10):2143–2153. doi: 10.1242/jcs.02334. [DOI] [PubMed] [Google Scholar]
- 91.Qian P, et al. Pivotal role of reduced let-7g expression in breast cancer invasion and metastasis. Cancer Res. 2011;71(20):6463–6474. doi: 10.1158/0008-5472.CAN-11-1322. [DOI] [PubMed] [Google Scholar]
- 92.Pandey V, et al. Artemin reduces sensitivity to doxorubicin and paclitaxel in endometrial carcinoma cells through specific regulation of CD24. Transl Oncol. 2010;3(4):218–229. doi: 10.1593/tlo.09325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miller LD, et al. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res. 2011;71(21):6728–6737. doi: 10.1158/0008-5472.CAN-11-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pawitan Y, et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7(6):R953–R964. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Loi S, et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007;25(10):1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- 97.Loi S, et al. Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genomics. 2008;9:239. doi: 10.1186/1471-2164-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miller LD, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005;102(38):13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.