

Abstract
Objective
An elevated plasma aldosterone level is an independent cardiovascular risk factor. Although excess aldosterone promotes cardiovascular disease, no studies have examined the effect of increased plasma aldosterone on the cerebral circulation. A major source of vascular reactive oxygen species (ROS) during cardiovascular disease is the NADPH oxidases. Because NOX2-containing NADPH oxidase (NOX2 oxidase) is highly expressed in cerebral endothelium, we postulated that it might contribute to ROS generation and vascular dysfunction in response to aldosterone. Here we examined the effect of aldosterone and NOX2 oxidase on ROS production and endothelial dysfunction in the cerebral circulation, and whether the effects of aldosterone are exacerbated in aged mice.
Methods and Results
In adult (average age ~24–25 wk) wild-type (WT) and Nox2-deficient (Nox2−/y) mice, neither vehicle nor aldosterone (0.28 mg/kg/day for 14 days) affected blood pressure (measured using tail-cuff). By contrast, aldosterone treatment reduced dilation of the basilar artery (measured using myography) to the endothelium-dependent agonist acetylcholine in WT mice (P<0.05), but had no such effect in NOX2−/y mice (P>0.05). Aldosterone increased basal and phorbol-dibutyrate stimulated superoxide production (measured using L-012-enhanced chemiluminesence) in cerebral arteries from WT but not Nox2−/y mice. In aged WT mice (average age ~70 wk), aldosterone treatment increased blood pressure, but had a similar effect on cerebral artery superoxide levels as in adult WT mice.
Conclusions
These data indicate that NOX2 oxidase mediates aldosterone-induced increases in ROS production and endothelial dysfunction in cerebral arteries from adult mice independently of blood pressure changes. Aldosterone-induced hypertension is augmented during aging.
Keywords: cerebral artery, cerebrovascular disease, endothelium, NOX2, aging
INTRODUCTION
Aldosterone acts on the mineralocorticoid receptor (MR) expressed in renal tubular epithelial cells to play an important role in sodium and water retention and potassium excretion, thus regulating fluid and electrolyte homeostasis and blood pressure [1]. Aldosterone synthesis and MR expression also occur in non-epithelial (non-renal) tissues including in the brain [2] and cerebral blood vessels [3].
Chronic hypertension exerts many adverse effects on the cerebral circulation and brain [4]. Patients with primary aldosteronism (characterized by an overproduction of aldosterone) suffer more strokes than patients with essential hypertension despite having lower blood pressure [5,6], and have much higher rates of stroke than age-, sex-, and blood pressure-matched essential hypertensives [7]. These observations suggest that elevated plasma aldosterone increases stroke risk in a blood pressure-independent manner. Such an effect could involve deleterious direct actions of aldosterone in the cerebral circulation. Thus, we first aimed to test the hypothesis that aldosterone increases cerebral vascular ROS production and causes endothelial dysfunction.
Aldosterone stimulates the production of reactive oxygen species (ROS) [8–12] and causes endothelial dysfunction [9, 11, 13–17] in systemic arteries. NADPH oxidases are a major source of vascular ROS, and aldosterone increases vascular NADPH oxidase activity [8, 11, 13] and expression of the NOX2 subunit [8]. However, the role of specific NADPH oxidases in mediating aldosterone-dependent vascular ROS production and dysfunction is not well characterized, and has received no study in the cerebral circulation. NOX2 is highly expressed in the endothelium of cerebral arteries [18, 19], and there is evidence for an involvement of NOX2 oxidase in mediating cerebrovascular dysfunction and oxidative stress in response to angiotensin II, and in models of diabetes, aging, hypercholesterolemia and Alzheimer’s disease [20–22]. Thus, our second aim was to test whether NOX2 oxidase mediates the deleterious actions of aldosterone in cerebral arteries.
While the frequency of stroke and Alzheimer’s disease increases with age [23], underlying age-dependent mechanisms leading to pathological changes in the cerebral circulation are poorly understood. Because there is evidence that mRNA expression of vascular MR is increased in aged (30 months) vs adult (8 months) rats [24], the third aim of our study was to test whether the cerebrovascular actions of aldosterone are exacerbated during aging.
METHODS
Experimental animals
Male mice were studied. Nox2−/y mice were originally generated in the laboratory of Professor Mary Dinauer [25] and have been backcrossed to the C57Bl/6J strain for at least 10 generations. Littermates and age-matched C57Bl/6J mice were used as wild-type (WT; i.e. Nox2+/y) controls. Mice had access to regular chow and water ad libitum. All protocols and procedures were approved by the Animal Ethics Committee at Monash University. Adult WT (average age: 24.1±0.8 wk, n=62), Nox2−/y (average age: 25.4±0.6 wk, n=27) and aged WT (average age: 69.8±2.4 wk, n=18) mice were used in this study.
Aldosterone administration and measurement of blood pressure
Following anesthesia with ketamine/xylazine, an osmotic minipump (Alzet, model 1004) was placed subcutaneously in the mid-scapular region to administer vehicle (87% propylene glycol; 9% ethanol; 4% distilled H2O) or aldosterone (0.28 mg/kg/d for 14 days). Systolic blood pressure was measured using an automated tail-cuff device (MC 4000, Hatteras Instruments Inc.). Prior to surgery, mice were trained for 2–5 days and baseline blood pressure was recorded, followed by implantation of pumps and subsequent measurements of arterial pressure.
Functional study of basilar artery
Isolation and preparation of cerebral arteries was performed as described previously [18, 21]. Basilar arteries were mounted between two microcannulae in a myograph (Living Systems Instrumentation Inc.) and superfused with carbogen-bubbled (95% O2, 5% CO2) Krebs-bicarbonate solution (composition in mmol/L; NaCl 118, KCl 4.5, MgSO4 0.45, KH2PO4 1.03, NaHCO3 25, glucose 11.1, CaCl2 2.5) maintained at 37°C. Intraluminal pressure was gradually increased to 60 mmHg and maintained at this level with a pressure servo unit without further intraluminal perfusion. Baseline diameters were recorded, and changes in artery diameter in response to KCl (50 mmol/L) were then measured. To examine responses to the endothelium-dependent vasodilator, acetylcholine, arteries were constricted submaximally (~40% of the response to KCl) using the thromboxane A2 analog, U46619 (100 – 400 nmol/L). The approach of constricting isolated vessels submaximally prior to examination of endothelium-dependent dilator response is very common in the literature.21, 26–31 This approach places the vessels in a state where dilation or constriction (additional constriction) can be detected. In that sense, it approximates a level of tone that would be seen commonly in vivo. After development of a stable baseline diameter, concentration-response curves to acetylcholine were obtained. Papaverine (100 μmol/L) was used as a control (i.e. endothelium-independent) vasodilator and its effect was assessed at the end of each experiment. In some cases, full dose response curves were tested for papaverine.
Measurement of ROS
L-012 (100 μmol/L) -enhanced chemiluminesence was used to measure superoxide levels in cerebral arteries (pooled samples of basilar artery, middle cerebral arteries, and the circle of Willis) in WT and Nox2−/y mice, as described previously [21]. Measurements were made under basal conditions and in the presence of phorbol-12,13-dibutyrate (PdB,10 μmol/L). PdB was used to compare the effect of vehicle and aldosterone chronic treatments on the capacity of NOX2 oxidase to acutely generate superoxide. All readings were corrected for background.
Drugs
Acetylcholine and papaverine were obtained from Sigma and were dissolved in saline. Aldosterone was obtained from Sigma and dissolved in 87% propylene glycol, 9% ethanol, 4% distilled H2O. U46619 was obtained from Cayman Chemical and dissolved in 100% ethanol, with subsequent dilutions being made with Krebs-bicarbonate buffer. PdB was purchased from Calbiochem and prepared at 10 mmol/L in dimethyl sulphoxide and diluted in Krebs-HEPES solution (composition in mmol/L; NaCl 99, KCl 4.7, KH2PO4 10, MgSO4 1.2, NaHCO3 25, glucose 11, CaCl2 2.5), such that the final concentration of dimethyl sulfoxide was ≤0.1%. L-012 was purchased from Wako Pure Chemicals and was prepared at 100 mmol/L in dimethyl sulfoxide and diluted in Krebs-HEPES solution such that the final concentration of dimethyl sulfoxide was ≤0.1%.
Statistical analysis
All data are expressed as mean±SE. Changes in blood pressure were calculated by subtracting baseline blood pressure (i.e. at Day 0) from the average blood pressure recorded over Days 5–7 (representing 1 week) and Days 12–14 (representing 2 weeks) of treatment. Kidney weight:body weight was expressed as kidney weight (mg)/body weight (g) at the end of the study. Vasoconstriction in response to KCl is expressed as % decrease in diameter from baseline. Vasodilator responses are expressed as the % increase in diameter from the pre-constricted level towards the resting artery diameter, which was equivalent to 100%. For measurements of superoxide, data were normalized to dry tissue weight, and expressed as counts/s/mg. Comparisons of vasodilator and vasoconstrictor responses, superoxide levels, and blood pressure were made using two-way ANOVA, or Students t-test, as appropriate. Statistical significance was accepted at P<0.05.
RESULTS
ADULT MICE
Blood pressure and kidney weights
Consistent with previous findings [21, 32], initial blood pressure was similar in WT and Nox2−/y mice (Table 1). In WT and Nox2−/y mice, two weeks of treatment with either vehicle or aldosterone had no significant effect on blood pressure (Table 1). Kidney weight:body weight ratio was markedly increased in aldosterone- vs vehicle-treated mice in both WT and Nox2−/y mice (Table 1).
Table 1. Adult mice.
Initial blood pressure, basilar artery diameter, kidney weight (KW): body weight (BW) ratio, and changes in systolic blood pressure (SBP) following vehicle or aldosterone treatment (number in brackets indicates n value) in adult mice.
| Initial SBP (mmHg) | Treatment Subgroup | ΔSBP (mmHg) Week 1 |
ΔSBP (mmHg) Week 2 |
Baseline Diameter (μm) | KW:BW (mg:g) | |
|---|---|---|---|---|---|---|
| WT | 115±2 | Vehicle | 4±3 (21) | 6±3 (21) | 151±10 (14) | 12.4±0.2 (30) |
| (41) | Aldo | 6±3 (20) | 8±4 (20) | 153±10 (12) | 16.2±0.2 (31)* | |
| Nox2−/y | 111±3 | Vehicle | 0±3 (10) | 8±4 (10) | 149±15 (6) | 12.5±0.3 (15) |
| (19) | Aldo | 0±6 (9) | 2±6 (9) | 157±18 (4) | 15.3±0.2 (12)* |
P<0.0001 vs corresponding vehicle
Functional responses
Baseline internal diameter of the basilar artery at 60 mmHg in the absence of any pharmacologically-induced tone was similar in WT and Nox2−/y mice, whether treated with either vehicle or aldosterone (Table 1). In WT mice, treatment with aldosterone resulted in marked and selective impairment of vasodilator responses to acetylcholine compared with vehicle treatment (Figure 1A), whereas vasomotor responses to papaverine (Figure 1B) and KCl (Figure 1C) were unaffected by aldosterone treatment. In vehicle-treated Nox2−/y mice, responses to acetylcholine were similar to responses in vehicle-treated WT mice (Figures 1A & 2A, P>0.05). By contrast, in Nox2−/y mice treatment with aldosterone had no effect on vasodilator responses to acetylcholine compared to vehicle treatment (Figure 2A). Responses to papaverine (Figure 2B) and KCl (Figure 2C) were also unaffected.
Figure 1.
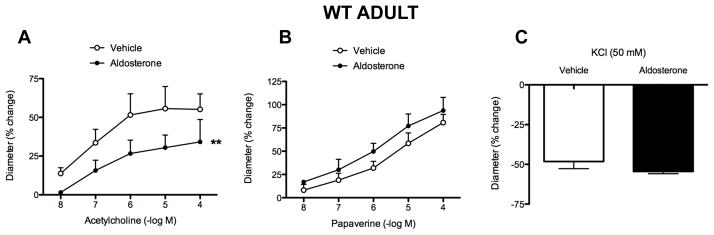
Vascular responses to acetylcholine (A: vehicle, n=5; aldosterone, n=5), papaverine (B: vehicle, n=4–9; aldosterone, n=4–9), and KCl (C: vehicle, n=5; aldosterone, n=3) in WT adult mice. All data are mean±SE. **P<0.01 vs vehicle.
Figure 2.

Vascular responses to acetylcholine (A: vehicle, n=6; aldosterone, n=4), papaverine (B: vehicle, n=6; aldosterone, n=4), and KCl (C: vehicle, n=4; aldosterone, n=3) in Nox2−/y adult mice. All data are mean±SE.
Superoxide levels
Under basal conditions, there was a small (~1.4-fold) increase in superoxide levels in arteries from aldosterone- versus vehicle-treated WT mice (P=0.056), whereas there was no such effect of aldosterone treatment in Nox2−/y mice (Figure 3). In cerebral arteries from vehicle-treated WT mice, superoxide levels were increased by ~7.5-fold following stimulation with PdB (Figure 3). Superoxide levels were found to be further increased (by ~1.3-fold) in cerebral arteries from aldosterone-treated WT mice (Figure 3). In contrast, PdB had little or no effect on superoxide levels produced in arteries from Nox2−/y mice treated with either vehicle or aldosterone, suggesting that effects of aldosterone on cerebrovascular superoxide levels occur in a NOX2-dependent manner.
Figure 3.
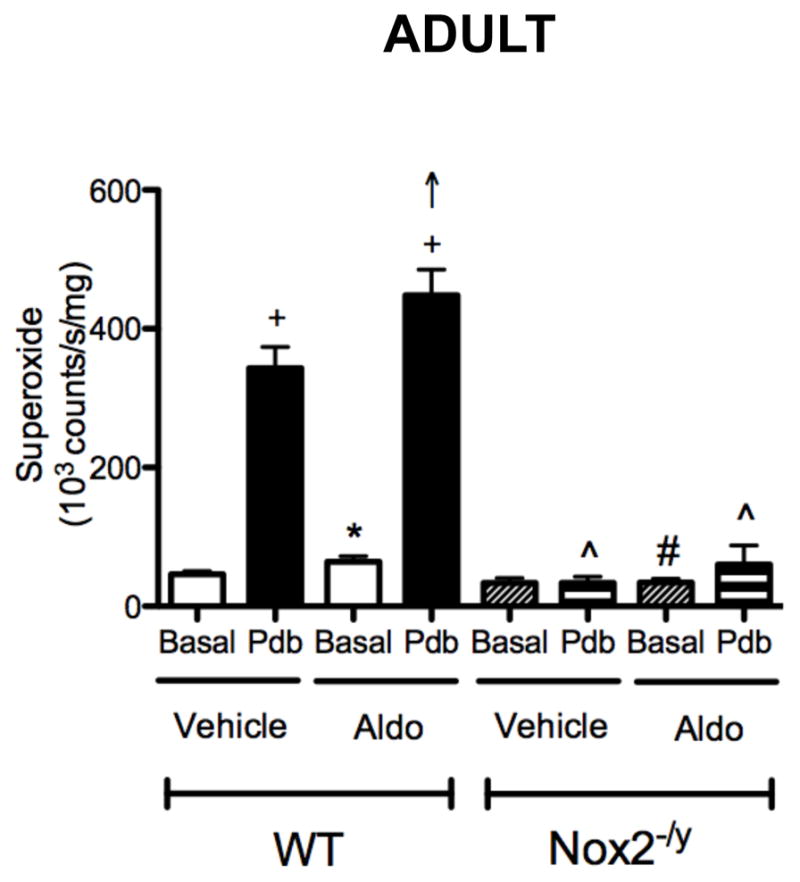
Superoxide levels in cerebral arteries from WT (vehicle, n=19; aldosterone, n=18) and Nox2−/y mice (vehicle, n=9; aldosterone, n=8) measured after vehicle or aldosterone treatment, under basal conditions or in the presence of 10 μmol/L PdB. All data are mean±SE. * P=0.056 vs vehicle-treated WT basal, + P<0.05 vs vehicle-treated WT basal, ↑ P<0.05 vs vehicle-treated WT PdB, # P<0.05 vs aldosterone-treated WT basal, ^ P<0.05 vs vehicle- and aldosterone-treated WT PdB.
AGED WT MICE
Blood pressure and kidney weights
Initial blood pressure was ~10 mmHg lower in aged WT mice than in adult WT mice (Tables 1 & 2). Whereas treatment with vehicle had no effect on blood pressure, treatment with aldosterone increased blood pressure by ~20 mmHg over 2 weeks (Table 2). Kidney weight:body weight ratio was markedly increased by aldosterone treatment in aged mice, in a similar manner to that observed in adult mice (Tables 1 & 2).
Table 2. Aged mice.
Initial blood pressure, kidney weight (KW): body weight (BW) ratio, and changes in systolic blood pressure (SBP) in response to vehicle and aldosterone treatment (number in brackets indicates n value) in aged mice.
| Initial SBP (mmHg) | Treatment subgroup | ΔSBP (mmHg) Week 1 |
ΔSBP (mmHg) Week 2 |
KW:BW (mg:g) | |
|---|---|---|---|---|---|
| WT | 103±4 (16)*** | Vehicle | 2±4 (6) | 3±4 (6) | 12.0±0.4 (9) |
| Aldo | 8±5 (7) | 19±4 (7) * | 16.9±0.7 (9) ** |
P<0.05 vs corresponding vehicle
P<0.0001 vs corresponding vehicle
P<0.05 vs adult WT
Superoxide levels
Under basal conditions, there was no difference in superoxide levels of cerebral arteries from aldosterone- versus vehicle-treated WT aged mice (Figure 4). Similar to adult mice, superoxide levels in cerebral arteries from WT aged mice were acutely increased by stimulation with PdB, and were ~1.4 fold greater following aldosterone versus vehicle treatment, although this difference did not reach statistical significance (Figure 4).
Figure 4.
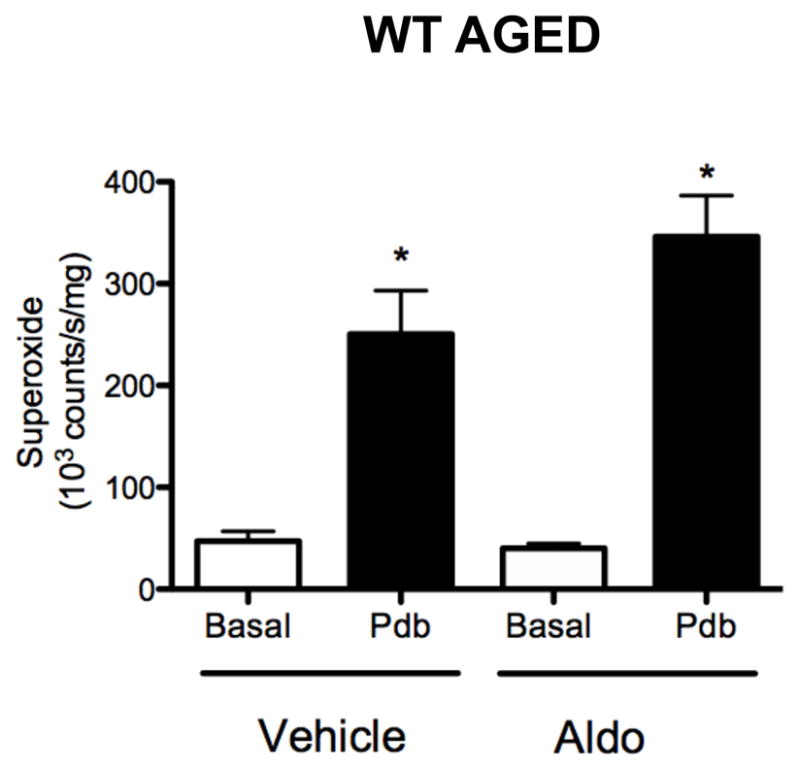
Superoxide levels in cerebral arteries from vehicle- and aldosterone-treated WT aged mice (both n=9) measured under basal conditions and in the presence of 10 μmol/L PdB. All data are mean±SE. *P<0.05 vs vehicle- and aldosterone-treated basal.
DISCUSSION
To our knowledge, this is the first study to report the effects of chronic aldosterone administration on cerebrovascular levels of ROS or endothelial function. Moreover, this is the first study to examine effects of aldosterone in any vascular bed using aged mice. There are four major new findings of this study. First, chronically increased systemic levels of aldosterone lead to increased ROS production and endothelial dysfunction in cerebral arteries. Second, these detrimental effects of aldosterone in the cerebral circulation occurred in the absence of a significant change in blood pressure. Third, aldosterone causes increased cerebral vascular ROS production and endothelial impairment in a NOX2-dependent manner. Fourth, we found no evidence for any further exacerbation of increased cerebral vascular ROS production by aldosterone in aged animals compared with adults, despite the presence of hypertension.
Increased ROS production and endothelial dysfunction caused by aldosterone
Chronic treatment with aldosterone causes oxidative stress and endothelial dysfunction in systemic vessels [9, 11, 13–15, 17], but this is the first study to test for such effects in the cerebral circulation. Increased ROS production involves elevated levels of superoxide anion, which can impair endothelium-dependent relaxation by decreasing bioavailability of nitric oxide [20, 33, 34]. NADPH oxidases are known to be a major source of vascular superoxide [35], and we found here that NOX2-deficient mice are indeed resistant to aldosterone-induced increases in ROS production and endothelial dysfunction. Our results also suggest that the increase in ROS production and endothelial dysfunction that occur in the cerebral circulation in response to aldosterone are likely to be independent of increases in blood pressure [13, 15, 16], analogous to previous reports of subpressor doses of angiotensin II (a major stimulus of aldosterone production) causing cerebrovascular oxidative stress and vascular dysfunction [36, 37]. The prevalence of primary aldosteronism in patients with hypertension ranges between 3% and 20%, constituting an important cause of unrecognized secondary hypertension [38]. The lower limit of aldosterone levels in such patients is more than 0.4 nM [39], with concentrations increasing to as high as 8 nM in patients with congestive heart failure [40]. Treatment of rodents with aldosterone for 2–4 weeks at doses equal or similar to that used in this study is reported to produce a serum aldosterone concentration of >4 nM [14, 41, 42]. Thus, the level produced in rodents with this protocol is in the range seen in patients.
Role of NOX2 oxidase
NOX2 is expressed in the endothelium of cerebral blood vessels [18, 19, 43]. Under control conditions, we found NOX2 deficiency to have no effect on endothelial function, consistent with earlier reports [21, 44]. One study has reported that chronic aldosterone treatment increased NOX2 expression in the aorta [8]. Here, we found that aldosterone treatment increased ROS levels and caused endothelial dysfunction in cerebral arteries of WT mice but not NOX2-deficient mice, indicating that superoxide generated by vascular NOX2 oxidase activity is a key mediator of cerebrovascular dysfunction [4]. Moreover, we used PdB to compare the effect of vehicle and aldosterone treatments on the capacity of NOX2 oxidase to acutely generate superoxide. PdB promotes formation of the NOX2 oxidase complex through protein kinase C-dependent phosphorylation of the p47 phox subunit [45] resulting in enhanced Nox2 oxidase activity. Using this approach, the PdB-stimulated increase in superoxide is entirely NOX2-dependent (Figure 3 and [21]), making it a useful tool to study the role of NOX2 oxidase in the vasculature. Increased superoxide levels following stimulation with PdB in arteries from aldosterone-treated mice thus reflects increased expression/activity of NOX2 following aldosterone treatment.
Aged mice
The frequency of stroke and Alzheimer’s disease increases with age [23], but mechanisms of ageing-related pathological changes in the cerebral circulation are poorly understood. As there is evidence that expression of vascular MR is higher in aged (30 months) versus adult (8 months) rats [24], and that this receptor mediates an augmented vascular contractile response in mice aged >9 months versus 3–4 months [46], we tested whether the pathological effects of chronic aldosterone that we observed in young adult mice (average age: 5–6 months old) might be further exacerbated in aged mice (average age: 16–17 months old). Such a finding would be suggestive of a role for aldosterone signaling through the MR as a contributing mechanism to worsening cerebral vascular pathology during aging. However, we found no evidence for exacerbated oxidative stress in the cerebral circulation in response to aldosterone in aged mice, although there was a greater hypertensive effect in these animals. Our findings are compatible with the conclusion that the cerebrovascular and pressor actions of aldosterone may be independent of one another. Indeed, other studies have reported vascular actions of aldosterone in peripheral blood vessels occur in the absence of an increase in blood pressure [13, 15, 16].
Conclusions
Our findings support the concept that chronically increased levels of plasma aldosterone may be an important contributor to cerebrovascular disease, and that such effects are likely to be mediated by increased ROS production due to increased NOX2 oxidase activity. Further studies are needed to examine the role of other sources of superoxide, including Nox isoforms, in the cerebrovascular actions of aldosterone, and whether the cerebrovascular actions of aldosterone are MR-dependent with a view to potential use of MR antagonists in the prevention and treatment of cerebrovascular disease and stroke [5, 47–50].
Acknowledgments
FUNDING SOURCES
This work was supported by a Grant-In-Aid from the National Heart Foundation of Australia (G 10M 5218). SC was supported by a CJ Martin Fellowship from the National Health and Medical Research Council (NHMRC) of Australia (359282) and a postdoctoral fellowship from the High Blood Pressure Research Council of Australia. CGS and GRD are Senior Research Fellows of the NHMRC. FMF was supported by the National Institutes of Health (NS-24621, HL-62984, HL-113863) and the Department of Veterans Affairs (BX001399).
Footnotes
DISCLOSURES/CONFLICT OF INTEREST
Sophocles Chrissobolis NONE
Grant R. Drummond NONE
Frank M. Faraci NONE
Christopher G. Sobey NONE
References
- 1.Herrada AA, Campino C, Amador CA, Michea LF, Fardella CE, Kalergis AM. Aldosterone as a modulator of immunity: Implications in the organ damage. J Hypertens. 2011;29:1684–1692. doi: 10.1097/HJH.0b013e32834a4c75. [DOI] [PubMed] [Google Scholar]
- 2.Han F, Ozawa H, Matsuda KI, Lu H, De Kloet ER, Kawata M. Changes in the expression of corticotrophin-releasing hormone, mineralocorticoid receptor and glucocorticoid receptor mRNAs in the hypothalamic paraventricular nucleus induced by fornix transection and adrenalectomy. J Neuroendocrinol. 2007;19:229–238. doi: 10.1111/j.1365-2826.2006.01519.x. [DOI] [PubMed] [Google Scholar]
- 3.Rigsby CS, Burch AE, Ogbi S, Pollock DM, Dorrance AM. Intact female stroke-prone hypertensive rats lack responsiveness to mineralocorticoid receptor antagonists. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1754–1763. doi: 10.1152/ajpregu.00145.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faraci FM. Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol. 2011;300:H1566–H1582. doi: 10.1152/ajpheart.01310.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Osmond JM, Rigsby CS, Dorrance AM. Is the mineralocorticoid receptor a potential target for stroke prevention? Clin Sci (Lond) 2008;114:37–47. doi: 10.1042/CS20070155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeda R, Matsubara T, Miyamori I, Hatakeyama H, Morise T. Vascular complications in patients with aldosterone producing adenoma in japan: Comparative study with essential hypertension. The research committee of disorders of adrenal hormones in Japan. J Endocrinol Invest. 1995;18:370–373. doi: 10.1007/BF03347840. [DOI] [PubMed] [Google Scholar]
- 7.Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, Mourad JJ. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. 2005;45:1243–1248. doi: 10.1016/j.jacc.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 8.Park YM, Lim BH, Touyz RM, Park JB. Expression of NAD(P)H oxidase subunits and their contribution to cardiovascular damage in aldosterone/salt-induced hypertensive rat. J Korean Med Sci. 2008;23:1039–1045. doi: 10.3346/jkms.2008.23.6.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL. Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension. 2002;40:504–510. doi: 10.1161/01.hyp.0000034738.79310.06. [DOI] [PubMed] [Google Scholar]
- 10.Xavier FE, Blanco-Rivero J, Avendano MS, Sastre E, Yela R, Velazquez K, Salaices M, Balfagon G. Aldosterone alters the participation of endothelial factors in noradrenaline vasoconstriction differently in resistance arteries from normotensive and hypertensive rats. Eur J Pharmacol. 2011;654:280–288. doi: 10.1016/j.ejphar.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, Neves MF, Laurant P, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012;59:324–330. doi: 10.1161/HYPERTENSIONAHA.111.181123. [DOI] [PubMed] [Google Scholar]
- 12.Keidar S, Kaplan M, Pavlotzky E, Coleman R, Hayek T, Hamoud S, Aviram M. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: A possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation. 2004;109:2213–2220. doi: 10.1161/01.CIR.0000127949.05756.9D. [DOI] [PubMed] [Google Scholar]
- 13.Leibovitz E, Ebrahimian T, Paradis P, Schiffrin EL. Aldosterone induces arterial stiffness in absence of oxidative stress and endothelial dysfunction. J Hypertens. 2009;27:2192–2200. doi: 10.1097/HJH.0b013e328330a963. [DOI] [PubMed] [Google Scholar]
- 14.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–197. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tasatargil A, Tekcan M, Celik-Ozenci C, Ece Gungor N, Dalkiran B. Aldosterone-induced endothelial dysfunction of rat aorta: Role of poly(ADP-ribose) activation. J Renin Angiotensin Aldosterone Syst. 2009;10:127–137. doi: 10.1177/1470320309343655. [DOI] [PubMed] [Google Scholar]
- 16.Xavier FE, Aras-Lopez R, Arroyo-Villa I, Campo LD, Salaices M, Rossoni LV, Ferrer M, Balfagon G. Aldosterone induces endothelial dysfunction in resistance arteries from normotensive and hypertensive rats by increasing thromboxane a2 and prostacyclin. Br J Pharmacol. 2008;154:1225–1235. doi: 10.1038/bjp.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanco-Rivero J, Cachofeiro V, Lahera V, Aras-Lopez R, Marquez-Rodas I, Salaices M, Xavier FE, Ferrer M, Balfagon G. Participation of prostacyclin in endothelial dysfunction induced by aldosterone in normotensive and hypertensive rats. Hypertension. 2005;46:107–112. doi: 10.1161/01.HYP.0000171479.36880.17. [DOI] [PubMed] [Google Scholar]
- 18.De Silva TM, Broughton BR, Drummond GR, Sobey CG, Miller AA. Gender influences cerebral vascular responses to angiotensin II through Nox2-derived reactive oxygen species. Stroke. 2009;40:1091–1097. doi: 10.1161/STROKEAHA.108.531707. [DOI] [PubMed] [Google Scholar]
- 19.Miller AA, Drummond GR, De Silva TM, Mast AE, Hickey H, Williams JP, Broughton BR, Sobey CG. NADPH oxidase activity is higher in cerebral versus systemic arteries of four animal species: Role of Nox2. Am J Physiol Heart Circ Physiol. 2009;296:H220–H225. doi: 10.1152/ajpheart.00987.2008. [DOI] [PubMed] [Google Scholar]
- 20.Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front Biosci. 2011;16:1733–1745. doi: 10.2741/3816. [DOI] [PubMed] [Google Scholar]
- 21.Chrissobolis S, Banfi B, Sobey CG, Faraci FM. Role of Nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J Appl Physiol. 2012;113:184–191. doi: 10.1152/japplphysiol.00455.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller AA, De Silva TM, Judkins CP, Diep H, Drummond GR, Sobey CG. Augmented superoxide production by nox2-containing nadph oxidase causes cerebral artery dysfunction during hypercholesterolemia. Stroke. 2010;41:784–789. doi: 10.1161/STROKEAHA.109.575365. [DOI] [PubMed] [Google Scholar]
- 23.Gorelick PB. Risk factors for vascular dementia and alzheimer disease. Stroke. 2004;35:2620–2622. doi: 10.1161/01.STR.0000143318.70292.47. [DOI] [PubMed] [Google Scholar]
- 24.Krug AW, Allenhofer L, Monticone R, Spinetti G, Gekle M, Wang M, Lakatta EG. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension. 2010;55:1476–1483. doi: 10.1161/HYPERTENSIONAHA.109.148783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 26.Channon KM, Qian H, Neplioueva V, Blazing MA, Olmez E, Shetty GA, Youngblood SA, Pawloski J, McMahon T, Stamler JS, George SE. In vivo gene transfer of nitric oxide synthase enhances vasomotor function in carotid arteries from normal and cholesterol-fed rabbits. Circulation. 1998;98:1905–1911. doi: 10.1161/01.cir.98.18.1905. [DOI] [PubMed] [Google Scholar]
- 27.Higueras J, Sarria B, Ortiz JL, Cortijo J, Maruenda A, Barbera M, Morcillo EJ. Halothane inhibits endothelium-dependent relaxation elicited by acetylcholine in human isolated pulmonary arteries. Eur J Pharmacol. 1997;326:175–181. doi: 10.1016/s0014-2999(97)85412-x. [DOI] [PubMed] [Google Scholar]
- 28.Munzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J Clin Invest. 1995;95:187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol. 2011;31:1368–1376. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 30.Pena Silva RA, Chu Y, Miller JD, Mitchell IJ, Penninger JM, Faraci FM, Heistad DD. Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke. 2012;43:3358–3363. doi: 10.1161/STROKEAHA.112.667063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papadopoulos P, Rosa-Neto P, Rochford J, Hamel E. Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of alzheimer’s disease with combined amyloid-beta and cerebrovascular pathology. PLoS One. 2013;8:e68612. doi: 10.1371/journal.pone.0068612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ, Cohen RA. Role of nadph oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res. 2001;88:947–953. doi: 10.1161/hh0901.089987. [DOI] [PubMed] [Google Scholar]
- 33.Chrissobolis S, Faraci FM. The role of oxidative stress and nadph oxidase in cerebrovascular disease. Trends Mol Med. 2008;14:495–502. doi: 10.1016/j.molmed.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faraci FM, Didion SP. Vascular protection: Superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- 35.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capone C, Faraco G, Park L, Cao X, Davisson RL, Iadecola C. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am J Physiol Heart Circ Physiol. 2011;300:H397–H407. doi: 10.1152/ajpheart.00679.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chrissobolis S, Faraci FM. Sex differences in protection against angiotensin II-induced endothelial dysfunction by manganese superoxide dismutase in the cerebral circulation. Hypertension. 2010;55:905–910. doi: 10.1161/HYPERTENSIONAHA.109.147041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chao CT, Wu VC, Kuo CC, Lin YH, Chang CC, Chueh SJ, Wu KD, Pimenta E, Stowasser M. Diagnosis and management of primary aldosteronism: An updated review. Ann Med. 2013;45:375–383. doi: 10.3109/07853890.2013.785234. [DOI] [PubMed] [Google Scholar]
- 39.Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, Young WF, Jr, Montori VM. Case detection, diagnosis, and treatment of patients with primary aldosteronism: An endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:3266–3281. doi: 10.1210/jc.2008-0104. [DOI] [PubMed] [Google Scholar]
- 40.Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345:1689–1697. doi: 10.1056/NEJMra000050. [DOI] [PubMed] [Google Scholar]
- 41.Jia Z, Aoyagi T, Kohan DE, Yang T. mPGES-1 deletion impairs aldosterone escape and enhances sodium appetite. Am J Physiol Renal Physiol. 2010;299:F155–F166. doi: 10.1152/ajprenal.90702.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schreier B, Rabe S, Schneider B, Ruhs S, Grossmann C, Hauptmann S, Blessing M, Neumann J, Gekle M. Aldosterone/NaCl-induced renal and cardiac fibrosis is modulated by TGF-β responsiveness of T cells. Hypertens Res. 2011;34:623–629. doi: 10.1038/hr.2011.16. [DOI] [PubMed] [Google Scholar]
- 43.Miller AA, Drummond GR, Schmidt HHW, Sobey CG. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97:1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- 44.Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through Nox-2-derived radicals. Arterioscler Thromb Vasc Biol. 2006;26:826–832. doi: 10.1161/01.ATV.0000205849.22807.6e. [DOI] [PubMed] [Google Scholar]
- 45.Gupte SA, Kaminski PM, George S, Kouznestova L, Olson SC, Mathew R, Hintze TH, Wolin MS. Peroxide generation by p47phox-src activation of Nox2 has a key role in protein kinase C-induced arterial smooth muscle contraction. Am J Physiol Heart Circ Physiol. 2009;296:H1048–H1057. doi: 10.1152/ajpheart.00491.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18:1429–1433. doi: 10.1038/nm.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dinh QN, Arumugam TV, Young MJ, Drummond GR, Sobey CG, Chrissobolis S. Aldosterone and the mineralocorticoid receptor in the cerebral circulation and stroke. Exp Transl Stroke Med. 2012;4:21. doi: 10.1186/2040-7378-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dorrance AM, Rupp NC, Nogueira EF. Mineralocorticoid receptor activation causes cerebral vessel remodeling and exacerbates the damage caused by cerebral ischemia. Hypertension. 2006;47:590–555. doi: 10.1161/01.HYP.0000196945.73586.0d. [DOI] [PubMed] [Google Scholar]
- 49.Frieler RA, Meng H, Duan SZ, Berger S, Schutz G, He Y, Xi G, Wang MM, Mortensen RM. Myeloid-specific deletion of the mineralocorticoid receptor reduces infarct volume and alters inflammation during cerebral ischemia. Stroke. 2011;42:179–185. doi: 10.1161/STROKEAHA.110.598441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwanami J, Mogi M, Okamoto S, Gao XY, Li JM, Min LJ, Ide A, Tsukuda K, Iwai M, Horiuchi M. Pretreatment with eplerenone reduces stroke volume in mouse middle cerebral artery occlusion model. Eur J Pharmacol. 2007;566:153–159. doi: 10.1016/j.ejphar.2007.03.043. [DOI] [PubMed] [Google Scholar]