

Abstract
Activation of M1-type muscarinic acetylcholine receptors excites neocortical pyramidal neurons, in part by gating a non-selective cation conductance that produces calcium-dependent “afterdepolarizing potentials” (ADPs) following short trains of action potentials. While the identity of the cation conductance mediating the ADP is not known, previous work has implicated canonical transient potential receptor (TRPC) channels, specifically of the TRPC5 and TRPC6 subtypes. Using pharmacological and genetic approaches, we tested the role of TRPC channels in generating cholinergic ADPs in layer 5 pyramidal neurons in the mouse medial prefrontal cortex (mPFC). A variety of compounds that block TRPC channels, including 2-aminoethoxydiphenyl borate (2-APB), flufenamic acid, lanthanum, SKF-96365, and Pyr-3, had little, if any, impact on cholinergic ADPs. Similarly, genetic deletion of several TRPC subunits, including TPRC1, TRPC5, and TRPC6 (single knockouts), or both TRPC5 and TRPC6 together (double knockout), failed to reduce the amplitude of cholinergic ADPs. These data suggest that TRPC5 and TRPC6 subunits are not required for cholinergic excitation of layer 5 pyramidal neurons in the mouse mPFC, and suggest that the focus of future work should be expanded to test the involvement of other potential ionic effectors.
Keywords: Neocortex, Pyramidal Neuron, Acetylcholine, Afterdepolarization, TRPC channel, TRPC5 channel, TRPC6 channel, TRPC1 channel, Mouse, Muscarinic receptor
Introduction
M1-type muscarinic acetylcholine receptors (M1Rs) play a central role in regulating the excitability of cortical pyramidal neurons [1]. One effect of M1R activation is the gating of a calcium-dependent cation conductance that generates afterdepolarizations (ADPs) following bursts of action potentials [1-4]. Although the subject of much investigation, the ionic mechanism underlying the cholinergic ADP remains unknown. Several recent studies have focused on members of the canonical transient receptor potential (TRPC) channel family. These cation channels are gated by Gq-coupled metabotropic receptors, facilitated by increases in cytosolic calcium [5], and widely expressed in the cerebral cortex, including the medial prefrontal cortex (mPFC) [6,7]. Specifically, Yan et al. [8] found in cultured cortical pyramidal neurons that genetic expression of a TRPC dominant negative subunit inhibits, while over-expression of TRPC5 or TRPC6 subunits enhances, the amplitude of cholinergic ADPs. Furthermore, pharmacological antagonists of TRPC channels, such as SKF-96365, 2-aminoethoxydiphenyl borate (2-APB), and flufenamic acid (FFA), are reported to block ADP genesis [8,9].
The aim of this study was to confirm a role for TRPC5 and/or TRPC6 channels in mediating cholinergic ADPs in neocortical pyramidal neurons from the mouse mPFC using pharmacological approaches and genetic deletions of specific TRPC subunits. Our results cast doubt on the “TRPC hypothesis” of cholinergic excitation of cortical pyramidal neurons, and suggest that alternative ionic mechanisms should be explored.
Materials and Methods
All procedures and experiments were conducted according to methods approved by the Institutional Animal Care and Use Committee of Dartmouth College. Experiments involved tissue from 4- to 6-week-old mice, including wild-type C57BL/6, wild-type crossbred 1:1 129SvEv:C57BL/6 (129Sv/C57) mice, or 129Sv/C57 mice lacking TRPC1, TRPC5, TRPC6, or both TRPC5 and TRPC6 channels (TRPC knockout mice). Genetic deletion of TRPC subunits was confirmed by PCR using genomic DNA extracted from mouse tail biopsies [10]. Animals were anesthetized with vaporized isoflurane, decapitated, and brains removed into an artificial cerebral spinal fluid (aCSF) composed of (in mM): 125 NaCl, 25 NaHCO3, 3 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 6 MgCl2, and 25 glucose (bubbled with 95% O2/5% CO2). Coronal slices (250 μm) of the mPFC were cut on a vibroslicer (Leica 1200), and slices transferred to a holding chamber containing heated (35 °C) aCSF in which CaCl2 and MgCl2 were 2 mM and 1 mM, respectively. To measure ADPs in layer 5 pyramidal neurons, whole-cell recordings were made with micropipettes (5-7 MΩ) containing (in mM): 140 potassium gluconate, 2 NaCl, 2 MgCl2, 10 HEPES, 3 Na2ATP, and 0.3 NaGTP (pH 7.2 with KOH).
Data were acquired with AxographX software (AxographX, Sydney, Australia) using a BVC-700 amplifier (Dagan, Minneapolis) and an ITC-18 digitizer (HEKA Instruments, Bellmore, New York). Data were filtered at 5 kHz and digitized at 25 kHz. Whole-cell series resistance (between 10 and 30 MΩ) was maximally compensated. Experiments were carried out at 35 °C, and membrane potentials were corrected for the liquid junction potential of 12 mV.
ADPs were evoked using trains of 10 action potentials generated at 40 Hz using short (2 ms), high-amplitude (3 nA) somatic current injections. Baseline ADP amplitudes were quantified as the peak depolarization (relative to resting membrane potential; Erest) occurring within the first 2 s following the spike train. When baseline ADP amplitudes were negative to Erest, they were quantified as 0 mV. ADP amplitudes were measured in baseline conditions and after 5- or 15-minutes of exposure to bath-applied carbachol (CCh, 10 μM; with or without TRPC antagonists present), and M1R-dependent ADP amplitudes quantified as the difference between the ADP amplitude occurring in the presence of CCh and the baseline ADP amplitude. Most drugs were obtained from Tocris Biosciences. Cadmium chloride (CdCl2), lanthanum chloride (LaCl3) and CCh were obtained from Sigma Aldrich. FFA was obtained from both Tocris Biosciences and Sigma. Stock solutions of CCh, Cd2+, 2-aminoethoxydiphenyl borate (2-APB), SKF-96365 (SKF), LaCl3 and Pyr-3 were made by dissolving compounds in water; FFA was dissolved in dimethyl sulfoxide (DMSO), with the final concentration of DMSO not exceeding 0.1%.
Results and Discussion
We tested the ability of TRPC antagonists to block cholinergic ADPs in layer 5 pyramidal neurons from the mPFC (Fig. 1a). Trains of 10 action potentials were generated at 40 Hz using brief (2 ms), high amplitude (3 nA) current injections. In the presence of bath-applied CCh (10 μM, for 5 minutes), spike trains generated ADPs of similar amplitude in all experimental groups (thick black traces in Fig. 1a; p = 0.16, ANOVA), which were comparable to M1R-dependent ADPs previously observed in pyramidal neurons in the neocortex [1] and hippocampus [11]. After ADPs were measured in the presence of CCh alone, TRPC antagonists were co-applied with CCh for an additional 10 minutes, at the end of which ADPs were measured again, before a 10-to-15-minute wash in drug-free aCSF (Fig. 1a). Antagonists tested included non-selective calcium-channel blocker Cd2+ (200 μM; n = 5), the broad-spectrum TRPC blockers FFA (30 μM; n = 11), 2-APB (100 μM; n = 6), and SKF-96365 (50 μM; n = 8), and blockers thought to be somewhat selective for TRPC6 (La3+; 4 μM, n = 6, or 100 μM, n = 6) or TRPC3 (Pyr-3; 10 μM, n = 5) channels [12,13]. As expected, ADPs were fully blocked in the presence of Cd2+ (Fig. 1; mean ADP amplitudes in CCh alone and CCh + Cd2+ conditions were 2.2 ± 0.4 mV and 0.1 ± 0.1 mV, respectively; n = 5; p < 0.01, paired t-test). Unexpectedly, however, most TRPC antagonists failed to block CCh-induced ADPs (Fig. 1). Only in the case of FFA was there a significant, but incomplete, reduction in ADP amplitude (mean change = -33 ± 9%; from 2.0 ± 0.2 mV in CCh alone, to 1.4 ± 0.2 mV with FFA present; n = 11; p < 0.01, paired t-test). However, FFA has non-specific effects independent of TPRC blockade, including modulation of voltage-gated sodium channels [14], an effect we confirmed using higher concentrations of FFA (100 μM) that consistently blocked action potential generation altogether (n = 4; data not shown). Therefore, the combined results from our pharmacological experiments fail to support a role for TRPC channels in mediating cholinergic ADPs in cortical pyramidal neurons.
Figure 1.
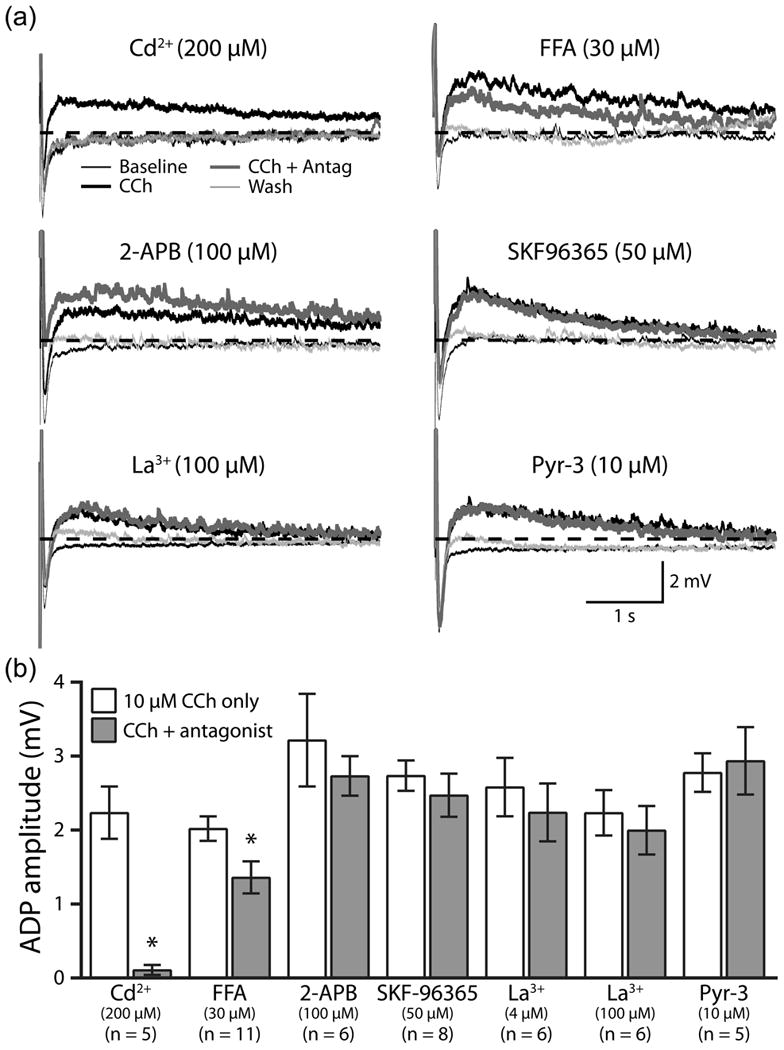
TRPC channel blockers fail to block cholinergic excitation. (a) Average responses (5 trials) to trains of 10 current-evoked action potentials (40 Hz) in baseline conditions (thin black traces), in the presence of bath-applied carbachol (CCh, 10 μM, for 5 minutes; thick black traces), after co-application of CCh with Cd2+ or TRPC channel antagonists (for 10 minutes; thick gray traces), and after 10 to 15 minutes of wash in drug-free aCSF (thin light gray traces). Dashed-lines indicate resting membrane potentials. (b) Comparison of cholinergic ADP amplitudes in CCh alone, and after co-application of TRPC antagonists. The number of neurons tested in each experimental group is indicated below the name of the corresponding antagonist.
A more direct approach to test the role of TRPC channels in generating cholinergic excitation is to utilize neurons from animals genetically lacking specific TRPC subunits (Fig. 2). Such animals have been developed for several TRPC isoforms, including the TRPC5 and TRPC6 subunits implicated in cholinergic ADPs [10,15,16]. Since these animals are maintained in a crossbred 129Sv/C57 mouse strain [10], we first confirmed that muscarinic receptor stimulation facilitates ADPs in prefrontal layer 5 pyramidal neurons from these animals (Fig. 2). Bath application of CCh (10 μM) reversibly revealed ADPs of comparable amplitude in neurons from C57BL/6 (Fig. 2a) and 129Sv/C57 (Fig. 2b) mice. Mean ADP amplitudes were 2.5 ± 0.3 mV (n = 7) and 1.7 ± 0.2 mV (n = 6) in neurons from C57BL/6 and 129Sv/C57 mice, respectively (p = 0.08; t-test), and therefore data from these two wild-type groups were pooled (mean ADP amplitude of 2.1 ± 0.2 mV; n = 13).
Figure 2.
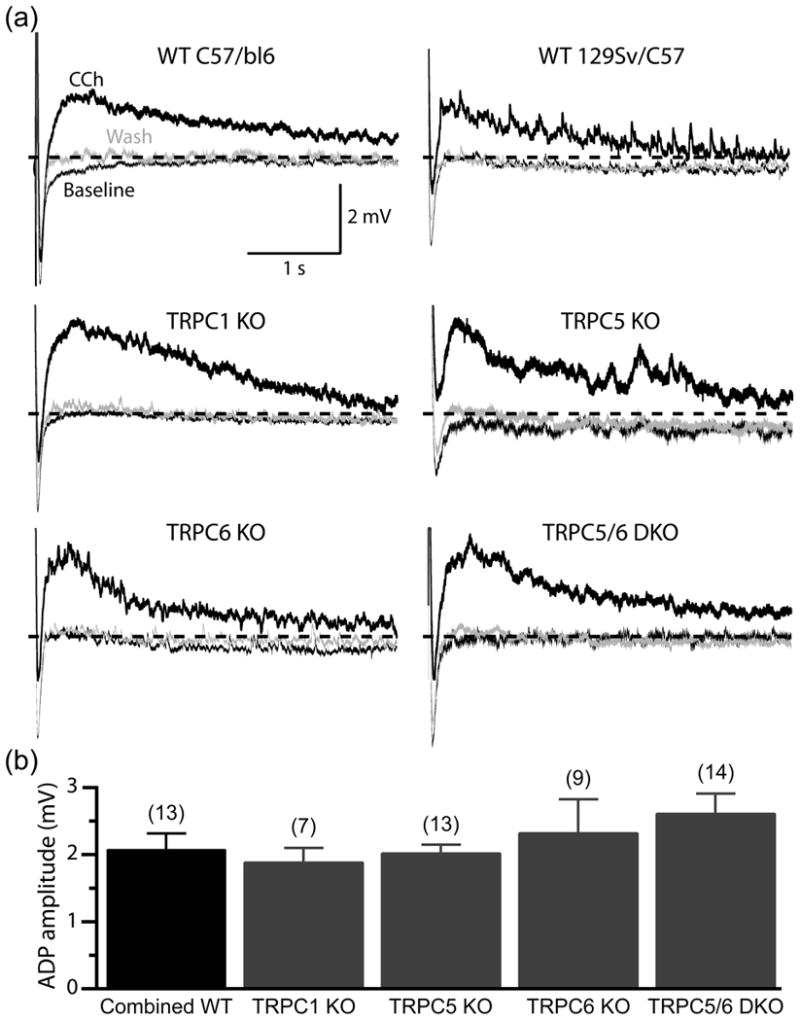
TRPC1, TRPC5, and TRPC6 are not necessary for generating cholinergic ADPs in layer 5 pyramidal neurons. (a) Average responses (5 trials) of layer 5 pyramidal neurons from wild-type (“WT”) C57/Bl6 and 129Sv/C57 mice, and neurons from 129Sv/C57 mice genetically lacking specific TRPC subunits (knockout, “KO”, or double-knockout, “DKO”, animals), to trains of 10 current-evoked action potentials (40 Hz) in baseline conditions (thin black traces), at the end of a 5-minute-long bath application of carbachol (CCh, 10 μM; thick black traces), and following 10 to 15 minutes of wash (thin gray traces). Dashed-lines indicate resting membrane potentials. (b) Comparison of the amplitudes of cholinergic ADPs occurring in neurons having different TRPC expression. Numbers in parentheses indicate the number of neurons in each experimental group.
Because previous work has specifically implicated TRPC5 and TRPC6 channels in mediating cholinergic ADPs [8,9], and given the presence of heteromeric TRPC1/5 channels in hippocampus and cortex [17], we compared the amplitude of cholinergic ADPs in layer 5 pyramidal neurons from wild-type mice and those lacking TPC1, TRPC5, or TRPC6 subunits (Fig. 2). CCh reversibly facilitated ADP generation in layer 5 pyramidal neurons from all experimental groups, with mean ADP amplitudes being 1.9 ± 0.2 mV, 2.0 ± 0.1 mV, and 2.3 ± 0.5 mV, respectively, for neurons from TRPC1-/- (n = 7), TRPC5-/- (n = 13), and TRPC6-/- (n = 9) animals. Finally, we tested tonic cholinergic responses in layer 5 pyramidal neurons from mice lacking both TRPC5 and TRPC6 (TRPC5/6 double KO neurons), as over-expression of these two TRPC subunits together can increase ADP amplitudes [8]. However, even in the absence of both of these TRPC subunits, CCh continued to reveal robust ADPs similar in amplitude (2.5 ± 0.3 mV; n = 14) to those observed in other experimental groups (Fig. 2). When analyzed with a one-way ANOVA, no significant differences in ADP amplitude were observed in neurons from wild-type or knockout animals (p = 0.44; ANOVA). These results are consistent with our pharmacological findings, and further suggest that TRPC channels, including both TRPC5 and TRPC6 channels, are not necessary for cholinergic excitation of layer 5 pyramidal neurons.
The prospect that TRPC5 and/or TRPC6 channels mediate cholinergic excitation has been extremely attractive, as these TRPC subunits can be activated by muscarinic receptor stimulation [8,18,19], are positively modulated by intracellular calcium [20,21], and are expressed in the cerebral cortex [6,7]. Further, because TRPC channels are calcium permeable [22], their activation could facilitate the refilling of intracellular calcium stores critical for phasic cholinergic inhibition of cortical and hippocampal neurons [23-25]. Indeed, TRPC5 and TRPC6 subunits have been implicated in mediating cholinergic excitatory responses, and over-expression of these channels enhances the amplitude of cholinergic ADPs in cortical pyramidal neurons [8].
We utilized both pharmacological and genetic approaches to test the role of TRPC channels in generating cholinergic ADPs in layer 5 pyramidal neurons from the mouse mPFC. Our results uniformly suggest that TRPC channels are not necessary for cholinergic excitation of cortical pyramidal neurons. Although the diversity of potential TRPC heteromeric channels, as well as the possibility of compensatory developmental expression of alternative subunits and/or families of TRP channels (e.g., TRPM or TRPV channels), leaves open a possible physiological role for TRPC channels in contributing to cholinergic responses, our results cast doubt on the “TRPC hypothesis” of cholinergic excitation, and suggest that alternative ionic mechanisms should be explored.
Acknowledgments
The authors thank Daniel Avesar, Genevieve St. Germain, and Alison Rudkin for technical assistance.
Funding: National Institute for Mental Health grant R01 MH83806 (A.T.G) and the Intramural Research Program of the NIH, project Z01-ES-101684 (L.B.).
Footnotes
Conflict of interest: None declared.
References
- 1.Gulledge AT, Bucci DJ, Zhang SS, Matsui M, Yeh HH. M1 receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J Neurosci. 2009;29:9888–9902. doi: 10.1523/JNEUROSCI.1366-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrade R. Cell excitation enhances muscarinic cholinergic responses in rat association cortex. Brain Res. 1991;548:81–93. doi: 10.1016/0006-8993(91)91109-e. [DOI] [PubMed] [Google Scholar]
- 3.Haj-Dahmane S, Andrade R. Muscarinic activation of a voltage-dependent cation nonselective current in rat association cortex. J neurosci. 1996;16:3848–3861. doi: 10.1523/JNEUROSCI.16-12-03848.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwindt PC, Spain WJ, Foehring RC, Chubb MC, Crill WE. Slow conductances in neurons from cat sensorimotor cortex in vitro and their role in slow excitability changes. J Neurophysiol. 1988;59:450–467. doi: 10.1152/jn.1988.59.2.450. [DOI] [PubMed] [Google Scholar]
- 5.Clapham DE, Julius D, Montell C, Schultz G. International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev. 2005;57:427–450. doi: 10.1124/pr.57.4.6. [DOI] [PubMed] [Google Scholar]
- 6.Fowler MA, Sidiropoulou K, Ozkan ED, Phillips CW, Cooper DC. Corticolimbic expression of TRPC4 and TRPC5 channels in the rodent brain. PLoS One. 2007;2:e573. doi: 10.1371/journal.pone.0000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunert-Keil C, Bisping F, Kruger J, Brinkmeier H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics. 2006;7:159. doi: 10.1186/1471-2164-7-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan HD, Villalobos C, Andrade R. TRPC Channels Mediate a Muscarinic Receptor-Induced Afterdepolarization in Cerebral Cortex. J Neurosci. 2009;29:10038–10046. doi: 10.1523/JNEUROSCI.1042-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tai C, Hines DJ, Choi HB, MacVicar BA. Plasma membrane insertion of TRPC5 channels contributes to the cholinergic plateau potential in hippocampal CA1 pyramidal neurons. Hippocampus. 2011;21:958–967. doi: 10.1002/hipo.20807. [DOI] [PubMed] [Google Scholar]
- 10.Dietrich A, Mederos YSM, Gollasch M, Gross V, Storch U, Dubrovska G, et al. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dasari S, Gulledge AT. M1 and M4 receptors modulate hippocampal pyramidal neurons. J Neurophysiol. 2011;105:779–792. doi: 10.1152/jn.00686.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci. 2009;106:5400–5405. doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boulay G. Ca2+-calmodulin regulates receptor-operated Ca2+ entry activity of TRPC6 in HEK-293 cells. Cell Calcium. 2002;32:201–207. doi: 10.1016/s0143416002001550. [DOI] [PubMed] [Google Scholar]
- 14.Yau HJ, Baranauskas G, Martina M. Flufenamic acid decreases neuronal excitability through modulation of voltage-gated sodium channel gating. J Physiol. 2010;588:3869–3882. doi: 10.1113/jphysiol.2010.193037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaudhuri P, Colles SM, Bhat M, Van Wagoner DR, Birnbaumer L, Graham LM. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol Biol Cell. 2008;19:3203–3211. doi: 10.1091/mbc.E07-08-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phelan KD, Shwe UT, Abramowitz J, Wu H, Rhee SW, Howell MD, et al. Canonical Transient Receptor Channel 5 (TRPC5) and TRPC1/4 Contribute to Seizure and Excitotoxicity by Distinct Cellular Mechanisms. Mol Pharmacol. 2013;83:429–438. doi: 10.1124/mol.112.082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 18.Lee YM, Kim BJ, Kim HJ, Yang DK, Zhu MH, Lee KP, et al. TRPC5 as a candidate for the nonselective cation channel activated by muscarinic stimulation in murine stomach. Am J Physiol-Gastr L. 2003;284:G604–616. doi: 10.1152/ajpgi.00069.2002. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Guo F, Kim JY, Saffen D. Muscarinic acetylcholine receptors activate TRPC6 channels in PC12D cells via Ca2+ store-independent mechanisms. J biochem. 2006;139:459–470. doi: 10.1093/jb/mvj065. [DOI] [PubMed] [Google Scholar]
- 20.Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, et al. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol. 2004;561:415–432. doi: 10.1113/jphysiol.2004.075051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blair NT, Kaczmarek JS, Clapham DE. Intracellular calcium strongly potentiates agonist-activated TRPC5 channels. J Gen Physiol. 2009;133:525–546. doi: 10.1085/jgp.200810153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem. 2000;275:17517–17526. doi: 10.1074/jbc.275.23.17517. [DOI] [PubMed] [Google Scholar]
- 23.Gulledge AT, Kawaguchi Y. Phasic cholinergic signaling in the hippocampus: functional homology with the neocortex? Hippocampus. 2007;17:327–332. doi: 10.1002/hipo.20279. [DOI] [PubMed] [Google Scholar]
- 24.Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc Natl Acad Sci. 2009;106:3202–3206. doi: 10.1073/pnas.0813346106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]