

Abstract
Developing new compounds targeting virulence factors (e.g., inhibition of pilus assembly by pilicides) is a promising approach to combating bacterial infection. A high-throughput screening campaign of a library of 17,500 small molecules identified 2-amino-3-acyl-tetrahydrobenzothiophene derivatives (hits 2 and 3) as novel inhibitors of pili-dependent biofilm formation in an uropathogenic Escherichia coli strain UTI89. Based on compounds 2 and 3 as a starting point, we designed and synthesized a series of structurally related analogs and investigated their activity against biofilm formation of E.coli UTI89. Systematic structural modification of the initial hits provided valuable information on their SARs for further optimization. In addition, small structural changes to the parent molecules resulted in low micromolar inhibitors (20–23) of E.coli biofilm development without effect on bacterial growth. The hit compound 3 and its analog 20 were confirmed to prevent pili formation in a hemagglutination (HA) titer assay and electron microscopy (EM) measurements. These findings suggest that 2-amino-3-acyl-tetrahydrobenzothiophenes may serve as a new class of compounds for further elaboration as antibacterial agents with antivirulence activity.
Introduction
The rise and spread of bacteria that are resistant to most of the commonly used antibiotics demand the discovery of new therapeutic approaches. Conventional antibiotics typically kill bacteria (bactericidal) or inhibit their growth (bacteriostatic) by interfering with essential functions of bacteria such as cell wall biosynthesis, protein synthesis, and DNA replication and repair, imposing a strong selective pressure on bacteria to acquire resistance. The chronic misuse and overuse of antibiotics nowadays has given rise to multiple antibiotic resistant bacteria. Targeting bacterial virulence factors to disarm pathogens is a promising alternative to classical antimicrobial therapy.1 This strategy has also been considered as a “second generation” antibiotic approach.2–4 Pathogenic bacteria produce virulence factors (e.g., adhesion molecules, secretion systems, toxins, and other factors), which are crucial for their ability to cause disease and damage the host’s tissues.3,4 It has been demonstrated that inhibiting the virulence factors can significantly attenuate infection and thus offers a potential approach to combating infection.5–7 Compared to conventional antibiotic treatment, this strategy would be more benign to the human microbiota as inhibiting bacterial virulence would lead to living but non-pathogenic bacteria that eventually will be cleared by the host’s innate defenses. Furthermore, given that most virulence factors are not essential for bacterial viability, blocking virulence factors would attenuate infection without threatening their survival and thus may reduce selective pressure for resistance.3
Urinary tract infections (UTIs) are among the most common bacterial infectious diseases in human population, and E. coli are the most predominant pathogens responsible for 80–90% of community-acquired and 30–50% of hospital acquired UTIs.8 Uropathogenic E. coli (UPEC) strains are equipped with a particular set of virulence factors allowing them to colonize distinct sites in the urinary system. Development of an UTI is a multi-step process that starts with bacteria recognizing and attaching to the host tissue. The first contact is mediated by hair-like surface proteins called pili or fimbriae expressed on the bacterial surface. Different strains of UPEC display various type of pili, but two of the most important types are type 1 and P pili, which mediate infections of bladder and kidneys, respectively.9–11 Each pilus rod is composed of a number of repeating protein subunits (called Pap in P pili and Fim in type 1 pili).12–14 Pili are assembled via a complex secretion system called the chaperone/usher pathway.15 Pili are important virulence factors for the bacteria, and they need these organelles to attach to the host cell, to withstand shear forces (in the urinary tract), to invade the host and to establish biofilm-like colonies.16–19 Type 1 pili have been implicated in mediating biofilm formation in E.coli.20,21 Treatment of UTIs, like other microbial infections, is exacerbated by increasing antibiotic resistance within biofilms,22 highlighting the need for alternative therapeutics. Accordingly, type 1 pili represent an excellent target for therapeutic intervention of bacterial infection.23
Using UPEC as a model pathogen we have developed a novel class of anti-virulence compounds containing a 2-pyridone scaffold, named pilicides (such as 1 shown in Figure 1), which inhibit the formation of type 1 pili by interfering with the chaperone/usher pathway.24 These substituted 2-pyridones bind to periplasmic chaperones (PapD/FimC) responsible for the folding and transport of pilus subunits to the outer membrane, and thus interfere with the essential chaperone function in the assembly mechanism. In a mouse model of UTI infection, the pilicides decreased the bacterial adhesion to the bladder and effectively prevented infection in the higher structures of the urogenital tract,25 suggesting that inhibition of pilus biogenesis by small molecules like pilicides is a promising strategy to combat infection. Considering that this assembly pathway is highly conserved, the pilicides could potentially be applicable against many Gram-negative pathogens. Inspired by these findings, a high-throughput screening campaign of a library of 17,500 compounds, which were purchased from ChemBridge and carefully selected to cover a large chemical space, was performed to discover new chemical classes of small molecules having pilicide-like activity. As a result, two compounds 2 and 3 containing a 2-amino-3-acyl-tetrahydrobenzothiophene scaffold were identified as novel inhibitors of pili-dependent biofilm formation in the UPEC strain (the clinical isolate E.coli UTI89).
Figure 1.

Structures of pilicide 1 and hit compounds 2 and 3 identified from HTS and related pharmaceutical agents under development or marketed containing a 2-amino-3-acylthiophene fragment.
Compounds containing the 2-amino-3-acylthiophene scaffold, commonly prepared via Gewald reaction,26 have been the subject of many chemical and biological studies due to their interesting pharmacological properties.27 For example, Tinoridine (4) is a nonsteroildal basic anti-inflammatory drug. Compound T-62 (5), a selective allosteric enhancer of adenosine A1 receptor, is currently under phase-II clinical trial for the treatment of neuropathic pain.28,29 Other allosteric enhancers (6 and 7) are also promising leads.30,31 AX20017 (8), a promising compound with antituberculosis activity, has also been identified as a specific inhibitor of protein kinase G (PknG),32 and the thiophene 3-carboxylic acid amide TPCA-1 (9) has recently been identified as a small-molecule IκB kinase β (IKKβ) inhibitor33 (Figure 1).
In spite of many studies on chemistry and bioactivity of 2-amino-3-acyl-tetrahydrobenzothiophene analogs, there is no report so far on this class of compounds being evaluated for anti-virulence activity. Therefore, a structure-activity relationship (SAR) study based on the hit compounds 2 and 3 was initiated with the primary aim to gain valuable information on their SAR for further optimization. In this paper, we describe the synthesis and biological activities of 2-amino-3-acyl-tetrahydrobenzothiophene derivatives (12–39), which were designed and prepared according to the modification strategy outlined in Figure 2.
Figure 2.
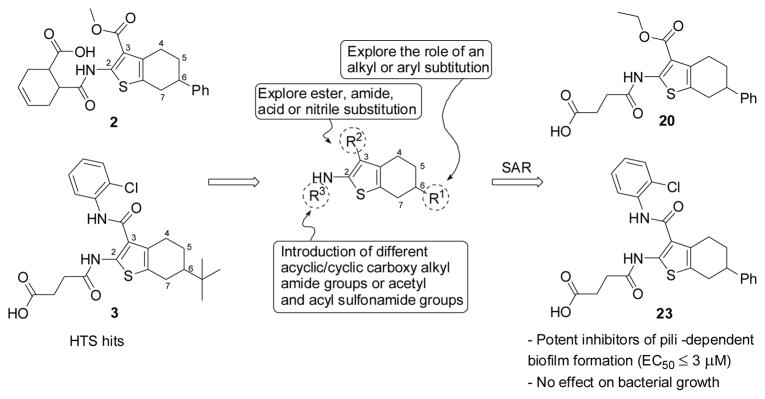
Structural modification strategy of hits 2 and 3 to investigate SAR for further optimization.
Results and discussion
Chemistry
The hit compounds 2 and 3 and their analogs (12–39) were synthesized according to a general pathway described in Scheme 1. Briefly the key intermediates, 2-amino-3-acyl-tetrahydrobenzothiophenes (11a–k), were prepared via the Gewald reaction,26 a three-component reaction between a carbonyl compound, an activated nitrile and elemental sulfur in the presence of a suitable base as a catalyst. Depending on the nature of activated nitriles (cyanoacetates or N-(2-haloaryl)cyanoacetamides), different conditions of Gewald reaction were employed (See experimental section for details). On the other hand, the intermediates (11m–p) were synthesized from the corresponding methyl ester 11f through a synthesis sequence, including N-Boc protection, saponification, amidation and then N-Boc deprotection. With the intermediates in hand, the hits (2 and 3) and target compounds (12–25, 27–31 and 36–39) were prepared with moderate to good yields (47–89%) by overnight refluxing of these precursors with a variety of acid anhydrides in dry CH2Cl2 under N2 gas. The target analogs (26, 32–35) were synthesized by using other reaction conditions as shown in the Scheme 1.
Scheme 1.

Synthesis of 2-amino-3-acyl-tetrahydrobenzothiophene derivatives. Reagents and conditions: (a) S8, amine base, alcohol or DMF, rt, 24 h; (b) S8, morpholine, basic Al2O3, MWI, 160 W, 20 min; (c) acid anhydride, CH2Cl2, N2 gas, reflux, overnight; (d) NH3, MeOH, MWI 110 °C, 1 h; (e) NaOH, H2O, MeOH/THF, 80 °C, 6 h; (f) acetyl chloride, pyridine, rt, 2 h; (g) tert-butyl 2-chloro-2-oxoacetate, TEA, CH2Cl2, 0 °C, 5 min, then TFA, CH2Cl2, rt, 21 h; (h) p-Toluenesulfonyl isocyanate, TEA, toluene, reflux, 20 h; (i) Boc2O, DMAP, dioxane, 80 °C, 3 h, then N2H4, 40 °C, 1.5 h; (j) amine, TBTU, TEA, EtOAc, rt, 3 h; (k) TFA, CH2Cl2, rt, 1 h. * Yields of isolated products.
Biological evaluation and SAR studies
All of the synthesized compounds, which included the hits 2 and 3 and their analogs 12–39 together with several intermediates 11a, 11b, 11f and 11g, were evaluated for their activity against biofilm formation of E.coli UTI89. The biofilm formation assay is based on the ability of E. coli to form type 1 pili-dependent biofilms on polyvinylchloride (PVC) surfaces.34 Blocking type 1 pili formation in this assay also prevents the ability of the bacteria to form biofilm and the amount of biofilm that is formed in the presence of tested compounds is thus related to the potency of the substances in blocking pili formation. Furthermore, the best compounds from the biofilm evaluation were then evaluated in a hemagglutination (HA) titer assay and electron microscopy (EM) measurements to verify that the observed biofilm inhibition is a result of reduced pili formation. In the biofilm assay, all compounds were first evaluated at 200 μM, 100 μM and 50 μM to measure their relative inhibitory activity. Subsequently, only active compounds with good inhibition rate (> 50% at 50 μM) were further tested at lower concentrations (25, 12.5, 6.3, 3.1 and 1.5 μM) to compare their bioactivity profile, and the biological data of active compounds (2, 3 and 14–23) was presented in Figure 3. In addition, a growth inhibition assay was also performed to validate that the active compounds are really true inhibitors of biofilm formation and not acting as bactericidal or bacteriostatic agents (Fig. S1 in ESI). A summary of structure-antibiofilm activity relationships (SAR) of this class of compounds is outlined in Figure 4.
Figure 3.

Antibiofilm activity of active compounds including hits 2 and 3 and their related derivatives (14–23), BL (blank, only UTI89 + DMSO). The data represent means ± SD of triplicate experiments.
Figure 4.

Structure-antibiofilm activity relationships of 2-amino-3-acyl-tetrahydrobenzothiophene analogs.
C-6 substituent effects on antibiofilm activity
The initial data showed that compounds 12 and 13 without a substitution in the C-6 position did not exhibit any antibiofilm activity at all tested concentrations, indicating that an alkyl- or aryl-substitution in this position is required for the activity. Further evaluation of active compounds revealed that 6-phenyl-substituted analogs (2 and 19–23) had higher potency than 6-tert-butyl-substituted compounds (3 and 14–18) as shown in Figure 3. Comparing the activity of the most active 6-tert-butyl-substituted derivatives (3 and 16) and 6-phenyl-substituted counterparts (20 and 23), it was observed that 6-phenyl-substituted analogs 20 and 23 inhibited biofilm formation in a concentration-dependent manner and both displayed stronger activity than 6-tert-butyl-substituted derivatives 3 and 16 at lower concentrations (Figure 3), suggesting that substitution by a phenyl group in the C-6 position is more favorable for the desired activity.
C-2 substituent effects on antibiofilm activity
The initial data showed that none of the precursors 11a, 11b, 11f and 11g with a free amine group in the C-2 position displayed activity, indicating that the presence of an amide substitution in the C-2 position of the hits 2 and 3 is important for the antibiofilm activity. Regardless of the effect of substitutions in C-3 and C-6 positions, the type of the amide substituent in the C-2 position strongly modulated the activity, as observed by the different activity of compounds 2 and 19; 3 and 18; 20 and 21. Comparison of the activity of compounds 2, 19 and 21 with their corresponding C-2 modified analogs (24–29 and 33–35) showed that no improvement in antibiofilm activity was obtained by replacing the parent amide substituent in the C-2 position of the hits 2 and 3 with other amide groups (Fig. S2 in ESI and Fig. 4), supporting the view that parent amide substituent in the hits 2 and 3 is more favorable for the antibiofilm activity.
C-3 substituent effects on antibiofilm activity
Regardless of the effect of substitutions in the C-2 and C-6 positions, many C-3 ester analogs (2, 16 and 19–22) exhibited significant activity with higher potency observed for ethyl- and isopropyl-esters (Figures 3 and 4), suggesting that other ester groups would also be well tolerated. Replacement of the C-3 ester group by other groups (such as acid, nitrile or alkyl amide group) resulted in inactive compounds (30–32, 36, 37 and 39) (Fig. S2 in ESI and Fig. 4), indicating that these groups are unfavorable for the activity. Among amide analogs, only phenyl amides (3 and 23) bearing a chlorine atom at the ortho position of the phenyl ring exhibited potent activity (Fig. 3). The difference of potency between compounds 3 (o-Cl) and 17 (o-F) or between 23 (o-Cl) and compound 38 (o-H) (Fig. 3 and Fig. S2 in ESI) indicated the requirement of a proper halogen atom or an EWG in the ortho position of the phenyl ring for the antibiofilm activity.
Hemagglutination (HA) titer assay and electron microscopy (EM) measurements
It is known that the pili-dependent biofilm assay is a good initial screen for identification of potential pilicides or pilicide-like compounds, but it is not conclusive since biofilm formation can be inhibited due to other factors than the absence of pili.24 Therefore, the hit compound 3 and its analog 20 were evaluated in a hemagglutination (HA) titer assay (Table 1) to verify that the observed biofilm inhibition is a result of reduced pili formation. In the hemagglutination (HA) titer assay, the relative hemagglutination ability reflects the amount of plili being expressed in the presence of tested compounds. A low HA-titer demonstrates that a high concentration of bacteria is required for agglutination to occur, and the pili-producing E.coli strain (UTI89) in this assay condition gave the HA-titer 7.5. In the presence of compounds 3 and 20, the HA titers were reduced to 5 by both compounds, confirming that agglutination and thus pilus assembly is partially blocked by these analogs and this correlated with their observed biofilm inhibition. As shown in table 1 one of the most potent pilicide EC240,35 which was used as a positive control and reduced the HA-titer to 2.5, exhibited a stronger effect on agglutination than both compounds 3 and 20 in the HA titer assay. However, the HA titers were only used to evaluate the relative potencies of the compounds in blocking pilus formation and reflected the average expression of pili in a bacterial population. Accordingly, to clearly verify the effects of the compounds on the degree of piliation, electron microscopy (EM) measurements were employed to quantitate the pili on the compounds-treated bacteria. Interestingly, both derivatives 3 and 20 displayed a more pronounced de-piliation effect than the pilicide EC240 as determined by pilus counts using EM (Figure 5). Further studies on the 2-amino-3-acyl-tetrahydrobenzothiophenes to improve their antivirulence activity and elucidate the underlying mechanisms of the observed biological results are currently underway in our laboratories.
Table 1.
HA Titer Data for Selected Compounds
| Compounds | HA titera |
|---|---|
| 3 | 5 |
| 20 | 5 |
| EC240 | 2.5 |
| UTI89 + DMSO | 7.5 |
| UTI89 | 8.5 |
Number of wells with agglutination (2n), E. coli was grown in the presence of tested compounds (250 μM) and HA titers were determined, average of duplicate runs. All compounds were dissolved in DMSO.
Figure 5.

Effects of compounds 3 and 20 on levels of piliation. The pili content of control or test samples (3, 20 and EC240)-treated UTI89 bacteria in triplicate was quantitated by electron microscopy (EM) in a blinded study. Bacteria were classified as having one of four degree of piliation: abundant, moderate, low, or bald (no pili). The percentage of the total number of bacteria counted in each category is displayed.
Conclusion
A high-throughput screening campaign of a library of 17,500 compounds resulted in the identification of 2-amino-3-acyl-tetrahydrobenzothiophene derivatives as new inhibitors of pili-dependent biofilm formation in the UPEC strain (UTI89). By systematic structural variation of the initial hit compounds 2 and 3, valuable information on their SARs was obtained for further optimization: 1) an aromatic substituent (e.g., a phenyl group) in the C-6 position is favorable for the antibiofilm activity; 2) other ester groups or 2-EWG-bearing aryl amides in the C-3 position may be employed to find out the best C-3 substituent; 3) the parent substituent in the C-2 position of the hits 2 and 3 should be kept constant. In addition to SAR establishment, structural modification of the parent compounds resulted in several analogs (20–23) with significant antivirulence activity. Biological evaluation using biofilm formation and hemagglutination (HA) titer assays together with pili quantitated by EM demonstrated that this class of compounds potently suppress the development of biofilm formation of E.coli by inhibiting pilus biogenesis without affecting bacterial growth. Thus, compounds having the 2-amino-3-acyl-tetrahydrobenzothiophene scaffold may serve as a new class of compounds for further development as antibacterial agents with antivirulence activity. At this point it is not clear whether the compounds are acting on pilus regulation or directly targeting the pilus assembly machinery. Further studies to delineate this are however underway in our laboratories in combination with studies to expand SAR knowledge and improve compound potency.
Experimental
General
All chemicals and solvents were purchased from commercial suppliers (Aldrich, Alfa Aesar, and Fisher Scientific) and used as received. Microwave-assisted reactions were conducted in sealed Emrys™ process vials with an Initiator™ 2.0 microwave instrument from Biotage. The reactions were monitored by thin layer chromatography (TLC) and LC-MS. TLC was performed on Silica Gel 60 F254 (Merck) with detection by UV light (254 nm). Flash column chromatography separations (eluents given in brackets) were performed on silica gel (Matrex, 60 Å, 35–70 μm, Grace Amicon) or Biotage SP1 flash purification system with mounted 230–400 mesh silica gel columns. Preparative reverse phase HPLC was performed on a C18 reversed-phase column (25 cm × 21.2 mm, 5 μm) with MeCN: H2O mixtures as eluent at a flow rate of 20 mL/min over 20 min. 1H NMR and 13C NMR spectra were recorded on a Bruker DRX-400 and calibrated using the residual peak of solvents as internal standard [CDCl3 (CHCl3 δH 7.26 ppm, CDCl3 δC 77.0 ppm), CD3OD (CD3OD δH 3.31 ppm, CD3OD δC 49.0 ppm), d6-DMSO (d5-DMSO δH 2.50 ppm, d6-DMSO δC 40.0 ppm), d3-AcOD (d2-AcOD δH 2.00 ppm and 11.70 ppm, d3-AcOD δC 20.0 ppm and 180.0 ppm)]. Melting points were determined using an Electrothermal IA 9000 series Digital Melting Point Apparatus and uncorrected. IR spectra were recorded on a Perkin Elmer FT-IR spectrometer equipped with Attenuated Total Reflection (ATR) capability. LC-MS was conducted on a Micromass ZQ mass spectrometer using ES+ ionization. HRMS was performed on a BrukermicrOTOF II mass spectrometer with positive electrospray ionization (ES+); sodium formate was used as the calibration chemical.
Synthesis of intermediates 11a–p
Synthesis of intermediates 11a–k
Method A
The method was used for the synthesis of intermediates derived from cyanoacetates. A substituted cycloketone 10 (1 equiv), an activated nitrile (1.1 equiv) and elemental sulfur (1.2 equiv) were added to a proper alcohol solvent or DMF (10 mL/g), which depends on the corresponding cyanoacetate used to avoid transesterification. An amine (diethylamine, 0.5 equiv or triethylamine, 1.1 equiv) was added dropwise to the solution, and the reaction mixture was stirred at room temperature for 24 h. The resultant precipitate was collected by filtration and washed with ice cold alcohol until the filtrate was colorless with recovery and recrystallization of the mother liquor. In case of no precipitate formed the reaction mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography or HPLC.
Method B36
The method was used for the synthesis of intermediates derived from N-(2-haloaryl)cyanoacetamides. A substituted cycloketone 10 (1 equiv), an activated nitrile (1.1 equiv), elemental sulfur (1.2 equiv), morpholine (0.8 equiv) and basic Al2O3 (100 mg/1 mmol) were mixed thoroughly in a microwave tube. The tube was sealed and irradiated at the power of 160 W for 20 min. After cooling CH2Cl2 was added to the reaction mixture and then Al2O3 was removed by filtration. The filtrate was concentrated under reduced pressure, and the residue was purified by flash column chromatography or HPLC.
Methyl 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11a)
Compound 11a (1.71 g, 85%) was obtained by following general procedure A using cyclohexanone (0.94 g), methyl cyanoacetate (0.93 mL), elemental sulfur (0.37 g) and diethylamine (0.49 mL) in methanol (10 mL). The product was purified by washing the resultant precipitate with ice cold methanol. Light yellow solid. M.p. 113–116 ºC. IR νmax/cm−1 (ATR) 3416 (NH), 3306 (NH), 1650 (C=O), 1576 (C=C) and 1268 (C-O) cm−1. 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 3H), 2.69-2.67 (m, 2H), 2.50-2.48 (m, 2H), 1.80-1.71 (m, 4H); 13C NMR (400 MHz, CDCl3) δ 166.4, 160.3, 132.5, 118.6, 106.6, 50.7, 26.8, 24.6, 23.2, 22.8. LC-MS Rf (min) = 4.97. LC-MS (ES) calcd for C10H14NO2S [M+ H]: 212, found: 212.
Methyl 2-amino-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11b)
Compound 11b (1.4 g, 82%) was obtained by following general procedure A using 4-tert-butylcyclohexanone (1.0 g), methyl cyanoacetate (0.63 mL), elemental sulfur (0.25 g) and diethylamine (0.33 mL) in methanol (10 mL). The product was purified by washing the resultant precipitate with ice cold methanol. Light yellow solid. M.p. 125–128 ºC. IR νmax/cm−1(ATR) 3475 (NH), 3339 (NH), 1663 (C=O), 1567 (C=C) and 1245 (C-O). 1H NMR (400 MHz, CDCl3) δ 3.77 (s, 3H), 2.95-2.91 (m, 1H), 2.50-2.44 (m, 2H), 2.43-2.33 (m, 1H), 1.94-1.92 (m, 1H), 1.49-1.43 (m, 1H), 1.30-1.21 (m, 1H), 0.91 (s, 9H); 13C NMR (400 MHz, CDCl3) δ 166.3, 160.3, 132.5, 119.5, 106.3, 50.7, 45.2, 32.4, 27.9, 27.3 (3C), 26.0, 24.3. LC-MS Rf (min) = 5.79. LC-MS (ES) calcd for C14H22NO2S [M+ H]: 268, found: 268.
Ethyl 2-amino-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11c)
Compound 11c (1.44 g, 79%) was obtained by following general procedure A using 4-tert-butylcyclohexanone (1.0 g), ethyl cyanoacetate (0.76 mL), elemental sulfur (0.25 mg) and diethylamine (0.33 mL) in ethanol (10 mL). The product was purified by column chromatography on silica gel using Heptane: EtOAc (10:1 → 1:1) as mobile phase. Light yellow oil. IR νmax/cm−1 (ATR) 3425 (NH), 3313 (NH), 1657 (C=O), 1537 (C=C) and 1241 (C-O). 1H NMR (400 MHz, CD3OD,) δ 4.23 (d, J = 7.2 Hz, 2H), 2.99-2.93 (m, 1H), 2.53-2.46 (m, 2H), 2.32–2.34 (m, 1H), 2.01-1.96 (m, 1H), 1.49-1.42 (m, 1H), 1.34 (t, J = 7.2 Hz, 3H), 1.29-1.21 (m, 1H), 0.95 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 166.2, 163.7, 131.6, 116.9, 103.4, 58.9, 45.2, 31.8, 27.8, 26.3 (3C), 25.5, 24.3, 13.5; LC-MS Rf (min) = 5.99. LC-MS (ES) calcd for C15H24NO2S [M+ H]: 282, found: 282.
N-(2-Chlorophenyl)-2-amino-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (11d)
Compound 11d (126 mg, 51%) was obtained by following general procedure B using 4-tert-butylcyclohexanone (83 mg), N-(2-chlorophenyl)-2-cyanoacetamide (116 mg), elemental sulfur (21 mg), morpholine (37 μL) and Al2O3 (70 mg). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Colorless oil. IR νmax/cm−1 (ATR) 3430 (NH), 3298 (NH), 1637 (C=O) and 1502 (C=C). 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 7.6 Hz, 1H), 8.04 (s, 1H), 7.40 (d, J = 7.4 Hz, 1H), 7.30 (m, 1H), 7.04 (m, 1H), 3.09-3.02 (m, 1H), 2.85-2.75 (m, 1H), 2.65-2.58 (m, 1H), 2.44-2.34 (m, 1H), 2.17-2.09 (m, 1H), 1.63-1.53 (m, 1H), 1.46-1.35 (m, 1H), 0.97 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 164.5, 160.7, 135.3, 129.0, 128.7, 127.7, 124.1, 122.8, 121.9, 119.9, 108.7, 44.8, 32.5, 28.5, 27.3 (3C), 26.2, 24.7. LC-MS Rf (min) = 6.32. LC-MS (ES) calcd for C19H24ClN2OS [M+ H]: 363, found: 363.
N-(2-Fluorophenyl)-2-amino-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (11e)
Compound 11e (117 mg, 48%) was obtained by following general procedure B using 4-tert-butylcyclohexanone (83 mg), N-(2-fluorophenyl)-2-cyanoacetamide (106 mg), elemental sulfur (21 mg), morpholine (37 μL) and Al2O3 (70 mg). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Ligh yellow oil. IR νmax/cm−1 (ATR) 3437 (NH), 3316 (NH), 1650 (C=O) and 1502 (C=C). 1H NMR (400 MHz, CDCl3) δ 8.46-8.40 (m, 1H), 7.83 (s, 1H), 7.19-7.09 (m, 2H), 7.07-7.01 (m, 1H), 3.02-2.94 (m, 1H), 2.78-2.68 (m, 1H), 2.65-2.56 (m, 1H), 2.44-2.34 (m, 1H), 2.17-2.08 (m, 1H), 1.61-1.52 (m, 1H), 1.47-1.34 (m, 1H), 0.96 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 164.5, 160.7, 152.6 (d, J = 260 Hz), 128.5, 126.9 (d, J = 10 Hz), 124.6, 123.5 (d, J = 8.5 Hz), 121.6, 120.0, 114.6 (d, J = 19 Hz), 108.5, 44.8, 32.4, 28.1, 27.3 (3C), 26.1, 24.7. LC-MS Rf (min) = 6.02. LC-MS (ES) calcd for C19H24FN2OS [M+ H]: 347, found: 347.
Methyl 2-amino-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11f)
Compound 11f (2.77 g, 84%) was obtained by following general procedure A using 4-phenylcyclohexanone (2.0 g), methyl cyanoacetate (1.11 mL), elemental sulfur (0.44 g) and diethylamine (0.59 mL) in methanol (20 mL). The product was purified by washing the resultant precipitate with ice cold methanol. White solid. M.p. 130–133 ºC. IR νmax/cm−1 (ATR) 3442 (NH), 3320 (NH), 1668 (C=O), 1568 (C=C) and 1260 (C-O). 1H NMR (400 MHz, CDCl3) δ 7.37-7.25 (m, 5H), 3.83 (s, 3H), 3.02-2.98 (m, 2H), 2.78-2.70 (m, 3H), 2.20-2.10 (m, 1H), 1.93-1.80 (m, 1H); 13C NMR (400 MHz, CDCl3) δ 166.5, 162.1, 146.1, 132.2, 128.5 (2C), 126.9 (2C), 126.3, 117.0, 105.4, 50.7, 40.9, 32.4, 30.1, 27.2. LC-MS Rf (min) = 5.42. LC-MS (ES) calcd for C16H18NO2S [M+ H]: 288, found: 288.
Ethyl 2-amino-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11g)
Compound 11g (0.96 g, 79%) was obtained by following general procedure A using 4-phenylcyclohexanone (1 g), ethyl cyanoacetate (0.67 mL), elemental sulfur (0.22 g) and diethylamine (0.29 mL) in ethanol (10 mL). The product was purified by column chromatography on silica gel using Heptane:EtOAc (10:1 → 1:1) as mobile phase. Light yellow oil. IR νmax/cm−1 (ATR) 3456 (NH), 3322 (NH), 1663 (C=O), 1578 (C=C) and 1264 (C-O). 1H NMR (400 MHz, CD3OD) δ 7.32-7.26 (m, 4H), 7.21-7.17 (m, 1H), 4.24 (q, J = 7.1 Hz, 2H), 2.99-2.91 (m, 2H), 2.74-2.59 (m, 3H), 2.07-2.01 (m, 1H), 1.93-1.83 (m, 1H), 1.34 (t, J = 7.1 Hz, 3H); 13C NMR (400 MHz, CD3OD) δ 166.2, 163.8, 146.2, 131.5, 128.0 (2C), 126.5 (2C), 125.9, 115.9, 103.5, 58.9, 40.9, 31.9, 30.1, 27.0, 13.4. LC-MS Rf (min) = 5.60. LC-MS (ES) calcd for C17H20NO2S [M+ H]: 302, found: 302.
Isopropyl 2-amino-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (11h)
Compound 11h (0.35 g, 65%) was obtained by following general procedure A using 4-phenylcyclohexanone (0.3 g), isopropyl cyanoacetate (0.24 mL), elemental sulfur (66 mg) and triethyl amine (0.22 mL) in DMF (5 mL). The mixture was diluted with EtOAc and washed with water to remove DMF after the reaction was complete. The product was purified by column chromatography on silica gel using Heptane:EtOAc (10:1 → 1:1) as mobile phase. Light yellow oil. IR νmax/cm−1 (ATR) 3416 (NH), 3307 (NH), 1678 (C=O), 1530 (C=C), and 1200 (C-O). 1H NMR (400 MHz, CD3OD) δ 7.30-7.16 (m, 5H), 5.16-5.09 (m, 1H), 3.01-2.91 (m, 2H), 2.75-2.58 (m, 3H), 2.07-2.01 (m, 1H), 1.92-1.82 (m, 1H), 1.32 (d, J = 7.5 Hz, 6H); 13C NMR (400 MHz, CD3OD) δ 165.8, 163.7, 146.2, 131.5, 128.1 (2C), 126.5 (2C), 125.8, 115.9, 103.9, 66.4, 40.9, 32.0, 30.1, 27.2, 21.1 (2C). LCMS Rf (min) = 5.95. LRMS (ES) calcd for C18H22NO2S [M+ H]: 316, found: 316.
N-(2-Chlorophenyl)-2-amino-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (11i)
Compound 11i (67 mg, 48%) was obtained by following general procedure B using 4-phenylcyclohexanone (67 mg), N-(2-chlorophenyl)-2-cyanoacetamide (87 mg), elemental sulfur (15 mg), morpholine (27 μL) and Al2O3 (50 mg). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Amorphous solid. IR νmax/cm−1 (ATR) 3424 (NH), 3310 (NH), 1631 (C=O) and 1505 (C=C). 1H NMR (400 MHz, CDCl3) δ 8.50 (dd, J = 8.3, 1.7 Hz, 1H), 8.08 (s, 1H), 7.42-7.25 (m, 7H), 7.08-7.02 (m, 1H), 3.15-2.97 (m, 3H), 2.91-2.84 (m, 1H), 2.82-2.72 (m, 1H), 2.30-2.23 (m, 1H), 2.08-1.98 (m, 1H); 13C NMR (400 MHz, CDCl3) δ 164.5, 160.9, 145.5, 135.3, 129.1, 128.6 (2), 128.5, 127.6, 126.9 (2C), 126.6, 124.0, 122.8, 121.9, 118.8, 108.7, 40.5, 32.6, 30.1, 27.8. LC-MS Rf (min) = 6.43. LC-MS (ES) calcd for C21H20ClN2OS [M+ H]: 383, found: 383.
2-Amino-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (11j)
Compound 11j (0.31 g, 70%) was obtained by following general procedure A using 4-phenylcyclohexanone (0.3 g), manonitrile (0.12 mL), elemental sulfur (66 mg) and triethyl amine (0.26 mL) in DMF (5 mL). The mixture was diluted with EtOAc and washed with water to remove DMF after the reaction was complete. The product was purified by trituration of the crude product with methanol. Brown solid. M.p. 178–181 ºC. IR νmax/cm−1 (ATR) 3440 (NH), 3313 (NH), 2202 (C≡N) and 1514 (C=C). 1H NMR (400 MHz, CDCl3) δ 7.28-7.23 (m, 2H), 7.20-7.14 (m, 3H), 2.98-2.89 (m, 1H), 2.75-2.68 (m, 1H), 2.66-2.48 (m, 3H), 2.08-1.98 (m, 1H), 1.91-1.80 (m, 1H); 13C NMR (400 MHz, CDCl3) δ 160.6, 145.2, 132.3, 128.5 (2C), 126.9 (2C), 126.6, 120.1, 115.4, 88.2, 41.2, 31.8, 29.4, 24.7. LC-MS Rf (min) = 5.12. LC-MS calcd for C15H15N2S [M+ H]: 255, found: 255.
2-Amino-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (11k)
Compound 11k (0.35 g, 75%) was obtained by following general procedure A using 4-phenylcyclohexanone (0.3 g), cyanoacetamide (0.16 g), elemental sulfur (66 mg) and triethyl amine (0.26 mL) in DMF (5 mL). The mixture was diluted with EtOAc and washed with water to remove DMF after the reaction was complete. The product was purified by trituration of the crude product with methanol. White solid. M.p. 195–198 ºC. IR νmax/cm−1 (ATR) 3443 (NH), 3306 (NH), 1684 (C=O) and 1556 (C=C). 1H NMR (400 MHz, d6-DMSO) δ 7.34-7.29 (m, 4H), 7.23-7.19 (m, 1H), 2.97-2.88 (m, 1H), 2.84-2.66 (m, 3H), 2.64-2.55 (m, 1H), 2.01-1.93 (m, 1H), 1.90-1.78 (m, 1H); 13C NMR (400 MHz, d6-DMSO) 168.4, 159.8, 146.5, 130.3, 128.8 (2C), 127.5 (2C), 126.7, 115.8, 107.8, 40.4, 32.2, 30.2, 26.8. LC-MS Rf (min) = 5.70. LC-MS (ES) calcd for C15H17N2OS [M+ H]: 273, found: 273.
Synthesis of intermediates 11l-p
2-[(tert-Butoxycarbonyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylic acid (11l)
To a 50 mL round bottom flask was added dioxane (30 mL), compound 11f (0.72 g), Boc2O (1.15 g, 2.1 equiv) and DMAP (30.5 mg, 0.1 equiv) and the reaction mixture was stirred at 80 ºC for 3 h. Then N2H4.H2O (0.36 mL, 3.0 equiv) was added and the mixture was stirred at 40 ºC for additional 1.5 h.37 After cooling to room temperature the solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel (EtOAc:Heptane 5 → 30%) to yield the methyl ester of 11l (0.79 g, 81%). The ester (0.67 g) was subsequently subjected to a base hydrolysis at 80 °C for 6 h with a solution of NaOH (0.27 g) in 25 mL of solvent mixture (MeOH/H2O/THF = 2: 2: 1). The reaction was cooled to room temperature and the organic solvents were removed in vacuo. The aqueous layer was acidified with 5% HCl to give a precipitate, which was triturated with methanol to afford 11l (0.55 g, 85%). Yellow solid. M.p. 218–220 ºC. IR νmax/cm−1 (ATR) 1720 (C=O), 1521 (C=C) and 1148 (C-O). 1H NMR (400 MHz, DMSO-d6) δ 13.0 (brs, 1H), 10.4 (brs, 1H), 7.33-7.18 (m, 5H), 2.99-2.90 (m, 2H), 2.88-2.80 (m, 1H), 2.74-2.63 (m, 2H), 2.02-1.94 (m, 1H), 1.90-1.79 (m, 1H), 1.48 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 151.7, 149.0, 146.2, 131.4, 128.8 (2C), 127.3 (2C), 126.7, 124.6, 110.8, 82.2, 40.2, 31.9, 30.0, 28.2, 26.8 (3C); LC-MS Rf (min) = 5.67. LC-MS (ES) calcd for C20H24NO4S [M+ H]: 374, found: 374.
N-Isopropyl 2-[(tert-butoxycarbonyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (11m)
To a solution of compound 11l (80 mg) and triethylamine (TEA: 60 μL, 2 equiv) in EtOAc (8 mL) was added 2-(1H-Benzotriazole-1-yl)-1,2,3,3-tetramethyluronium tetrafluoroborate (TBTU: 69 mg, 1 equiv), and the mixture was stirred at room temperature for 10 min. Then isopropyl amine (34 μL, 2 equiv) was added and the reaction was stirred at room temperature for 3 h. The mixture was washed with water and extracted with EtOAc. The organic layer was separated, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using EtOAc:Heptane (1:10) as eluent to give 11m (48 mg, 54%). White solid. M.p. M.p. 211–215 ºC IR νmax/cm−1 (ATR) 1702 (C=O), 1523 (C=C) and 1154 (C-O). 1H NMR (400 MHz, CDCl3) δ 11.02 (s, 1H), 7.37-7.29 (m, 2H), 7.28-7.26 (m, 1H), 7.25-7.21 (m, 2H), 5.67 (brs, 1H), 4.28-4.17 (m, 1H), 3.07-2.89 (m, 2 H), 2.86-2.71 (m, 3H), 2.24-2.15 (m, 1H), 2.04-1.92 (m, 1H), 1.50 (s, 9H), 1.27 (t, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 165.4, 152.5, 147.4, 145.3, 128.6 (2C), 126.8 (3C), 126.5, 125.6, 112.5, 81.3, 41.4, 40.2, 32.1, 30.1, 28.2 (3C), 26.9, 23.0, 22.9; LC-MS Rf (min) = 6.10. LC-MS (ES) calcd for C23H31N2O3S [M+ H] : 415, found: 415.
[2-(tert-Butoxycarbonylamino)-4,5,6,7-tetrahydrobenzo[b]thien-3-yl]-4-morpholinyl-methanone (11n)
Compound 11n (44.4 mg, 47%) was obtained by following the same procedure for the preparation of 11m using 11l (80 mg), TEA (60 μL), TBTU (69 mg) and morpholine (36 μL) in EtOAc (8 mL). The product was purified by column chromatography on silica gel using EtOAc:Heptane (1:4 → 1:1) as mobile phase. White solid. M.p. 213–217 ºC. IR νmax/cm−1 (ATR) 1715 (C=O), 1535 (C=C) and 1153 (C-O). 1H NMR (400 MHz, CDCl3) δ 8.05 (brs, 1H), 7.34-7.29 (m, 2H), 7.28-7.26 (m, 1H), 7.24-7.19 (m, 2H), 3.75-3.41 (m, 8H), 3.09-2.91 (m, 2H), 2.84-2.74 (m, 1H), 2.63-2.49 (m, 2H), 2.13-2.05 (m, 1H), 1.96-1.85 (m, 1H), 1.51 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 166.7, 152.4, 145.5, 139.8, 129.0, 128.5 (2C), 127.4, 126.8 (2C), 126.4, 117.1, 81.7, 67.1, 67.0, 45.9, 44.9, 40.8, 32.0, 29.8, 28.2 (3C), 24.9; LC-MS Rf (min) = 5.55. LC-MS (ES) calcd for C24H31N2O4S [M+ H]: 443, found: 443.
N-Phenyl 2-[(tert-butoxycarbonyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (11o)
Compound 11o (41.6 mg, 43%) was obtained by following the same procedure for the preparation of 11m using 11l (80 mg), TEA (60 μL), TBTU (69 mg) and aniline (40 μL) in EtOAc (8 mL). The product was purified by column chromatography on silica gel using EtOAc:Heptane (1:15) as mobile phase. White solid. M.p. 207–210 ºC. IR νmax/cm−1 (ATR) 1709 (C=O), 1518 (C=C) and 1149 (C-O). 1H NMR (400 MHz, CDCl3) δ 10.82 (s, 1H), 7.58-7.47 (m, 3H), 7.41-7.30 (m, 4H), 7.30-7.25 (m, 2 H), 7.19-7.11 (m, 1H), 3.13-3.03 (m, 1H), 3.03-2.88 (m, 3H), 2.87-2.78 (m, 1H), 2.44-2.17 (m, 1H), 2.16-1.89 (m, 1H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 164.4, 152.5, 148.7, 145.1, 137.3, 129.1 (2C), 128.6 (2C), 126.9 (2C), 126.6, 126.5, 126.1, 124.7, 120.8 (2C), 112.6, 81.7, 40.2, 32.1, 30.1, 28.2 (3C), 27.0; LC-MS Rf (min) = 5.75. LC-MS (ES) calcd for C26H24N2O3S [M+ H]: 449, found: 449.
N-Benzyl 2-[(tert-butoxycarbonyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (11p)
Compound 11p (64 mg, 65%) was obtained by following the same procedure for the preparation of 11m using 11l (80 mg), TEA (60 μL), TBTU (69 mg) and benzylamine (46 μL) in EtOAc (8 mL). The product was purified by column chromatography on silica gel using EtOAc:Heptane (1:10) as mobile phase. Colorless oil. IR νmax/cm−1 (ATR) 1709 (C=O), 1508 (C=C) and 1149 (C-O). 1H NMR (400 MHz, CDCl3) δ 11.06 (s, 1H), 7.39-7.26 (m, 7H), 7.25-7.24 (m, 2H), 7.24-7.20 (m, 1H), 6.21-6.13 (m, 1H), 4.61 (d, J = 2.8 Hz, 2H), 3.05-2.90 (m, 2H), 2.86-2.69 (m, 3H), 2.20-2.12 (m, 1H), 2.03-1.91 (m, 1H), 1.51 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 166.1, 152.5, 148.1, 145.2, 138.1, 128.8 (2C), 128.6 (2C), 127.6, 127.5 (2C), 126.8 (2C), 126.5, 125.7, 112.0, 81.5, 43.5, 40.1, 32.0, 30.0, 28.2 (4C), 26.9; LC-MS Rf (min) = 6.01. LC-MS (ES) calcd for C27H31N2O3S [M+ H]: 463, found: 463.
Synthesis of the hits (2 and 3) and target compounds 12–39
Methyl 2-[[ (6-carboxy-3-cyclohexen-1-yl)carbonyl]amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (2)
The intermediate 11f (50 mg) was dissolved in dry CH2Cl2 (7 mL) and cis-1,2,3,6-tetrahydrophthalic anhydride (32 mg, 1.2 equiv) was added. The mixture was kept at refluxing overnight under N2 gas. After cooling to room temperature the reaction mixture was concentrated under reduced pressure and the residue was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase to afford compound 2 (51 mg, 67% yield). White solid. M.p. 223–226 ºC. IR νmax/cm−1 (ATR) 1690 (C=O), 1656 (C=O), 1534 (C=C) and 1207 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.34 (s, 1H), 11.26 (s, 1H) 7.34-7.31 (m, 4H), 7.25-7.19 (m, 1H), 5.71 (s, 2H), 3.84 (s, 3H), 3.18-3.13 (m, 1H), 3.11-3.07 (m, 1H), 3.01-2.84 (m, 3H), 2.78-2.68 (m, 2H), 2.52-2.43 (m, 3H), 2.40-2.31 (m, 1H), 2.05-1.98 (m, 1H), 1.93-1.81 (m, 1H); 13C NMR (100 MHz, d6-DMSO) δ 174.8, 170.8, 166.3, 147.4, 146.1, 130.4, 128.9 (2C), 127.3 (2C), 126.7, 126.2, 126.0, 124.9, 111.0, 52.2, 40.3, 40.1 (2C), 39.4, 31.8, 30.0, 26.6, 25.7. LC-MS Rf (min) = 5.47. HRMS (ES) calcd for C24H25NNaO5S [M + Na]: 462.1351, found: 462.1347.
N-(2-Chlorophenyl)-2-[(3-carboxy-1-oxopropyl) amino]-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (3)
Compound 3 (30 mg, 47% yield) was obtained by following the same procedure for the preparation of 2 using 11d (50 mg) and succinic anhydride (17 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 213–216 ºC. IR νmax/cm−1 (ATR) 3418 (NH), 1702 (C=O), 1682 (C=O), 1503 (C=C) and 1198 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.20 (s, 1H), 10.98 (s, 1H), 9.11 (s, 1H), 7.95 (d, J = 7.4 Hz, 1H, ArH), 7.54 (d, J = 7.4 Hz, 1H, ArH), 7.38 (m, 1H, ArH), 7.24 (m, 1H, ArH), 2.99-2.90 (m, 1H), 2.82-2.71 (m, 2H), 2.67 (t, J = 7.5 Hz, 2H), 2.54 (t, J = 7.5 Hz, 2H), 2.44-2.37 (m, 1H), 2.07-1.99 (m, 1H), 1.52-1.44 (m, 1H), 1.32-1.21 (m, 1H), 0.93 (s, 9H, t-Bu); 13C NMR (100 MHz, d6-DMSO) δ 174.1, 169.5, 164.0, 141.9, 135.3, 129.9, 129.5, 127.9, 127.6 (2C), 127.0, 126.7, 117.6, 45.0, 32.7, 31.0, 29.2, 27.6 (3C), 26.7, 25.9, 24.6. LC-MS Rf (min) = 5.89. HRMS (ES) calcd for C23H27ClN2NaO4S [M + Na] : 485.1278, found: 485.1256.
Methyl 2-[(3-carboxy-1-oxopropyl)amino]-4,5,6,7-tetra-hydrobenzo[b]thiophene-3-carboxylate (12)
Compound 12 (55 mg, 75% yield) was obtained by following the same procedure for the preparation of 2 using 11a (50 mg) and succinic anhydride (24 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 133–136 ºC. IR νmax/cm−1 (ATR) 1692 (C=O), 1555 (C=C) and 1205 (C-O). 1H NMR (400 MHz, CDCl3) δ 11.31 (s, 1H), 3.84 (s, 3H), 2.82-2.72 (m, 6H), 2.62-2.60 (m, 2H), 1.77-1.65 (m, 4H); 13C NMR (100 MHz, CDCl3,) δ 176.9, 168.3, 167.0, 147.4, 130.7, 126.9, 111.5, 51.5, 31.0, 28.8, 26.3, 24.3, 23.0, 22.8. LC-MS Rf (min) = 4.75. HRMS (ES) calcd for C14H17NNaO5S [M + Na]: 334.0725, found: 334.0724.
Methyl 2-[[(6-carboxy-3-cyclohexen-1-yl)carbonyl]amino]-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (13)
Compound 13 (52 mg, 60% yield) was obtained by following the same procedure for the preparation of 2 using 11a (50 mg) and cis-1,2,3,6-tetrahydrophthalic anhydride (43 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 161–163 ºC. IR νmax/cm−1 (ATR) 1669 (C=O), 1558 (C=C) and 1242 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.36 (s, 1H), 11.26 (s, 1H), 5.67 (s, 2H), 3.85 (s, 3H), 3.16-3.06 (m, 2H), 2.72-2.67 (m, 2H), 2.62-2.56 (m, 2H), 2.50-2.43 (m, 3H), 2.41-2.31 (m, 1H), 1.77-1.67 (m, 4H); 13C NMR (100 MHz, d6-DMSO) δ 174.7, 170.9, 166.3, 147.1, 130.8, 126.2 (2C), 124.9, 111.2, 52.1, 40.4, 40.2, 26.6, 26.3, 25.7, 24.2, 22.9, 22.7. LC-MS Rf (min) = 4.85. HRMS calcd for C18H21NNaO5S [M + Na]: 386.1038, found: 386.1028.
Methyl 2-[ (3-carboxy-1-oxopropyl)amino]-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (14)
Compound 14 (54 mg, 78%) was obtained by following the same procedure for the preparation of 2 using 11b (50 mg) and succinic anhydride (23 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 137–139 ºC. IR νmax/cm−1 (ATR) 1672 (C=O), 1537 (C=C) and 1209 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.25, 10.96, 3.81 (s, 3H), 2.98-2.90 (m, 1H), 2.74 (t, J = 7.6 Hz, 2H), 2.65-2.59 (m, 1H), 2.57 (t, J = 7.6 Hz, 2H), 2.49-2.41 (m, 2H), 2.37-2.27 (m, 1H), 1.98-1.91 (m, 1H), 1.44-1.34 (m, 1H), 1.25-1.12 (m, 1H), 0.90 (s, 9H); 13C NMR (100 MHz, d6-DMSO) δ 174.1, 170.0, 165.8, 147.0, 130.8, 127.2, 111.1, 51.5, 44.9, 32.4, 31.2, 29.1, 27.5 (3C), 27.2, 25.8, 24.4. LC-MS Rf (min) = 5.45. HRMS (ES) calcd for C18H25NNaO5S [M + Na]: 390.1351, found: 390.1332.
Methyl 2-[[(6-carboxy-3-cyclohexen-1-yl)carbonyl]amino]-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (15)
Compound 15 (56 mg, 71%) was obtained by following the same procedure for the preparation of 2 using 11b (50 mg) and cis-1,2,3,6-tetrahydrophthalic anhydride (34 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Light yellow solid. M.p. 123–126 ºC. IR νmax/cm−1 (ATR) 1695 (C=O), 1662 (C=O), 1537 (C=C) and 1235 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.32 (s, 1H), 11.12 (s, 1H), 5.71 (s, 2H), 3.89 (s, 3H), 3.16-3.05 (m, 2H), 3.01-2.92 (m, 1H), 2.68-2.60 (m, 1H), 2.50-2.41 (m, 4H), 2.40-2.29 (m, 2H), 2.01-1.93 (m, 1H), 1.46-1.35 (m, 1H), 1.27-1.13 (m, 1H), 0.91 (s, 9H); 13C NMR (100 MHz, d6-DMSO) δ 174.7, 170.9, 166.2, 147.3, 130.8, 126.8, 126.2, 124.9, 110.8, 55.4, 52.1, 44.9, 40.4, 40.2, 32.6, 27.6 (3C), 27.4, 26.6, 25.7, 24.4. LC-MS Rf (min) = 5.79. LC-MS (ES) calcd for C22H29NNaO5S [M + Na]: 442.1664, found: 442.1641.
Ethyl 2-[ (3-carboxy-1-oxopropyl)amino]-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (16)
Compound 16 (44 mg, 65%) was obtained by following the same procedure for the preparation of 2 using 11c (50 mg) and succinic anhydride (22 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Light yellow oil. IR νmax/cm−1 (ATR) 1673 (C=O), 1540 (C=C) and 1207 (C-O). 1H NMR (400 MHz, CD3OD) δ 4.35 (q, J = 7.2 Hz, 2H), 3.09-3.03 (m, 1H), 2.81-2.68 (m, 5H), 2.62-2.51 (m, 1H), 2.45-2.37 (m, 1H), 2.08-2.03 (m, 1H), 1.52-1.44 (m, 1H), 1.40 (t, J = 7.2 Hz, 3H), 1.35-1.28 (m, 1H), 0.98 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 174.8, 169.5, 166.1, 146.7, 130.7, 127.1, 111.3, 60.2, 45.0, 31.9, 30.8, 28.4, 27.3, 26.3 (3C), 25.3, 24.3, 13.3. LC-MS Rf (min) = 5.65. HRMS (ES) calcd for C19H27NNaO5S [M + Na]: 404.1508, found: 404.1502.
N-(2-Fluorophenyl)-2-[(3-carboxy-1-oxopropyl)amino]-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (17)
Compound 17 (32 mg, 51%) was obtained by following the same procedure for the preparation of 2 using 11e (50 mg) and succinic anhydride (17 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 212–215 ºC. IR νmax/cm−1 (ATR) 3423 (NH), 1687 (C=O), 1523 (C=C) and 1267 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 9.41 (brs, 1H) 7.90-7.86 (m, 1H), 7.30-7.19 (m, 3H), 2.87-2.69 (m, 3H), 2.65 (t, J = 7.7 Hz, 2H), 2.53 (t, J = 7.7 Hz, 2H), 2.44-2.35 (m, 1H), 2.04-1.96 (m, 1H), 152-1.42 (m, 1H), 1.31-1.17 (m, 1H), 0.95 (s, 9H); 13C NMR (100 MHz, d6-DMSO) δ 174.1, 169.5, 163.9, 155.2 (d, J = 241 Hz), 141.5, 129.8, 127.5, 126.4, 126.3, 125.9, 124.7 (d, J = 9 Hz), 118.1, 115.9 (d, J = 23 Hz), 45.1, 32.8, 30.9, 29.3, 27.6 (3C), 26.2, 25.8, 24.7. LC-MS Rf (min) = 5.60. HRMS (ES) calcd for C23H27FN2NaO4S [M + Na]: 469.1573, found: 469.1551.
N-(2-Chlorophenyl)-2-[[(6-carboxy-3-cyclohexen-1-yl)carbonyl]amino]-6-tert-butyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (18)
Compound 18 (45 mg, 63%) was obtained by following the same procedure for the preparation of 2 using 11d (50 mg) and cis-1,2,3,6-tetrahydrophthalic anhydride (25 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 150–153 ºC. IR νmax/cm−1 (ATR) 3419 (NH), 1706 (C=O), 1513 (C=C) and 1299 (C-O). 1H NMR (400 MHz, CDCl3) δ 12.34 (s, 1H), 8.46 (t, J = 7.9 Hz, 1H), 8.19 (m, 1H), 7.43 (t, J = 7.9 Hz, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.11 (t, J = 8.0 Hz, 1H), 5.77 (s, 2H), 3.27-3.22 (m, 1H), 3.19-3.09 (m, 2H), 2.92-2.82 (m, 1H), 2.79-2.63 (m, 3H), 2.60-2.35 (m, 3H), 2.22-2.12 (m, 1H), 1.62-1.52 (m, 1H), 1.48-1.36 (m, 1H), 0.98 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 177.0, 170.9, 164.5, 146.9, 134.5, 129.3, 129.0, 127.7, 126.8, 126.0, 125.0, 124.4, 123.4, 122.4, 114.0, 44.6, 41.2, 39.7, 32.5, 28.0, 27.3 (3C), 26.6, 26.2, 25.9, 24.7. LC-MS Rf (min) = 6.22. HRMS (ES) calcd for C27H31ClN2NaO4S [M + Na]: 537.1591, found: 537.1599.
Methyl 2-[ (3-carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (19)
Compound 19 (54 mg, 80%) was obtained by following the same procedure for the preparation of 2 using 11f (50 mg) and succinic anhydride (21 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White amorphous solid. IR νmax/cm−1 (ATR) 1686 (C=O), 1664 (C=O), 1534 (C=C) and 1216 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.26 (s, 1H), 10.98 (s, 1H), 7.33-7.22 (m, 5H), 3.84 (s, 3H), 3.30-2.85 (m, 3H), 2.77-2.69 (m, 4H), 2.58 (t, J = 7.1 Hz, 2H), 2.04-1.99 (m, 1H), 1.92-1.82 (m, 1H); 13C NMR (100 MHz, d6-DMSO) δ 173.9, 169.7, 165.7, 146.8, 146.2, 130.6, 128.8 (2C), 127.3 (2C), 126.7, 126.0, 111.3, 52.1, 40.2, 31.8, 31.2, 30.1, 29.1, 26.6. LC-MS Rf (min) = 5.17. HRMS (ES) calcd for C20H21NNaO5S [M + Na]: 410.1038, found: 410.1031.
Ethyl 2-[(3-carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (20)
Compound 20 (49 mg, 73%) was obtained by following the same procedure for the preparation of 2 using 11g (50 mg) and succinic anhydride (20 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 156–159 ºC. IR νmax/cm−1 (ATR) 1736 (C=O), 1644 (C=O), 1523 (C=C) and 1230 (C-O). 1H NMR (400 MHz, CD3OD) δ 7.33-7.28 (m, 4H), 7.24-7.19 (m, 1H), 4.37 (t, J = 6.8 Hz, 2H), 3.10-2.71 (m, 9H), 2.14-2.09 (m, 1H), 1.98-1.88 (m, 1H), 1.41 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, d6-DMSO) δ 174.0, 169.7, 165.4, 146.9, 146.2, 130.1, 128.8 (2C), 127.4 (2C), 126.7, 126.1, 111.3, 60.8, 40.5, 31.8, 31.3, 30.1, 29.1, 26.7, 14.6. LC-MS Rf (min) = 5.35. HRMS (ES) calcd for C21H23NNaO5S [M + Na]: 424.1195, found: 424.1196.
Ethyl 2-[[(6-carboxy-3-cyclohexen-1-yl)carbonyl]amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (21)
Compound 21 (53 mg, 71%) was obtained by following the same procedure for the preparation of 2 using 11g (50 mg) and cis-1,2,3,6-tetrahydrophthalic anhydride (30 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 192–194 ºC. IR νmax/cm−1 (ATR) 1703 (C=O), 1659 (C=O), 1523 (C=C) and 1245 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.33 (s, 1H), 11.29 (s, 1H), 7.33-7.30 (m, 4H), 7.26-7.20 (m, 1H), 4.31 (t, J = 7.0 Hz, 2H), 3.17-2.68 (m, 9H), 2.55-2.34 (m, 2H), 2.07-2.01 (m, 1H), 1.93-1.83 (m, 1H), 1.33 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, d6-DMSO) δ 174.7, 170.9, 165.9, 147.4, 146.2, 130.6, 128.9 (2C), 127.3 (2C), 126.7, 126.2, 126.0, 124.9, 111.1, 60.9, 40.2, 40.0 (2C), 39.6, 31.8, 30.2, 26.6, 25.7, 14.6. LC-MS Rf (min) = 5.67. HRMS (ES) calcd for C25H27NNaO5S [M + Na]: 476.1508, found: 476.1517.
Isopropyl 2-[(3-carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (22)
Compound 22 (48 mg, 73%) was obtained by following the same procedure for the preparation of 2 using 11h (50 mg) and succinic anhydride (19 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 150–153 ºC. IR νmax/cm−1 (ATR) 1685 (C=O), 1539 (C=C) and 1233 (C-O). 1H NMR (400 MHz, CD3OD) δ 7.31-7.21 (m, 5H), 5.28-5.23 (m, 1H), 3.08-2.73 (m, 9H), 2.13-2.06 (m, 1H), 1.98-1.87 (m, 1H), 1.31 (d, J = 7.2 Hz, 6H); 13C NMR (400 MHz, CD3OD) δ 174.5, 169.5, 165.5, 147.0, 145.8, 130.4, 128.1 (2C), 126.5 (2C), 126.1, 125.9, 111.6, 68.3, 40.5, 31.6, 30.8, 30.1, 28.4, 26.7, 20.9 (2C). LC-MS Rf (min) = 5.50. HRMS (ES) calcd for C22H25NNaO5S [M + Na]: 438.1351, found: 438.1336.
N-(2-Chlorophenyl) 2-[(3-carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (23)
Compound 23 (35 mg, 56%) was obtained by following the same procedure for the preparation of 2 using 11i (50 mg) and succinic anhydride (16 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. White solid. M.p. 197–199 ºC. IR νmax/cm−1 (ATR) 3436 (NH), 1710 (C=O), 1714 (C=O), 1510 (C=C) and 1204 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.26 (s, 1H), 11.06 (s, 1H), 7.95 (d, J = 7.4, 1H), 7.54 (dd, J = 8.0 1.3 Hz, 1H), 7.41-7.31 (m, 5H), 7.27-7.21 (m, 2H), 3.06-2.91 (m, 4H), 2.81-2.74 (m, 1H), 2.68 (t, J = 6.8 Hz, 2H), 2.54 (t, J = 6.8 Hz, 2H), 2.10-2.05 (m, 1H), 1.97-1.87 (m, 1H); 13C NMR (100 MHz, d6-DMSO) δ 174.0, 169.7, 163.9, 146.2, 141.8, 135.4, 129.9, 129.5, 128.9 (2C), 127.9, 127.7, 127.4 (2C), 127.0, 126.8, 126.7, 126.6, 117.9, 40.2, 32.1, 30.9, 30.2, 29.2, 26.0. LC-MS Rf (min) = 5.92. HRMS (ES) calcd for C25H23ClN2NaO4S [M + Na]: 505.0965, found: 505.0962.
Methyl 2-[(3-carboxy-1-oxo-2-propen-1-yl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (24)
Compound 24 (53 mg, 65%) was obtained by following the same procedure for the preparation of 2 using 11f (60 mg) and maleic anhydride (21 mg) in dry CH2Cl2 (7 mL). The compound was purified by trituration of the crude product with methanol. White solid. M.p. 191–194 ºC. IR νmax/cm−1 (ATR) 1710 (C=O), 1677 (C=O), 1556 (C=C) and 1218 (C-O). 1H NMR (400 MHz, CDCl3) δ 12.1 (s, 1H), 7.37-7.22 (m, 5 H), 6.55 (d, 1H, J = 16.2 Hz), 6.42 (d, 1H, J = 16.2 Hz), 3.93 (s, 3H), 3.10-2.96 (m, 3H), 2.89-2.75 (m, 2H), 2.20-2.11 (m, 1H), 2.01-1.87 (m, 1H); 13C NMR (100MHz, CDCl3) δ 167.1, 163.8, 162.0, 145.2, 144.6, 131.8, 130.3, 129.6, 128.6 (2C), 126.8 (2C), 126.6, 114.5, 77.2, 52.1, 40.3, 32.2, 29.8, 26.5. LC-MS Rf (min) = 4.95. HRMS (ES) calcd for C20H19NNaO5S [M + Na]: 408.0882, found: 408.0898.
Methyl 2-[ (3-carboxy-1-oxobutyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (25)
Compound 25 (75 mg, 89%) was obtained by following the same procedure for the preparation of 2 using 11f (60 mg) and glutanic anhydride (24 mg) in dry CH2Cl2 (7 mL). The compound was purified by trituration of the crude product with methanol. White amorphous solid. IR νmax/cm−1 (ATR) 1688 (C=O), 1665 (C=O), 1534 (C=C) and 1216 (C-O). 1H NMR (CDCl3, 400 MHz) δ 11.28 (s, 1H), 7.35-7.19 (m, 5H), 3.86 (s, 3H), 3.04-2.87 (m, 3H), 2.83-2.70 (m, 2H), 2.60 (t, J =7.4 Hz, 2H), 2.51 (t, J =7.4 Hz, 2H), 2.16-2.03 (m, 3H), 1.96-1.83 (m, 1H); 13C NMR (100 MHz, CDCl3,) δ 178.5, 169.2, 167.0, 147.8, 145.7, 130.4, 128.5 (2C), 126.9 (2C), 126.4, 126.3, 111.1, 51.5, 40.5, 35.4, 32.9, 32.1, 30.1, 26.6, 20.6. LC-MS Rf (min) = 5.30. HRMS (ES) calcd for C21H23NNaO5S [M + Na]: 424.1195, found: 424.1182.
Methyl 2-[(3-carbamoyl-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (26)
A suspended methanol solution of 19 (60 mg) in a sealed tube was bubbled with saturated NH3 for 30 min, and then the reaction mixture was subjected to microwave irradiation for 1 h at 110 ºC. After cooling and removal of solvent under reduce pressure the residue was purified by column chromatography on silica gel using MeOH:CH2Cl2 (5:95) as mobile phase to afford 26 (48.5 mg, 81%). Yellow solid. M.p. 196–199 ºC. IR νmax/cm−1 (ATR) 1689 (C=O), 1665 (C=O), 1533 (C=C) and 1216 (C-O). 1H NMR (400 MHz, CDCl3) δ 11.30 (s, 1H), 7.35-7.20 (m, 5H), 3.87 (s, 3H), 3.05-2.88 (m, 3H), 2.71-2.85 (m, 6H), 2.17-2.08 (m, 1H), 1.96-1.85 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 177.3, 168.3, 166.9, 147.7, 145.7, 130.5, 128.5 (2C), 126.9 (2C), 126.4, 126.3, 111.2, 51.5, 40.5, 32.1, 30.9, 30.0, 28.8, 26.6. LC-MS Rf (min) = 5.07. HRMS (ES) calcd for C20H22N2NaO4S [M + Na]: 409.1198, found: 409.1206.
Ethyl 2-[(2-carboxybenzoyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (27)
Compound 27 (46 mg, 62%) was obtained by following the same procedure for the preparation of 2 using 11g (50 mg) and phthalic anhydride (30 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Yellow solid. M.p. 155–158 ºC. IR νmax/cm−1 (ATR) 3422, 1665 (C=O), 1555 (C=C) and 1258 (C-O). 1H NMR (400 MHz, CD3OD) δ 8.03-8.01 (m, 1H), 7.74-7.64 (m, 3H), 7.32 (d, J = 4.4 Hz, 1H), 7.24-7.20 (m, 4H), 4.32 (q, J = 7.1 Hz, 2H), 3.16-2.80 (m, 5H), 2.17-2.11 (m, 1H), 2.02-1.91 (m, 1H), 1.36 (t, J = 7.1 Hz, 3H); 13C NMR (400 MHz, CD3OD) δ 168.0, 166.4, 166.1, 147.1, 145.8, 136.3, 131.9, 130.8, 130.5, 130.4, 130.1, 128.1 (2C), 127.4, 126.7, 126.5 (2C), 126.0, 111.0, 60.5, 40.6, 31.7, 30.0, 26.5, 13.2. LC-MS Rf (min) = 6.30. HRMS (ES) calcd for C25H23NNaO5S [M + Na]: 472.1195, found: 472.1200.
Ethyl 2-[(2-carboxy-3,4,5,6-tetrachlorobenzoyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (28)
Compound 28 (75 mg, 77%) was obtained by following the same procedure for the preparation of 2 using 11g (50 mg) and 4,5,6,7-tetrachlorophthalic anhydride (57 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Yellow solid. M.p. 178–181 ºC. IR νmax/cm−1 (ATR) 1719 (C=O), 1660 (C=O), 1517 (C=C) and 1251 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 11.72 (s, 1H), 7.37-7.29 (m, 5H), 4.34 (t, J = 6.8 Hz, 2H), 3.20-2.87 (m, 5H), 2.19-2.11 (m, 1H), 2.02-1.90 (m, 1H), 1.37 (t, J = 6.8 Hz, 3H); 13C NMR (400 MHz, d6-DMSO) δ 165.0, 164.8, 160.8, 145.9, 144.4, 134.7, 134.4, 134.2, 133.9, 131.3, 130.0, 129.4, 128.9 (2C), 127.8, 127.4 (2C), 126.8, 113.8, 61.3, 40.4, 32.2, 30.0, 26.8, 14.7. LC-MS Rf (min) = 6.34. LC-MS (ES) calcd for C25H19Cl4NNaO5S [M + Na]: 607.9636, found: 607.9601.
Ethyl 2-[(2-carboxy-3,4,5,6-tetrafluorobenzoyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (29)
Compound 29 (60 mg, 69%) was obtained by following the same procedure for the preparation of 2 using 11g (50 mg) and 4,5,6,7-tetrafluorophthalic anhydride (44 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Yellow solid. M.p. 185–187 ºC. IR νmax/cm−1 (ATR) 1717 (C=O), 1674 (C=O), 1558 (C=C) and 1250 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 11.96 (s, 1H), 7.37-7.20 (m, 5H), 4.38 (t, J = 6.8 Hz, 2H), 3.20-2.80 (m, 5H), 2.19-2.08 (m, 1H), 2.07-1.90 (m, 1H), 1.38 (t, J = 6.8 Hz, 3H); 13C NMR (400 MHz, d6-DMSO) δ 165.1, 162.8, 157.9 (4C), 146.0, 144.8, 131.1, 128.5 (3C), 127.7, 127.4 (2C), 126.8, 118.9 (2C), 113.6, 61.0, 40.6, 31.7, 30.0, 26.8, 14.4. LC-MS Rf (min) = 5.97. HRMS (ES) calcd for C25H19F4NNaO5S [M + Na]: 544.0818, found: 544.0824.
2-[(3-Carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (30)
Compound 30 (54 mg, 77%) was obtained by following the same procedure for the preparation of 2 using 11j (50 mg) and succinic anhydride (24 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Brown solid. M.p. 205–208 ºC. IR νmax/cm−1 (ATR) 2213 (C≡N), 1715 (C=O), 1683 (C=O), 1552 (C=C) and 1168 (C-O). 1H NMR (400 MHz, d3-AcOD) δ 10.67 (s, 1H), 7.36-7.20 (m, 5H), 3.15-2.74 (m, 9H), 2.15-2.06 (m, 1H), 2.01-1.91 (m, 1H); 13C NMR (400 MHz, d3-AcOD) δ 172.9, 169.6, 147.2, 145.6, 130.5, 128.5 (2C), 127.0, 126.9 (2C), 126.4, 113.6, 92.5, 40.8, 31.3, 30.1, 29.4, 28.1, 24.1. LC-MS Rf (min) = 4.90. HRMS (ES) calcd for C19H18N2NaO3S [M + Na]: 377.0936, found: 377.0916.
2-[(3-Carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (31)
Compound 31 (53 mg, 78%) was obtained by following the same procedure for the preparation of 2 using 11k (50 mg) and succinic anhydride (22 mg) in dry CH2Cl2 (7 mL). The product was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase. Brown solid. M.p. 204–207 ºC. IR νmax/cm−1 (ATR) 1712 (C=O), 1683 (C=O), 1537 (C=C) and 1167 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.21 (brs, 1H), 11.60 (s, 1H), 7.33 (d, J = 4.3 Hz, 1H) 7.25-7.21 (m, 4H), 3.01-2.70 (m, 5H), 2.66 (t, J = 6.2 Hz, 2H), 2.55 (t, J = 6.2 Hz, 2H), 2.05-2.01 (m, 1H), 1.94-1.84 (m, 1H); 13C NMR (400 MHz, d6-DMSO) δ 173.9, 169.0, 167.7, 146.2, 142.6, 129.1, 128.9 (2C), 127.4 (2C), 126.7, 125.9, 116.4, 40.2, 32.2, 31.1, 30.1, 29.2, 26.0. LC-MS Rf (min) = 4.59. HRMS (ES) calcd for C19H20N2NaO4S [M + Na]: 395.1041, found: 395.1011.
2-[(3-carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylic acid (32)
To a solution of compound 19 (50 mg) in 8 mL of solvent mixture (MeOH/THF = 3:1, 9 mL) was added a solution of NaOH (20 mg) in water (5 mL) and the reaction mixture was stirred at 80 ºC for 6 h. The reaction was cooled to room temperature and the organic solvents were removed in vacuo. The aqueous layer was acidified with 5% HCl and extracted with EtOAc. The organic layer was separated, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase to afford 32 (31 mg, 65%). White solid. M.p. 214–217 ºC. IR νmax/cm−1 (ATR) 1699 (C=O), 1674 (C=O), 1534 (C=C) and 1220 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.70 (s, 1H), 11.31 (s, 1H), 7.33-7.22 (m, 5H), 3.03-2.83 (m, 3H), 2.77-2.67 (m, 4H), 2.57 (t, J = 7.2 Hz, 2H), 2.04-1.97 (m, 1H), 1.93-1.82 (m, 1H); 13C NMR (400 MHz, d6-DMSO) δ 174.0, 169.5, 167.4, 146.9, 146.3, 131.1, 128.9 (2C), 127.3 (2C), 126.7, 125.5, 112.1, 40.3, 31.8, 32.3, 30.1, 29.1, 26.7. LC-MS Rf (min) = 4.89. HRMS (ES) calcd for C19H19NNaO5S [M + H]: 396.0882, found: 396.0864.
Ethyl 2-(acetylamino)-6-phenyl-4,5,6,7-tetrahydro-benzo[b]thiophene-3-carboxylate (33)
To an ice-cold solution of compound 11g (60 mg) and pyridine (18 μL) in dry CH2Cl2 (5 mL) was added dropwise a solution of acetyl chloride (16.0 μL in 2 mL CH2Cl2). After stirring at RT for 2 h, the reaction was quenched by adding 5 mL of water. The organic layer was separated, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by reversed phase HPLC using MeCN:H2O (gradient 30% → 100%) as mobile phase to afford 33 (52 mg, 76%). White amorphous solid. IR νmax/cm−1 (ATR) 1681 (C=O), 1536 (C=C) and 1233 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 10.98 (s, 1H), 7.31-7.29 (m, 4H), 7.24-7.19 (m, 1H), 4.30 (q, J = 7.1 Hz, 2H), 2.99-2.83 (m, 3H), 2.79-2.66 (m, 2H), 2.23 (s, 3H), 2.05–2.98 (m, 1H), 1.94-1.82 (m, 1H), 1.32 (t, J = 7.1 Hz, 3H); 13C NMR (400 MHz, d6-DMSO) δ 167.9, 165.5, 147.0, 145.8, 130.5, 128.7 (2C), 127.3 (2C), 126.8, 126.1, 111.2, 61.0, 40.6, 31.9, 30.0, 26.5, 23.9, 14.7. LC-MS Rf (min) = 6.24. HRMS (ES) calcd for C19H21NNaO3S [M + Na]: 366.1140, found: 366.1127.
Methyl 2-[ (2-carboxy-1-oxomethyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (34)
To an ice-cold solution of tert-butanol (148 mg) in dry ether (5 mL) was added dropwise an ice-cold solution of oxalyl chloride (0.17 mL) in dry ether (5 mL). The reaction mixture was stirred at 0 ºC for 20 min and then at RT overnight. Removal of solvent under vacuum gave tert-butyl 2-chloro-2-oxoacetate, which was used for the next step without purification. To an ice-cold solution of compound 11f (60 mg) and TEA (58 μL) in dry CH2Cl2 (5 mL) was added dropwise a solution of tert-butyl 2-chloro-2-oxoacetate obtained above in CH2Cl2 (5 mL). The mixture was stirred at 0 ºC for 5 min, diluted with CH2Cl2 and then quenched with a saturated solution of NaHCO3 (10 mL). The organic layer was separated, dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using CH2Cl2:MeOH (99:1) as mobile phase to yield the tert-butyl ester of 34 (66 mg, 76%). Subsequently, cleavage of the tert-butyl ester group of the precursor (63 mg) with TFA (2 mL) in CH2Cl2 (6 mL) at RT for 21 h gave the desired product 34 (33 mg, 60%), which was purified by trituration of the crude product with methanol. White solid. M.p. 185–188 ºC. IR νmax/cm−1 (ATR) 1765 (C=O), 1675 (C=O), 1560 (C=C) and 1171 (C-O). 1H NMR (400 MHz, CDCl3) δ 12.5 (s, 1H), 7.35-7.20 (m, 5H), 3.93 (s, 3H), 3.11-2.93 (m, 3H), 2.87-2.74 (m, 2H), 2.19-2.11 (m, 1H), 1.98-1.87 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 165.9, 158.5, 153.8, 145.3, 144.1, 132.1, 129.3, 128.6 (2C), 126.8 (2C), 126.5, 115.0, 52.0, 40.4, 32.3, 29.8, 26.5. LC-MS Rf (min) = 4.75. HRMS (ES) calcd for C18H17NNaO5S [M + Na]: 382.0725, found: 382.0743.
Methyl 2-[[[[(4-methylphenyl)sulfonyl]amino]carbo-nyl]amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (35)
To a solution of compound 11f (30 mg) and TEA (29 μL) in dry toluene (5 mL) p-toluenesulfonyl isocyanate (16 μL) was added, and the reaction mixture was heated at 110 ºC in an oil bath for 20 h. After cooling and removal of solvent under reduced pressure the residue was triturated with methanol to afford compound 35 (38 mg, 75%). White solid. M.p. 175–178 ºC. IR νmax/cm−1 (ATR) 1668 (C=O), 1535 (C=C) and 1155 (C-O). 1H NMR (400 MHz, CDCl3) δ 11.64 (s, 1H), 8.07 (s, 1H), 7.92 (s, 1H), 7.90 (s, 1H), 7.35-7.28 (m, 4H), 7.25-7.19 (m, 2 H), 3.96 (s, 3H), 3.09-2.85 (m, 3H), 2.85-2.69 (m, 2H), 2.42 (s, 3H), 2.18-2.09 (m, 1H), 1.95-1.84 (m, 1H); 13C NMR (400 MHz, CDCl3) δ 165.9, 148.0, 146.9, 145.6, 145.2, 136.1, 131.3, 130.1 (2C), 128.5 (2C), 127.4 (2C), 126.8 (2C), 126.7, 126.4, 112.2, 51.8, 40.4, 32.0, 30.0, 26.6, 21.6. LC-MS Rf (min) = 5.47. HRMS (ES) calcd for C24H24N2NaO5S2 [M + Na]: 507.1024, found: 507.1036.
N-Isopropyl 2-[ (3-carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (36)
Compound 36 (13 mg, 47%) was obtained by following the same procedure for the preparation of 2 using the Boc-deprotected product of 11m, which was prepared by treatment of 11m (28 mg) with TFA in dry CH2Cl2 at rt for 1 h, and succinic anhydride (8.0 mg) in dry CH2Cl2 (7 mL). The product was purified by column chromatography on silica gel using CH2Cl2:MeOH (98:2) as mobile phase. Yellow oil. IR νmax/cm−1 (ATR) 1709 (C=O), 1681 (C=O), 1519 (C=C) and 1213 (C-O). 1H NMR (400 MHz, CDCl3) δ 12.01 (s, 1H), 7.30-7.13 (m, 5H), 5.72 (brs, 1H), 4.20-4.09 (m, 1H), 3.01-2.85 (m, 2H), 2.80-2.65 (m, 7H), 2.18-2.09 (m, 1H), 1.98-1.86 (m, 1H) 1.20 (t, J = 7.2 Hz, 6H); 13C NMR (400 MHz, CDCl3) δ 176.2. 168.6, 165.4, 145.1, 128.9, 128.6 (2C), 127.4, 126.8 (2C), 126.6, 126.5, 113.9, 41.7, 40.1, 32.0, 31.0, 30.1, 28.9, 26.7, 22.9 (2C). LC-MS Rf (min) = 5.24. HRMS (ES) calcd for C22H26N2NaO4S [M + Na]: 437.1511, found: 437.1526.
[2-[(3-Carboxy-1-oxopropyl)amino]-4,5,6,7-tetrahydrobenzo[b]thien-3-yl]-4-morpholinyl-methanone (37)
Compound 37 (22 mg, 50%) was obtained by following the same procedure for the preparation of 2 using the Boc-deprotected product of 11n, which was prepared by treatment of 11n (44 mg) with TFA in CH2Cl2 at rt for 1 h, and succinic anhydride (12 mg) in dry CH2Cl2 (7 mL). The product was purified by column chromatography on silica gel using CH2Cl2:MeOH (98:2) as mobile phase. Colorless oil. IR νmax/cm−1 (ATR) 1705 (C=O), 1678 (C=O), 1541 (C=C) and 1111 (C-O). 1H NMR (400 MHz, CDCl3) δ 9.51 (s, 1H), 7.35-7.18 (m, 5H), 3.74-3.42 (m, 8H), 3.06-2.91(m, 2H), 2.82-2.74 (m, 4H), 2.68-2.58 (m, 1H), 2.57-2.46 (m, 2H), 2.15-2.05 (m, 1H), 1.99-1.85 (m, 1H); 13C NMR δ (400 MHz, CDCl3) 176.8, 168.9, 167.4, 145.4, 136.6, 128.9, 128.5 (3C), 128.2, 126.8 (2C), 126.5, 66.9 (2C), 51.9, 45.0, 40.8, 31.9, 30.7, 29.7, 28.7, 24.8. LC-MS Rf (min) = 5.14. LC-MS (ES) calcd for C23H26N2NaO5S [M + Na]: 465.1460, found: 465.1479.
N-Phenyl 2-[(3-carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (38)
Compound 38 (22 mg, 52%) was obtained by following the same procedure for the preparation of 2 using the Boc-deprotected product of 11o, which was prepared by treatment of 11o (42 mg) with TFA in dry CH2Cl2 for 1 h at rt, and succinic anhydride (11 mg) in dry CH2Cl2 (7 mL). The product was purified by column chromatography on silica gel using CH2Cl2:MeOH (98:2) as mobile phase. Yellow solid. M.p. 191–194ºC. IR νmax/cm−1 (ATR) 1719 (C=O), 1666 (C=O), 1521 (C=C) and 1201 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.14 (brs, 1 H), 10.76 (brs, 1 H), 9.94 (s, 1H), 7.72 (s, 1 H), 7.70 (s, 1 H), 7.37-7.29 (m, 6H), 7.26-7.19 (m, 1H), 7.10-7.04 (t, J = 7.38 Hz, 1H), 3.02-2.70 (m, 5H), 2.68-2.60 (m, 2H), 2.57-2.50 (m, 2H), 2.05-1.97 (m, 1H), 1.93-1.81 (m, 1H); 13C NMR (400 MHz, d6-DMSO) δ 174.1, 169.6, 163.5, 146.2, 139.6, 129.8, 129.0 (2C), 128.8 (2C), 127.3 (2C), 126.7, 126.5, 126.3, 123.8, 120.4 (2C), 120.0, 40.7, 32.1, 30.7, 30.1, 29.3, 25.4. LC-MS Rf (min) = 5.35. HRMS (ES) calcd for C25H24N2NaO4S [M + Na]: 471.1354, found: 471.1342.
N-Benzyl 2-[(3-carboxy-1-oxopropyl)amino]-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (39)
Compound 39 (31.5 mg, 49%) was obtained by following the same procedure for the preparation of 2 using the Boc-deprotected product of 11p, which was prepared by treatment of 11p (64 mg) with TFA in CH2Cl2 at rt for 1 h, and succinic anhydride (17 mg) in dry CH2Cl2 (7 mL). The product was purified by column chromatography on silica gel using CH2Cl2: MeOH (99:2) as mobile phase. White solid. M.p. 179–183 ºC IR νmax/cm−1 (ATR) 1726 (C=O), 1681 (C=O), 1518 (C=C) and 1161 (C-O). 1H NMR (400 MHz, d6-DMSO) δ 12.21 (brs, 1 H), 11.24 (brs, 1H), 8.09 (t, J = 5.8 Hz, 1H), 7.37-7.28 (m, 7H), 7.28-7.17 (m, 2H), 4.56-4.39 (m, 2H), 3.01-2.70 (m, 5H), 2.66-2.60 (m, 2H), 2.55-2.51 (m, 2H), 2.05-1.97 (m, 1H), 1.95-1.81 (m, 1H); 13C NMR (400 MHz, d6-DMSO) δ 174.0, 169.3, 165.4, 146.1, 141.2, 139.9, 129.1, 128.3 (2C), 128.7(2C), 127.6 (2C), 127.3 (2C), 127.1, 126.7, 126.3, 117.4, 43.0, 40.4, 32.0, 31.0, 30.1, 29.2, 25.8. LC-MS Rf (min) = 5.89. HRMS (ES) calcd for C26H26N2NaO4S [M + Na]: 485.1511, found: 485.1505.
Supplementary Material
Acknowledgments
We are grateful for financial support from the Swedish Research Council, the Knut and Alice Wallenberg Foundation, VINNOVA and the Kempe Foundation (SJCKMS). The authors thank Lianpao Wu for synthesizing several analogs used in this study. SJH is supported from NIH grants AI048689 and AI029549.
Footnotes
Electronic supplementary information (ESI) available: Effects of compounds (2, 3, and 12–39) on bacterial growth, antibiofilm activity of compounds 24–39, and 1H and 13C NMR spectra of all synthesized compounds. See DOI:
References and notes
- 1.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. Nat Rev Microbiol. 2008;6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Travis J, Potempa J. Biochim Biophys Acta. 2000;1477:35–50. doi: 10.1016/s0167-4838(99)00278-2. [DOI] [PubMed] [Google Scholar]
- 3.Clatworthy AE, Pierson E, Hung DT. Nat Chem Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 4.Barczak AK, Hung DT. Curr Opin Microbiol. 2009;12:490–496. doi: 10.1016/j.mib.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finlay BB, Falkow S. Microbiol Mol Biol Rev. 1997;61:136–169. doi: 10.1128/mmbr.61.2.136-169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee YM, Almqvist F, Hultgren SJ. Curr Opin Pharmacol. 2003;3:513–519. doi: 10.1016/j.coph.2003.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Marra A. Expert Rev Anti Infect Ther. 2004;2:61–72. doi: 10.1586/14787210.2.1.61. [DOI] [PubMed] [Google Scholar]
- 8.Ejrnaes K, Stegger M, Reisner A, Ferry S, Monsen T, Holm SE, Lundgren B, Frimodt-Moller N. Virulence. 2011;2:528–537. doi: 10.4161/viru.2.6.18189. [DOI] [PubMed] [Google Scholar]
- 9.Dodson KW, Pinkner JS, Rose T, Magnusson G, Hultgren SJ, Waksman G. Cell. 2001;105:733–743. doi: 10.1016/s0092-8674(01)00388-9. [DOI] [PubMed] [Google Scholar]
- 10.Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ. Science. 1998;282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 11.Roberts JA, Marklund BI, Ilver D, Haslam D, Kaack MB, Baskin G, Louis M, Möllby R, Winberg J, Normark S. Proc Natl Acad Sci U S A. 1994;91:11889–11893. doi: 10.1073/pnas.91.25.11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bullitt E, Makowski L. Nature. 1995;373:164–167. doi: 10.1038/373164a0. [DOI] [PubMed] [Google Scholar]
- 13.Hahn E, Wild P, Hermanns U, Sebbel P, Glockshuber R, Häner M, Taschner N, Burkhard P, Aebi U, Müller SA. J Mol Biol. 2002;323:845–857. doi: 10.1016/s0022-2836(02)01005-7. [DOI] [PubMed] [Google Scholar]
- 14.Jones CH, Pinkner JS, Roth R, Heuser J, Nicholes AV, Abraham SN, Hultgren SJ. Proc Natl Acad Sci U S A. 1995;92:2081–2085. doi: 10.1073/pnas.92.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geibel S, Procko E, Hultgren SJ, Baker D, Waksman G. Nature. 2013;496:243–246. doi: 10.1038/nature12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ. Science. 1998;282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 17.Mulvey MA, Schilling JD, Hultgren SJ. Infect Immun. 2001;69:4572–4579. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schilling JD, Lorenz RG, Hultgren SJ. Infect Immun. 2002;70:7042–7049. doi: 10.1128/IAI.70.12.7042-7049.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Science. 2003;301:105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 20.Pratt LA, Kolter R. Mol Microbiol. 1998;30:285–293. doi: 10.1046/j.1365-2958.1998.01061.x. [DOI] [PubMed] [Google Scholar]
- 21.Reisner A, Haagensen JA, Schembri MA, Zechner EL, Molin S. Mol Microbiol. 2003;48:933–946. doi: 10.1046/j.1365-2958.2003.03490.x. [DOI] [PubMed] [Google Scholar]
- 22.Litwin MS, Saigal CS, Yano EM, Avila C, Geschwind SA, Hanley JM, Joyce GF, Madison R, Pace J, Polich SM, Wang M. J Urol. 2005;173:933–937. doi: 10.1097/01.ju.0000152365.43125.3b. [DOI] [PubMed] [Google Scholar]
- 23.Aberg V, Almqvist F. Org Biomol Chem. 2007;5:1827–1834. doi: 10.1039/b702397a. [DOI] [PubMed] [Google Scholar]
- 24.Pinkner JS, Remaut H, Buelens F, Miller E, Åberg V, Pemberton N, Hedenström M, Larsson A, Seed P, Waksman G, Hultgren SJ, Almqvist F. Proc Natl Acad Sci USA. 2006;103:17897–17902. doi: 10.1073/pnas.0606795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Aberg V, Walker JN, Seed PC, Almqvist F, Chapman MR, Hultgren SJ. Nat Chem Biol. 2009;5:913–919. doi: 10.1038/nchembio.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gewald K, Schinke E, Bottcher H. Chem Ber. 1966;99:94–100. [Google Scholar]
- 27.Huang Y, Domling A. Mol Divers. 2011;15:3–33. doi: 10.1007/s11030-010-9229-6. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Conklin D, Pan HL, Eisenach JC. J Pharmacol Exp Ther. 2003;305:950–955. doi: 10.1124/jpet.102.047951. [DOI] [PubMed] [Google Scholar]
- 29.147939A1. WO Pat. 2008
- 30.Mudumbi RV, Montamat SC, Bruns RF, Vestal RE. Am J Physiol. 1993;264:1017–1022. doi: 10.1152/ajpheart.1993.264.3.H1017. [DOI] [PubMed] [Google Scholar]
- 31.Figler H, Olsson RA, Linden J. Mol Pharmacol. 2003;64:1557–1564. doi: 10.1124/mol.64.6.1557. [DOI] [PubMed] [Google Scholar]
- 32.Walburger A, Koul A, Ferrari G, Nguyen L, Prescianotto-Baschong C, Huygen K, Klebl B, Thompson C, Bacher G, Pieters J. Science. 2004;304:1800–1804. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- 33.Podolin PL, Callahan JF, Bolognese BJ, Li YH, Carlson K, Davis TG, Mellor GW, Evans C, Roshak AK. J Pharmacol Exp Ther. 2005;312:373–381. doi: 10.1124/jpet.104.074484. [DOI] [PubMed] [Google Scholar]
- 34.O’Toole GA, Kolter R. Mol Microbiol. 1998;28:449– 461. doi: 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- 35.Chorell E, Pinkner JS, Phan G, Edvinsson S, Buelens F, Remaut H, Waksman G, Hultgren SJ, Almqvist F. J Med Chem. 2010;53:5690–5695. doi: 10.1021/jm100470t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang W, Li J, Tang J, Liu H, Shen J, Jiang H. Synth Commun. 2005;35:1351–1357. [Google Scholar]
- 37.Aurelio L, Flynn BL, Scammells PJ. Org Biomol Chem. 2011;9:4886–4902. doi: 10.1039/c0ob01156h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.