

Abstract
Integrin-linked kinase (ILK) is a multidomain focal adhesion protein implicated in signal transduction between integrins and growth factor/extracellular receptors. We have previously shown that ILK expression is increased in liver fibrosis and that ILK appears to be a key regulator of fibrogenesis in rat hepatic stellate cells, effectors of the fibrogenic response. Here we hypothesized that the mechanism by which ILK mediates the fibrogenic phenotype is by engaging the small GTPase, Rho in a signal transduction pathway linked to fibrogenesis.
Methods
ILK function in quiescent (non fibrogenic) and activated (fibrogenic) stellate cells was examined in cells isolated from rat livers. ILK, Rho, and Gα12/13 signaling were manipulated using established chemical agents or specific adenoviral constructs.
Results
ILK activity was minimal in quiescent stellate cells, but prominent in activated stellate cells; inhibition of ILK activity had no effect in quiescent cells, but had prominent effects in activated cells. Overexpression of ILK in activated stellate cells increased Rho activity, but had no effect in quiescent cells. Further, endothelin-1 (ET-1) stimulated Rho activity in activated stellate cells, but not in quiescent cells. Rho, RhoGEF and Gα12/13 expression were increased after stellate cell activation. Inhibition of Gα12/13 signaling, by expression of the RGS domain of the p115-Rho- specific guanine nucleotide exchange factor (p115-RGS) in activated stellate cells, significantly inhibited type I collagen and smooth muscle α actin expression, both classically upregulated after stellate cell activation. The data suggest that ILK mediates Rho dependent functional effects in activated stellate cells, and raise the possibility that ILK is important in cross talk with the GPCR system.
Introduction
It is well established that activated hepatic stellate cells play an important role in the development of liver fibrosis/cirrhosis (1–4). In the normal liver, stellate cells exhibit a quiescent phenotype, but after liver injury, they undergo differentiation into myofibroblast-like cells with subsequent proliferation, synthesis of extracellular matrix, and de novo expression of smooth muscle α-actin (4, 5). Upregulation of smooth muscle α-actin in particular, appears to have a number of important functional consequences, including in cytoskeletal maintenance, cellular contraction, and cell motility.
The family of Rho GTPases play an important role in the cellular cytoskeleton, and therefore are essential components of important cellular processes such as contractility and motility. RhoA, a prominent GTPase, promotes formation of actin structures such as stress fibers, while Cdc42 and Rac1 family members form filopodia and lamellipodia, respectively (6). Further, there appears to be a link between Rho family GTPases and the extracellular matrix, via integrin binding proteins (7, 8). RhoA may also be activated in response to GPCR ligands (9–11), and appears to be coupled to the heterotrimeric G12/13 proteins (12). This response is catalyzed by guanine nucleotide exchange factors (GEFs), several of which are direct targets (effectors) of G12/13 alpha subunits (13, 14).
A key signaling component downstream of integrin engagement is integrin-linked kinase (ILK) (15, 16). ILK is a PI3-kinase-dependent, serine/threonine protein kinase that interacts with the cytoplasmic domains of both β1 and β3 integrins, possesses bonafide kinase activity, and regulates diverse signaling pathways (17–19). This modular protein, consisting of four amino-terminal ankyrin repeats followed by a pleckstrin homology (PH)-like domain and a protein kinase catalytic domain near its carboxyl-terminus (20, 21), plays an important functional role in cell motility and other cellular processes (22).
Given that integrin signaling depends on the effects of RhoA on the actin cytoskeleton, and that ILK is likewise important in activation of RhoA and the actin cytoskeleton, we have hypothesized here that ILK serves as a critical upstream regulator of Rho GTPases in a primary rat stellate cells, and that it is likely to be inactive in quiescent cells, but active in activated cells. We have also postulated that ILK is important in cross talk with the G protein coupled receptor (GPCR) system, and moreover that it has effects in stellate cells that are likely to be related to their function during activation. In the present study, we have demonstrated that ILK is differentially active in quiescent and activated stellate cells, and moreover that it appears to at least in part serve as a regulator of Rho GTPase activation in this cell type. We have also characterized a downstream ILK signaling pathway that includes the GPCR system, which mediates critical functional stellate cell attributes.
Materials and Methods
Materials
Anti-Gα12, -Gα13 and -RhoGEF antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); monoclonal anti-ILK antibody was purchased from BD Transduction Laboratories (Lexington, KY). Endothelin-1 (ET-1) was from American Peptide Co. Inc. (Sunnyvale, CA). Recombinant human PDGF-BB was from R&D Systems (Minneapolis, MN), polyclonal phospho-Akt (Ser-473), total-Akt, and GSK3β antibodies were from Cell Signaling (Beverly, MA). Anti-rabbit IgG/horseradish peroxidase conjugate or anti-mouse IgG/horseradish peroxidase conjugate were from Promega (Madison, WI). Anti-β-actin, and anti-smooth muscle α-actinantibodies were from Sigma (St. Louis, MO). The G-LISA Rho and Rac1 activation assay kits were from Cytoskeleton (Denver, CO). QLT was a gift of QLT, Inc. (Vancouver, BC, Canada) and Y-27632 was purchased from Sigma (St. Louis, MO).
Cell Isolation and Culture
Hepatic stellate cells were isolated from male Sprague-Dawley rats (450 to 500g, Harlan, Indianapolis, IN) as previously described (23, 24). In brief, after in situ perfusion of the liver with 0.20 mg/100 mL of pronase (Roche Molecular Biochemicals, Indianapolis, IN), followed by 0.013 mg/100 mLof collagenase (Crescent Chemical, Hauppauge, NY), dispersed cell suspensions were layered on a discontinuous density gradient of 8.2% and 15.6% accudenz (Accurate Chemical and Scientific, Westbury, NY); the resulting upper layer consists of more than 95% stellate cells, confirmed by microscopic analysis. Cells were suspended in modified medium 199, containing 20% serum (10% horse serum and 10% calf serum; Life Technologies, Inc., Gaithersburg, MD) at a densityof 1×106 cells/mL. Cultures were incubated in a humidified incubator containing 95% O2/2.5% CO2. Cell viability was greater than 95% in all of the cultures used for study.
Immunoblotting
Cell lysates were prepared in a buffer containing 1% Triton X-100, 150 mM NaCl, 20 mM Tris, pH 7.5, 1mM EDTA, 50 mM NaF, 50 mM sodium-2-glycerophosphate, 0.05 mM Na3VO4, 10 mg/mL leupeptin, 10% glycerol, and 100 mM phenylmethylsulfonyl fluoride. Samples containing 50 mg of total protein were subjected to SDS-PAGE, after which proteins were transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). Membranes were incubated for 1 h at room temperature in blocking buffer (10 mM sodium phosphate, 0.5MNaCl, 0.05% Tween 20, and 2.5% dry milk) and then with primary antibody (1:1000) overnight at 4 °C. Membranes were then washed of excess primary antibody at room temperature in a phosphate-buffered saline Tween buffer (TBST: 10 mM 0.05%, Tris pH 8, 0.9% sodium chloride, Tween 20 0.05%) and incubated for 1 h at room temperature with secondary antibody. After washing, specific signals were visualized using enhanced chemiluminescence detection pursuant to the manufacturer’s instructions (Pierce). Specific bands were scanned and data collected over a narrow range of x-ray film (Eastman Kodak Co., Rochester, NY) linearity and quantitated by scanning densitometry.
Rho and Rac assays
The G-LISA Rho activation assay is an ELISA based system that measures the level of Rho activation, which was used per the manufacturer’s instructions (Cytoskeleton, Denver, CO). In brief, the active GTP bound form of Rho is captured on a substrate and then detected by incubation with specific Rho primary antibody followed by a secondary antibody conjugated to HRP. Controls include both excluded and non-activated lysates.
Active Rac was determined with a pull-down assay according to the manufacturer’s instructions (Cytoskeleton, Denver, CO). Briefly, stellate cells were stimulated and then collected in 300 μL of lysis buffer (50 mM Tris, pH 7.5, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mM NaCl, and 10 mM MgCl2) and centrifugedto remove cellular debris. The supernatant was then mixed with PAK-PBD beads and rotated at 4 °C for 45 min. The beads were then centrifuged, washed multiple times, and the proteins were eluted in SDS sample buffer and separated on a 12% SDS-PAGE gel. After transfer to nitrocellulose membranes, immunoblot analysis was performed with Rac1 antibody and specific signals were detected by immunoblotting as above.
Cell Migration Assay
Cell migration was assessed by measuring the repair of a linear wound generated in the confluent monolayer of cells. Hepatic stellate cells were isolated, plated at an equivalent density on chamber slides (Lab-Tek, Westmont, IL), and allowed to undergo activation for 7 days. They were then infected with constructs (all at a multiplicity of infection of 100) or went untreated for 24 h. Some cells were exposed to Y-27632 (10 μM) for 60 min prior to the application of a linear scratch in cell monolayers using a sterile plastic pipette tip. Cell migration was recorded for 24 h using an Applied Precision deconvolution microscope (UT Southwestern imaging center).
Cell Adhesion Assay
Hepatic stellate cells were isolated, plated at an equivalent density, and allowed to undergo culture-induced activation for 5 days. They were then infected with Ad-GFP and Rho A (V14) (at a multiplicity of infection of 100) or went untreated. Forty-eight hours after viral infection, the cells were detached from the culture dishes with trypsin. Some cells were exposed to Y-27632 (10 μM) for 60 min. The detached cells were suspended in Dulbecco’s modified Eagle’s medium, 20% serum was added, and the cells were then replated on fresh tissue culture dishes. Two hours after replating, the cells were washed twice with PBS to remove unattached cells, trypsinized, and the adherent cells were counted.
Real-Time PCR
Total RNA was extracted with TRIzol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). One microgram of RNA was reverse-transcribed using an oligo (dt) primer and Superscript RNase H-reverse transcriptase as per the manufacturer’s directions (Invitrogen, Carlsbad, CA). Amplification reactions were performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). Primer sequences were as follows: GAPDH - forward, 5′-ATT GAC CAC TAC CTG GGC AA -3′ and reverse, 5′-GAG ATA CAC TTC AAC ACT TTG ACC T -3′; collagen I a1 forward, 5′-GAG TGA GGC CAC GCA TGA 3′; and reverse, 5′-AGC CGG AGG TCC ACA AAG -3′; smooth muscle a-actin forward, 5′-CCG AGA TCT CAC CGA CTA CC -3′ and reverse, 5′-TCC AGA GCG ACA TAG CAC AG -3′; ET-1 forward, 5′-GCT CGG AGT TCT TTG TCT GC -3′; and reverse, 5′-ACT TCT GCC ACC TGG ACA TC -3′; Rho GEF forward, 5′-ATA CCC AGG CTT CCC TTC CG -3′; and reverse, 5′-GCC GCT GGT AAT CCT TGA GC -3′; Rho GAP forward, 5′-CCA CTA TCG AGA CAT TGC GC -3′; and reverse, 5′-CGC TGT TCA CAG GTT GTA AAG G -3′;Rho GDI forward, 5′-GAA GGA AGG TGT GGA GTA CCG --3′; and reverse, 5′-GCC TGA CAC GAT CTC TCT GTT CA --3′. Five ml of diluted cDNA samples (1:5 dilution) were used in a quantitative two-step PCR (a 10-min step at 95 °C followed by 50 cycles of 15 s of 95 °C and 1 s at 65 °C) in the presence of 400 nM specific forward and reverse primers and SYBR Green PCR Master Mix. Each sample was analyzed in triplicate using an ABI system (7900HT Fast Real-Time PCR). As negative controls, water was used as a template for each reaction.
Adenoviral Gene Transfer
Stellate cells were infected with constitutively active Rho (V14), a dominant negative Rho (N19) (each kindly provided by Dr. Aviv Hassid, University of Tennessee), p115-RGS (a gift of Dr. Patrick Casey, Duke University Medical Center), or a matched adenovirus containing GFP as a control, at multiplicity infection of (MOI) of 100. Adenoviral constructs encoding ILK and a short hairpin inhibiting ILK were as previously described (25). The infection efficiency of the adenovirus was monitored by the expression of green fluorescent protein (GFP) and typically reached 80–90% within 48 h. Viral titers were measured by standard plaque assay using 293 cells.
Immunohistochemistry
Cells were cultured as above, washed, and fixed and permeabilized with 2% paraformaldehyde and 0.5% Triton X-100 in PBS for 10 min. For detection of ILK, cells were exposed to monoclonal anti-ILK antibody (1:1,000) in PBS for 1 hour at RT. After washing, primary antibody was detected with Alexa Fluor- 488 (Molecular Probes)-conjugated anti-mouse and Dapi (Alexis Corp., San Diego, CA) to identify nuclei. After washing and mounting, signals were visualized with a Zeiss LSM 510 META confocal microscope. A solution of bovine serum albumin/PBS at 3% and a non-immune mouse IgG were employed as a negative control. For detection of actin, cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 followed by labeling with AL-488-Phalloidin (Molecular Probes). After washing and mounting, signals were visualized as described above.
Statistical Analysis
Data are expressed as means ± SEM. Statistical analysis was performed by using an independent Student t test or 1-way analysis of variance with the Tukey post hoc test when appropriate. A P value less than 0.05 was considered to be statistically significant.
Results
ILK kinase activity in activated and quiescent stellate cells
We have hypothesized in the current study that there is an intrinsic difference in ILK kinase activity in normal (quiescent) and activated stellate cells, consistent with an inherent divergent biology in the 2 states of cell differentiation. We initially found that ILK expression appeared to be remarkably different in quiescent and activated cells (Figure 1a/b) and that it increased progressively during classic cell culture induced stellate cell activation (26), suggesting the possibility of differential signaling and function in these two states of activation. To further explore the possible differential activity of ILK in quiescent and activated cells, we used two methods. In initial experiments, we used a second-generation ILK inhibitor, known as QLT-0267 (Figure 1), which specifically inhibits the kinase activity of ILK (27). We found that inhibition of ILK kinase activity in activated stellate cells caused a concentration dependent decrease in phosphorylation of GSK-3β and AKT (both known ILK substrates), but had no effect in quiescent stellate cells (Figure 1d–g).
Figure 1. Differential ILK activity in quiescent and activated stellate cells.
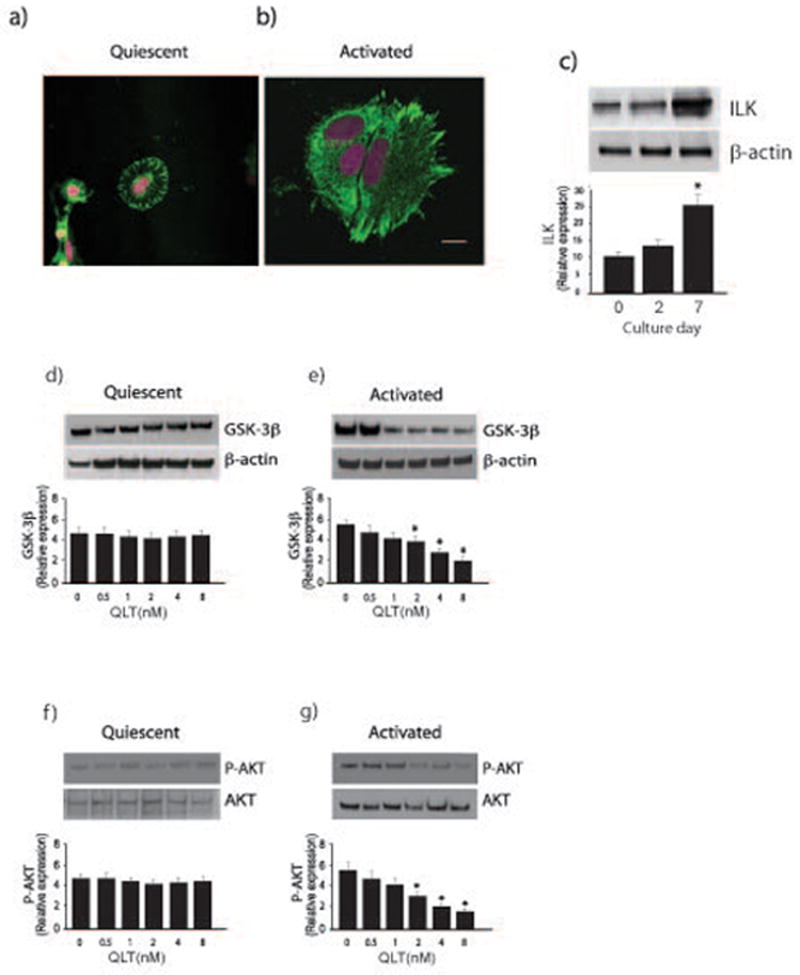
In (a) and (b), stellate cells were isolated from normal rats livers as in Methods, allowed to adhere to glass-coated culture dishes, and grown in 20% serum-containing medium for 1 day (a, quiescent) or 7 days (b, activated). Cells were fixed, and immunohistochemistry was performed as described in Methods. Representative images are shown (ILK is labeled with AL-488 (green)), and nuclei are labeled with DRAQ5 (pink)). The bar shown in the lower right hand corner of (a) represents 10 microns. In (c), stellate cells were as in (a/b); cell lysates were subjected to immunoblotting (50 μg total protein) with anti-ILK antibody. Representative blots are shown in the top portion of the panel and in the bottom portion of the panel, specific bands were scanned and quantitated, and presented graphically (n = 4; *p < 0.05, vs. day 0 or day 2). In (d), quiescent cells were exposed to serum overnight and were exposed to the indicated concentrations of QLT-0267 in medium containing 0.1% serum for 18 hours. Lysates were harvested and GSK3-β phosphorylation was detected by immunoblotting as in Methods. Signals were scanned, normalized to the control value, and displayed in the graph below the immunoblot (n=3). In (e), activated cells and sample processing were as in (d), (n=3, *p < 0.05, compared to control - without QLT). In (f/g), cells were as in (d/e) (i.e. quiescent and activated cells, respectively), and phospho-Akt was detected by immunoblotting as in Methods (n=3, *p < 0.05, compared to control - without QLT).
Rho- GTPase activity is increased during activation of stellate cells
Previous data have linked ILK and Rho in a variety of signaling pathways (28). We therefore further postulated that in stellate cells, ILK and Rho signaling might be connected to a functional endpoint or endpoints. To investigate Rho activity during stellate cell activation, we used a model in which freshly isolated cells from livers of normal rats were grown in serum containing medium and allowed to undergo culture induced activation for 3, 5, 7 and 14 days. In this model, essentially all cells become activated by day 7. We found that Rho activity increased during the stellate cell activation process (Figure 2b), and moreover, that in quiescent cells, Rho activity was stimulated in a dose dependent manner by ILK (Figure 2b). Of note, Rho activity was also inhibited by blocking ILK (in a “dose-dependent” fashion) (Figure 2c). Since the wounding response is characterized by production of endothelin-1, which helps drive the wounding response, we also examined whether the endothelin-1 might regulate Rho activity. Rho activity was stimulated by ET-1 (2 nM), prominently after 30 minutes (Figure 2d). As a control, LPA, which is known to activate Rho, rapidly stimulated Rho activity in stellate cells.
Figure 2. Rho-GTPase activity is increased during stellate cell activation.

Hepatic stellate cells were isolated as in Figure 1. In (a), stellate cells were grown for the time periods shown, cell lysates were harvested and Rho activity measured as in Methods (n=3, *p < 0.05, compared to day 0). In (b/c), stellate cells (cultured for 5 days) were exposed to the indicated concentrations of adenovirus-ILK (Ad-ILK) (b) or adenovirus Sh-ILK (c), and Rho activity was measured (n=3, *p < 0.05, compared to no adenovirus). In (d), stellate cells were grown for 6 days and serum starved over night and ET-1 (20 nM) and LPA (10 mg/mL) were added for the times shown. Cell lysates were harvested and Rho activity measured.
ILK differentially regulates Rho and Rac in quiescent and activated stellate cells
To better understand the role of ILK in regulation of Rho and Rac activity in quiescent and activated stellate cells, we utilized adenoviruses previously shown to either overexpress or block ILK expression (25). In quiescent stellate cells, either inhibition of or overexpression of ILK had no effect on either Rho or Rac activity (Figure 3a, c). In contrast, in activated stellate cells, overexpression of ILK led to enhanced Rho and Rac activity, while ILK knockdown was inhibitory (Figure 3b, d).
Figure 3. ILK differentially regulates Rho and Rac activity in quiescent and activated stellate cells.

In (a) and (b), stellate cells were isolated from the livers of normal rats as in methods and in (a), cells were studied after growth for 24 hours, while in (b), cells were allowed to undergo spontaneous culture induced activation for 6 days. Cells were exposed to the indicated adenoviral constructs (adenovirus-expressing ILK (Ad-ILK), adenovirus-expressing ILK small interfering short hairpin RNA (Ad-shILK), or a control adenovirus-expressing GFP (Ad-GFP) as in Methods and stimulated with ET-1 (20 nM) for 30 minutes prior to harvest. Cell lysates were harvested and Rho activity was measured as in Methods (n=3, *p<0.05 compared to Ad-GFP, #p<0.05 compared to Ad-GFP + ET-1). In (c) and (d), cells were isolated and treated same as above and Rac1 activity was measured as in Methods. In the upper panel of each (c/d), a Rac activity is visualized in cell lysates subjected to immunoblotting to detect active Rac. Specific signals were scanned, normalized to appropriate control values, and displayed in the graph below the immunoblot (n=3, *p<0.05 compared to Ad-GFP, #p<0.05 compared to Ad-GFP + PDGF).
The relationship between Rho and ILK in regulating fibrogenic fingerprints
Important functional features of stellate cell activation include expression of type I collagen and/or other matrix proteins as well as expression of the cytoskeletal protein smooth muscle α-actin (the latter, a well-accepted marker of stellate cell activation that not only imparts cellular contractility on stellate cells, but also labels stellate cells as hepatic myofibroblasts). To clarify the role of ILK and the Rho-ROCK pathway in regulating these stellate cell functional fingerprints, we employed several tools including QLT and Y-27632, specific ILK kinase and ROCK inhibitors, respectively as well as a dominant active Rho construct (29). Stimulation of stellate cells with ET-1 led to an increase in smooth muscle α-actin and type I collagen mRNA expression (Figure 4a/b), while and inhibition of either ILK or Rho alone led to a decrease in smooth muscle α-actin and type I collagen mRNA expression, respectively (Figure 4a/b). Interestingly, when both ILK and Rho were blocked, we found an additive effect on smooth muscle α-actin and type I collagen mRNA expression, respectively. To further evaluate the relationship of Rho and ILK in the functional regulation of smooth muscle α-actin, we over expressed Rho in isolated stellate cells. Overexpression of RhoA (RhoA V14) overexpression led to up-regulation of smooth muscle α-actin mRNA expression (Figure 4c). When RhoA was overexpressed, and followed by inhibition of ILK, there was a reduction in smooth muscle α-actin to control values (Figure 4c), suggesting that Rho A and ILK signaling were linked. Additionally, blocking both ILK and RhoA led to a greater reduction in smooth muscle α-actin than blocking ILK alone, but not than blocking RhoA alone. Although the results for collagen I mRNA paralleled smooth muscle α-actin, there were subtle differences (Figure 4c). Stimulation of RhoA led to an increase in collagen I mRNA expression, and following, inhibition of ILK following activation of RhoA led to reduction in collagen I mRNA to levels below control values, indicating a particularly prominent effect of ILK (Figure 4d). Further, blocking both ILK and RhoA led to a greater reduction in collagen I mRNA than blocking either ILK or RhoA alone (Figure 4d). Finally, we examined smooth muscle α-actin levels after manipulation of Rho and ILK. Initial immunoblotting experiments revealed that, analogous to ET-1’s effect on smooth muscle α-actin, inhibition or ILK or Rho reduced serum induced smooth muscle α-actin expression (Figure 4e). Blocking ILK and Rho together reduced smooth muscle α-actin to a greater degree than either alone. When Rho was overexpressed, inhibition of ILK was able to reduced smooth muscle α-actin, and combined Rho and ILK inhibition reduced smooth muscle α-actin to below those for Rho inhibition alone (Figure 4f). Immunocytochemical results paralleled those for immunoblotting (Figure 4g). Serum, ILK, and Rho each stimulated stress fibers, while ILK inhibition partially blocked these responses. Inhibition of Rho led to a similar response while inhibition of both ILK and Rho caused and even greater disruption in actin filaments, leading to cell rounding (Figure 4g).
Figure 4. ILK and Rho regulate smooth muscle β-actin and collagen mRNA expression.

In (a) and (b), stellate cells were isolated from the livers of normal rats as in Methods and were allowed to undergo spontaneous culture induced activation. Cells were serum starved over night and infected with Ad-ShILK, and exposed to ET-1 (20nM), QLT (16 nM), and/or Y-27632 (10μM). Smooth muscle α-actin (a), and type I collagen mRNA (b) were measured by RT-PCR as in Methods and the data presented graphically (n=3; *p<0.05 vs. control (i.e. no serum); #p< 0.01 vs. ET-1; †p<0.05 vs. ET-1 plus QLT and Y-27632 alone). In (c) and (d), stellate cells were as above. Cells were serum starved overnight, infected with the indicated adenoviral constructs, and exposed to ET-1 (20nM), QLT (16 nM), and/or Y-27632 (10μM). Smooth muscle α-actin (c), and collagen (d) were measured by RT-PCR as in Methods and the data presented graphically (n=3; * p<0.05, vs. control (i.e. no serum); #, p< 0.01 vs. Rho A (V14); † p<0.05 vs. ET-1 plus QLT and Y-27632 alone). In (e), stellate cells as above were serum starved over night, infected with Ad-ShILK, and exposed to QLT (16 nM), and/or Y-27632 (10μM) for a further 24 hours. One sample of cells was exposed to serum as a control. Cell lysates were harvested and subjected to immunoblotting to detect smooth muscle α-actin and β-actin and a representative immunoblot is shown in top panel. Specific signals were scanned, normalized to control, and displayed graphically (n=3, *p<0.05, versus serum; #, p< 0.01 versus QLT and Y-27632 alone). In (f), cells were as in (e), with the exception that adenovirus expressing active Rho A (V14) was introduced 24 hours prior to other compounds (n=3, *p<0.05, versus Rho A V14; #, p< 0.01 versus QLT and Y-27632 alone. In (g), cells, adenoviruses, and inhibitors were as in (e), and cells were fixed and actin was detected as in Methods. Images are representative of 10 others. The bar represents 10 microns.
Migration of stellate cells is also a prominent cell phenotype during wound healing. Therefore we tested weather Rho could mediate stellate cell migration. We found that overexpression of Rho with Rho A (V14) led to a marked increase in stellate cell migration, whereas inhibition of Rho with y-27632 led to reduced cell migration (Figure S1a). Cell adhesion, also appeared to be Rho dependent, because exposure of cells to Rho A (V14) increased cell adhesion and inhibition of Rho with y-27632 reduced cell adhesion (Figure S1b).
Regulation of Rho-GEF, Rho-GAP and Rho-GDI during stellate cell activation
We found that Rho kinase activity was increased in activated stellate cells (Figure 2). Thus, since Rho GTPase cycling is regulated by guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide-dissociation inhibitors(GDIs) (30), we postulated that these upstream regulators of RhoA might be regulated during stellate cell activation. We found that the level of RhoGDI and RhoGAP mRNA decreased with activation, while RhoGEF mRNA levels increased during activation (Figure 5a). We verified that the level of RhoGEF (protein) increased during activation using immunoblot analysis (Figure 5b), and interestingly, ILK appeared to regulate RhoGEF expression (Figure 5c).
Figure 5. Regulation of Rho proteins during stellate cell activation.

In (a), stellate cells were isolated from the livers of normal rats as in Methods and were allowed to undergo spontaneous culture induced activation for the indicated number of days. Rho GAP (left), Rho GDI (center), and Rho GEF (right) mRNA were measured by RT-PCR as in Methods and the data presented graphically (n=3, *p<0.05, compared to the level for day 1). In (b), stellate cells as above were subjected to immunoblotting (50 μg of total protein) with anti-Rho-GEF antibody. A representative immunoblot is shown in the upper panel, and below it, a stripped blot re-probed for β-actin; subsequently, specific bands were quantified, normalized to the signal for β-actin and the data presented graphically below (n = 5; *p<0.05, compared with the signal for day 1). In (c), cells were allowed to grow for 6 days, and cells were exposed to the indicated concentrations of QLT-0267 in medium containing 0.1% serum for 18 hours. Lysates were harvested and RhoGEF was detected by immunoblotting as in Methods. Signals were scanned, normalized to the control value, and displayed in the graph below the immunoblot (n=3, *p<0.05, compared to control - without QLT).
Gα12/13 are up regulated during stellate cell activation
RhoA is activated via GPCRs that couple to heterotrimeric Gα12/13 proteins; this response is catalyzed by GEFs, several of which are direct targets (effectors) of Gα12/13 (10, 31). Thus, we further hypothesized that RhoA signaling in stellate cells might be mediated by Gα12/13. To evaluate this possibility, we examined the effect of stellate cell activation on induction of Gα12/13 expression; there was gradual spontaneous increase of Gα12/13 expression during the activation process (Figure 6a, b), and manipulation of ILK using specific adenoviruses as above led to changes in Gα12/13 expression (Figure 6c, d), including that differential ILK activity modulated the levels of Gα12 and Gα13 (Figure 6c, d). Of note, overexpression of ILK had no effect on Gα12 or Gα13 expression in quiescent cells (Figure 7a, c), but had significant effects on activated cells (Figure 7b, d). Likewise, inhibition of ILK played a role in Gα12/13 expression in activated cells (Figure 7e, f).
Figure 6. Ga12 and Ga13 expression is stimulated during stellate cell activation.

In (a) and (b), stellate cells were isolated from normal rat livers and were allowed to undergo spontaneous culture induced activation for the indicated number of days. Cell lysates were subjected to immunoblotting to detect Gα12 and Gα13, respectively as in Methods. A representative immunoblot is shown in the upper panel, and below it, a stripped blot re-probed for β-actin; subsequently, specific bands were quantified, normalized to the signal for β-actin and presented graphically (n = 3; *p<0.05, compared with the signal for day 0). In (c) and (d), stellate cells were allowed to grow for 5 days in culture, and were then infected with the indicated adenoviral constructs as in Figure 3. Forty-eight hours later, cell lysates (50 μg total protein) were subjected to immunoblotting to detect Gα12 and Gα13. A representative immunoblot is shown in the top panels, and immediately below it, a stripped blot re-probed for β-actin; subsequently, specific bands were quantified, normalized to the signal for β-actin and presented graphically (n = 3; *p<0.05, compared to control or Ad-GFP).
Figure 7. ILK modulates expression of Ga12 and Ga13 in activated stellate cells.

In (a) through (d), stellate cells were as in Figure 1; each quiescent (cultured for one day) and activated cells (cultured for 7 days) were exposed to adenovirus-expressing ILK (Ad-ILK) for 24 hours. Forty-eight hours later, cell lysates (50 μg total proteins) were subjected to immunoblotting to detect Gα12 (a/b) or Gα13 (c/d). Representative immunoblots are shown in the upper panels, and below them, a stripped blot re-probed for β-actin; subsequently, specific bands were quantified, normalized to the signal for β-actin and presented graphically (n = 3; *p<0.05, compared to “0”). In (e) and (f), activated stellate cells were exposed to different concentrations of QLT for 18 hours in medium containing 0.1% serum, cell lysates were harvested (50 μg total protein) and subjected to immunoblotting to detect Gα12 and Gα13. A representative immunoblot is shown in the upper panel, and below it, a stripped blot re-probed for β-actin; specific bands were quantified, normalized to the signal for β-actin and presented graphically (n = 3; *p<0.05, compared to “0”).
A functional effect for Ga12/13 in stellate cells
To determine whether Gα12/13 could be important in mediating functional effects on activated stellate cells, we inhibited these G protein subunits with the RGS domain of p115RhoGEF which is an efficient GAP specifically for Gα12/13, and thus blocks only Gα12/13 signaling (32). We found that inhibition of Gα12/13 together caused a significant decrease in smooth muscle a-actin mRNA at all-time points after day 3 of the activation process (Figure 8a). Similar results were identified for type I collagen mRNA (Figure 8b). As before, ILK stimulated smooth muscle α-actin mRNA; inhibition of Gα12/13 partially inhibited the ILK mediated smooth muscle a-actin mRNA stimulation (Figure 8c). Analogous results were found for type I collagen (Figure 8d). These data suggest that ILK and Gα12/13 are linked in a signaling pathway to functional effectors in stellate cells.
Figure 8. A functional effect for Ga12 and Ga13 activated stellate cells.

In (a) and (b), stellate cells were isolated from normal rat livers and were allowed to undergo spontaneous culture induced activation for 48 hours. Cells were serum starved overnight and infected with the adenoviral p115-RGS construct as in Methods. Cells at the indicated day in culture were harvested and smooth muscle α actin and collagen mRNA were measured by RT-PCR as in Methods and the data presented graphically (n=3, *p<0.05, compared to day 3; # p < 0.05 compared to control). In (c & d), cells were as in (a & b), with the exception that adenovirus over expressing ILK (Ad-ILK) was introduced 24 hours after the p115-RGS construct. Cells were harvested 24 hours later and smooth muscle α actin and collagen mRNA were measured by RT-PCR as in Methods and the data presented graphically (n=3, *p<0.05, compared to control (no viral constructs; # p < 0.05 compared to Ad-ILK alone).
Discussion
A novel finding of this study was that at a cellular level, ILK had prominent functional effects that are likely to be of critical importance in wound-healing. Most importantly, ILK stimulated smooth muscle α actin and type I collagen expression in activated, but not in quiescent cells. We speculate that this latter phenomena may be related to the presence (or absence) of intermediate molecules important in executing ILK’s functional effects (for example, multiple signaling and transcriptional partners) (16). For example, ILK deficient fibroblasts appear to exhibit abnormalities in the actin cytoskeleton, apparently restricting their capacity to differentiate into myofibroblasts (33, 34), which would be expected to alter their fibrogenic phenotype. Additionally, we clearly demonstrated that Gα12/13 is important in mediating ILK activity (Figure 7), and this signaling partner appears to also be differentially regulated during stellate cell activation. What we don’t know at this point is whether ILKs effect on smooth muscle actin expression is necessary for the fibrogenic phenotype observed or whether ILK’s effect on fibrogenesis is independent of the actin cytoskeleton.
An explanation for the differential ILK activity in quiescent and activated cells is simply that the ILK activating machinery is missing in quiescent stellate cells. For example, it is possible that a critical extracellular signal is required to activate ILK in stellate cells; integrins or molecules linking integrins to the ILK signaling cascade are either in low affinity or are absent in quiescent cells, but are present and/or are in high affinity in activated cells (35). Another possibility is that ligand binding in quiescent stellate cells does not initiate the required conformational change in integrins that are needed to activate ILK (36). Finally, since integrin binding to ECM leads the integrin cytoplasmic domain to form a complex with ILK and isoforms of PINCH and parvin to subsequently form an IPP complex in the cytoplasm (37), it is possible that quiescent cells lack these downstream components of a multiprotein complex that would be required for activation of downstream signaling (Figure 9).
Figure 9. A working model for ILK signaling in quiescent and activated hepatic stellate cells.
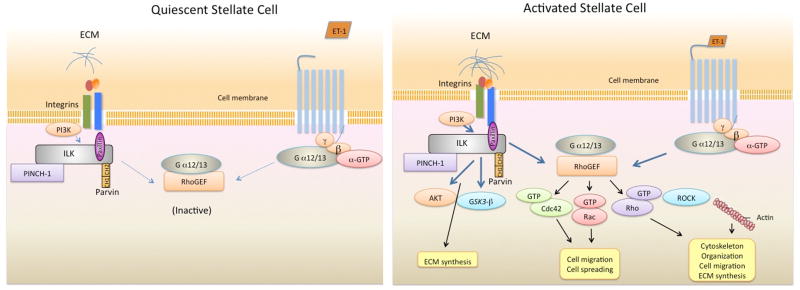
This figure highlights previously established ILK signaling partners and emphasizes data from the current study. In quiescent cells, integrins likely have low affinity for extracellular matrix (ECM) molecules (i.e. such as collagen I, fibronectin, etc... - which are in low abundance) and are therefore in a low affinity or inactive confirmation. Thus, ILK is relatively inactive. In addition, the Gα12/13 pathway is inactive (delineated by dotted lines). Upon ligand binding (i.e., collagen I, fibronectin, etc), the confirmation of the integrin changes and ILK binds to the cytoplasmic tail of β-integrin, resulting in formation of PINCH-ILK-parvin ternary complexes. ILK subsequently recruits other signaling molecules. In stellate cells, ILK is engaged in G-protein receptor signaling cross talk, where it appears to amplify Gα12/13 mediated signals. Activation of this ILK/Gα12/13 signaling pathway has several functional consequences, including in actin reorganization (and actin synthesis), cell spreading, cell migration, and extracellular matrix synthesis. The pathways leading to synthesis of ECM are likely to be multiple, and likely include both ILK direct pathways, as well as those that cross talk through the Gα12/13 and the Rho pathway. The activated stellate cell cartoon is meant to depict these both.
Another novel finding of this work was that the vasoactive peptide, ET-1, appeared to be a critical upstream mediator of ILK activation. We showed that ET-1 is capable of inducing ILK activation and subsequent smooth muscle α actin and type I collagen expression. This unique finding is consistent with previous data that showed that ET-1 was able to stimulate ILK dependent tumor cell migration, synthesis of MMP, and tumor invasion/growth in vivo (38).
We also found Rho-GTPase activity to be stimulated during stellate cell activation. Interestingly, not only Rho itself appeared to be upregulated by stellate cell activation, but also, ILK appeared to modify Rho’s activity (Figure 3B). Further, ILK also modified the activity of Rac (Figure 3D), consistent with a broad effect of ILK on Rho GTPases. We also found that Rho-GEF, Rho-GAP and Rho-GDI - all capable of modifying Rho activity - were regulated during stellate cell activation (Figure 5), and likely facilitated Rho’s activity. These data suggest that ILK and Rho are tightly linked in stellate cells.
A final novel finding of this work was that Gα12/13 was up regulated during stellate cell activation and further, Gα12/13 also mediated functional effects on stellate cells. Inhibition of Gα12/13 function specifically inhibited smooth muscle α actin and type I collagen expression in stellate cells (Figure 7A/B). Further, inhibition of Gα12/13 blocked ILK’s stimulation of smooth muscle α actin and type I collagen (Figure 7C/D), suggesting crosstalk between Gα12/13 and ILK in execution of stellate cell functional effects.
In summary, we have shown that ILK, rho and Gα12/13 interact to facilitate important functional effects in activated stellate cells. Further, these molecules appear to be predominantly (if not exclusively) active in the activated state. The findings lead us to speculate the a G-protein coupled receptor signaling pathway involving ET-1 and Gα12/13 converges to mediate critical functional effects important in the wounding milieu.
Supplementary Material
In (a), stellate cells from normal rats were isolated, plated at equivalent density, and allowed to undergo culture-induced activation for 7 days as described in Methods. Cells were transduced with a control adenovirus-expressing GFP, Rho A (V14) as described under “Experimental Procedures.” Twenty-four hours after infection, a wound was created on confluent cell monolayers as described under “Experimental Procedures,” and the distance that the wound edge migrated was assessed for 24 h using time lapse microscopy at 37 °C as described under “Experimental Procedures” (n = 3; *p < 0.05 vs. serum free control, #p < 0.05 vs. Ad-GFP;†, p <0.05 vs. Rho A (V14). In (b), cells as in (a) were detached and resuspended in Dulbecco’s modified Eagle’s medium with 20% serum, and then replated as described under “Experimental Procedures.” Two hours later, cells were washed twice with PBS to remove the unattached cells, and the remaining cells were counted (n = 3; *p < 0.05 versus serum free control, #p < 0.05 vs. Ad-GFP;†, p <0.05 vs. Rho A (V14).
Acknowledgments
Grant support: This work was supported by the National Institutes of Health, grant R01 DK 50574, to DCR.
Footnotes
Disclosure
The authors certify that we have no financial arrangements (e.g., consultancies, stock ownership, equity interests, patent-licensing arrangements, research support, major honoraria, etc.) with a company whose product figures prominently in this manuscript or with a company making a competing product.
References
- 1.Maher JJ, McGuire RF. Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Invest. 1990;86(5):1641–1648. doi: 10.1172/JCI114886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL, Rockey DC, Bissell DM. Hepatic fibrosis 2006: report of the Third AASLD Single Topic Conference. Hepatology. 2007;45(1):242–249. doi: 10.1002/hep.21459. [DOI] [PubMed] [Google Scholar]
- 3.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wells RG. Cellular sources of extracellular matrix in hepatic fibrosis. Clin Liver Dis. 2008;12(4):759–768. viii. doi: 10.1016/j.cld.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rockey DC, Boyles JK, Gabbiani G, Friedman SL. Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. JSubmicroscCytolPathol. 1992;24:193–203. [PubMed] [Google Scholar]
- 6.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 7.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18(3):578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hotchin NA, Hall A. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J Cell Biol. 1995;131(6 Pt 2):1857–1865. doi: 10.1083/jcb.131.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zohn IM, Campbell SL, Khosravi-Far R, Rossman KL, Der CJ. Rho family proteins and Ras transformation: the RHOad less traveled gets congested. Oncogene. 1998;17(11 Reviews):1415–1438. doi: 10.1038/sj.onc.1202181. [DOI] [PubMed] [Google Scholar]
- 10.Seasholtz TM, Majumdar M, Brown JH. Rho as a mediator of G protein-coupled receptor signaling. Mol Pharmacol. 1999;55(6):949–956. doi: 10.1124/mol.55.6.949. [DOI] [PubMed] [Google Scholar]
- 11.Walsh CT, Stupack D, Brown JH. G protein-coupled receptors go extracellular: RhoA integrates the integrins. Mol Interv. 2008;8(4):165–173. doi: 10.1124/mi.8.4.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki N, Nakamura S, Mano H, Kozasa T. Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc Natl Acad Sci U S A. 2003;100(2):733–738. doi: 10.1073/pnas.0234057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2(2):133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 14.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6(2):167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 15.Loer B, Bauer R, Bornheim R, Grell J, Kremmer E, Kolanus W, et al. The NHL-domain protein Wech is crucial for the integrin-cytoskeleton link. Nat Cell Biol. 2008;10(4):422–428. doi: 10.1038/ncb1704. [DOI] [PubMed] [Google Scholar]
- 16.Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5(1):51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- 17.Maydan M, McDonald PC, Sanghera J, Yan J, Rallis C, Pinchin S, et al. Integrin-linked kinase is a functional Mn2+-dependent protein kinase that regulates glycogen synthase kinase-3beta (GSK-3beta) phosphorylation. PLoS ONE. 2010;5(8):e12356. doi: 10.1371/journal.pone.0012356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155(4):505–510. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lange A, Wickstrom SA, Jakobson M, Zent R, Sainio K, Fassler R. Integrin-linked kinase is an adaptor with essential functions during mouse development. Nature. 2009;461(7266):1002–1006. doi: 10.1038/nature08468. [DOI] [PubMed] [Google Scholar]
- 20.Wu C. PINCH, N(i)ck and the ILK: network wiring at cell-matrix adhesions. Trends Cell Biol. 2005;15(9):460–466. doi: 10.1016/j.tcb.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A. 1998;95(19):11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian Y, Zhong X, Flynn DC, Zheng JZ, Qiao M, Wu C, et al. ILK mediates actin filament rearrangements and cell migration and invasion through PI3K/Akt/Rac1 signaling. Oncogene. 2005;24(19):3154–3165. doi: 10.1038/sj.onc.1208525. [DOI] [PubMed] [Google Scholar]
- 23.Rockey DC, Chung JJ. Endothelin antagonism in experimental hepatic fibrosis. Implications for endothelin in the pathogenesis of wound healing. J Clin Invest. 1996;98(6):1381–1388. doi: 10.1172/JCI118925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maschmeyer P, Flach M, Winau F. Seven steps to stellate cells. J Vis Exp. 2011;(51) doi: 10.3791/2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shafiei MS, Rockey DC. The role of integrin-linked kinase in liver wound healing. J Biol Chem. 2006;281(34):24863–24872. doi: 10.1074/jbc.M513544200. [DOI] [PubMed] [Google Scholar]
- 26.Rockey DC, Housset CN, Friedman SL. Activation-dependent contractility of rat hepatic lipocytes in culture and in vivo. JClinInvest. 1993;92:1795–1804. doi: 10.1172/JCI116769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards LA, Woo J, Huxham LA, Verreault M, Dragowska WH, Chiu G, et al. Suppression of VEGF secretion and changes in glioblastoma multiforme microenvironment by inhibition of integrin-linked kinase (ILK) Mol Cancer Ther. 2008;7(1):59–70. doi: 10.1158/1535-7163.MCT-07-0329. [DOI] [PubMed] [Google Scholar]
- 28.Khyrul WA, LaLonde DP, Brown MC, Levinson H, Turner CE. The integrin-linked kinase regulates cell morphology and motility in a rho-associated kinase-dependent manner. J Biol Chem. 2004;279(52):54131–54139. doi: 10.1074/jbc.M410051200. [DOI] [PubMed] [Google Scholar]
- 29.Chang Y, Ceacareanu B, Dixit M, Sreejayan N, Hassid A. Nitric oxide-induced motility in aortic smooth muscle cells: role of protein tyrosine phosphatase SHP-2 and GTP-binding protein Rho. Circ Res. 2002;91(5):390–397. doi: 10.1161/01.res.0000033524.92083.64. [DOI] [PubMed] [Google Scholar]
- 30.Hakoshima T, Shimizu T, Maesaki R. Structural basis of the Rho GTPase signaling. J Biochem. 2003;134(3):327–331. doi: 10.1093/jb/mvg149. [DOI] [PubMed] [Google Scholar]
- 31.Kranenburg O, Poland M, van Horck FP, Drechsel D, Hall A, Moolenaar WH. Activation of RhoA by lysophosphatidic acid and Galpha12/13 subunits in neuronal cells: induction of neurite retraction. Mol Biol Cell. 1999;10(6):1851–1857. doi: 10.1091/mbc.10.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meigs TE, Juneja J, DeMarco CT, Stemmle LN, Kaplan DD, Casey PJ. Selective uncoupling of G alpha 12 from Rho-mediated signaling. J Biol Chem. 2005;280(18):18049–18055. doi: 10.1074/jbc.M500445200. [DOI] [PubMed] [Google Scholar]
- 33.Colwell AS, Yun R, Krummel TM, Longaker MT, Lorenz HP. Keratinocytes modulate fetal and postnatal fibroblast transforming growth factor-beta and Smad expression in co-culture. Plast Reconstr Surg. 2007;119(5):1440–1445. doi: 10.1097/01.prs.0000256049.53562.39. [DOI] [PubMed] [Google Scholar]
- 34.Nakrieko KA, Vespa A, Mason D, Irvine TS, D’Souza SJ, Dagnino L. Modulation of integrin-linked kinase nucleo-cytoplasmic shuttling by ILKAP and CRM1. Cell Cycle. 2008;7(14):2157–2166. doi: 10.4161/cc.7.14.6241. [DOI] [PubMed] [Google Scholar]
- 35.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110(5):599–511. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 36.Takagi J, Springer TA. Integrin activation and structural rearrangement. Immunol Rev. 2002;186:141–163. doi: 10.1034/j.1600-065x.2002.18613.x. [DOI] [PubMed] [Google Scholar]
- 37.Legate KR, Montanez E, Kudlacek O, Fassler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006:20–31. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- 38.Rosano L, Spinella F, Di Castro V, Dedhar S, Nicotra MR, Natali PG, et al. Integrin-linked kinase functions as a downstream mediator of endothelin-1 to promote invasive behavior in ovarian carcinoma. Mol Cancer Ther. 2006;5(4):833–842. doi: 10.1158/1535-7163.MCT-05-0523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In (a), stellate cells from normal rats were isolated, plated at equivalent density, and allowed to undergo culture-induced activation for 7 days as described in Methods. Cells were transduced with a control adenovirus-expressing GFP, Rho A (V14) as described under “Experimental Procedures.” Twenty-four hours after infection, a wound was created on confluent cell monolayers as described under “Experimental Procedures,” and the distance that the wound edge migrated was assessed for 24 h using time lapse microscopy at 37 °C as described under “Experimental Procedures” (n = 3; *p < 0.05 vs. serum free control, #p < 0.05 vs. Ad-GFP;†, p <0.05 vs. Rho A (V14). In (b), cells as in (a) were detached and resuspended in Dulbecco’s modified Eagle’s medium with 20% serum, and then replated as described under “Experimental Procedures.” Two hours later, cells were washed twice with PBS to remove the unattached cells, and the remaining cells were counted (n = 3; *p < 0.05 versus serum free control, #p < 0.05 vs. Ad-GFP;†, p <0.05 vs. Rho A (V14).