

Abstract
Purpose
The ER chaperone GRP78 translocates to the surface of tumor cells and promotes survival, metastasis, and resistance to therapy. An oncogenic function of cell surface GRP78 has been attributed to the activation of phosphoinositide 3-kinase (PI3K) pathway. We intend to use a novel anti-GRP78monoclonal antibody (MAb159) to attenuate PI3K signaling and inhibit tumor growth and metastasis.
Experimental Design
MAb159 was characterized biochemically. Anti-tumor activity was tested in cancer cell culture, tumor xenograft models, tumor metastasis models, and spontaneous tumor models. Cancer cells and tumor tissues were analyzed for PI3K activity. MAb159 was humanized and validated for diagnostic and therapeutic application.
Results
MAb159 specifically recognized surface GRP78, triggered GRP78 endocytosis, and localized to tumors but not normal organs in vivo. MAb159 inhibited tumor cell proliferation and enhanced tumor cell death both in vitro and in vivo. In MAb159 treated tumors, PI3K signaling was inhibited without compensatory MAPK pathway activation. Furthermore, MAb159 halted or reversed tumor progression in the spontaneous PTEN loss driven prostate and leukemia tumor models, and inhibited tumor growth and metastasis in xenograft models. Humanized MAb159, which retains high affinity, tumor specific localization, and the anti-tumor activity, was non-toxic in mice and had desirable pharmacokinetics.
Conclusions
GRP78 specific antibody MAb159 modulates PI3K pathway and inhibits tumor growth and metastasis. Humanized MAb159 will enter human trials shortly.
Keywords: Surface GRP78, monoclonal antibody, targeted cancer therapy, PI3K
INTRODUCTION
Cancer cells are characterized by metabolic alterations and the tumor microenvironment is often marked with impaired blood flow and hypoxia, all of which can elicit endoplasmic reticulum (ER) stress. Tumor cells adapt to these adverse conditions by activating the unfolded protein response (UPR), with induction of GRP78 as a major pro-survival arm of the UPR signaling pathways (1, 2). GRP78, also referred to as BiP/HSPA5, is a 78 kilodalton glucose regulated protein with potent anti-apoptotic properties that plays critical roles in cancer cell survival, tumor progression, metastasis and resistance to therapy (3–5). Knockdown of GRP78 by siRNA in cancer cells as well as tumor associated endothelial cells reduced their proliferative rate and sensitize them to chemotherapeutic treatment (6, 7). Genetically altered GRP78 mouse models further demonstrated the critical role of GRP78 in cancer in vivo. For instance, GRP78 haploinsufficiency delayed tumor development, growth and inhibited metastasis. (8, 9). In mice harboring biallelic conditional knockout of both GRP78 and tumor suppressor PTEN in the prostate epithelium, prostate tumorigenesis was potently arrested (10). Inducible heterozygous knockout of GRP78 in the hematopoietic system also suppresses PTEN-null leukemogenesis with no harmful effect on hematopoiesis (11). Strikingly, in both the prostate and leukemia models, PI3K/AKT signaling resulting from the loss of PTEN was greatly impaired with only partial loss of GRP78 (10, 11). Collectively, these studies establish GRP78 as a novel regulator of the PI3K/AKT oncogenic signaling and a target for anti-cancer therapy.
While traditionally GRP78 has been regarded as an ER lumenal protein, evidence has accumulated that a fraction of GRP78 can exist on the plasma membrane of specific cell types (12–14) and that ER stress actively promotes cell surface localization of GRP78 (15). While the physiological function of GRP78 on the cell surface is still emerging, recent studies show that cell surface GRP78 forms complexes with specific protein partners, regulating both proliferation and viability (14, 16), suggesting that cell surface GRP78 presents an opportunity for therapeutic targeting (17–19).
Here we report the generation of a monoclonal antibody (MAb159) against human GRP78 that shows anti-tumor activity with no toxicity and also inhibits PI3K signaling. This antibody opens up a unique opportunity to study both the biology of cell surface GRP78 and its therapeutic potential.
MATERIALS AND METHODS
Antibodies, reagents, and cell lines
A549, HT29, Colo205, LNCap, MCF7, 4T1, and 293T cell lines were obtained from American Type Culture Collection (Manassas, VA). C4-2B cell was kindly provided by Michael Stallcup (USC) and H249 was kindly provided by Dr. Ravi Salgia in University of Chicago. The generation of the B16-Fluc-A1 melanoma cell line has been described (9). All these cells were propagated in RPMI-1640 supplemented with 10% fetal bovine serum, 100 units/ml of penicillin, and 100 μg/ml streptomycin from Cellgro (Manassas, VA). CE1 cell line was kindly provided by Dr. Pradip Roy-Burman and cultured as previously described (20). These cell lines have been validated by HLA typing and molecular phenotyping relative to the respective primary tumors. Information about antibodies and reagents used in this study can be found in the “Supplementary Methods”.
Generation of monoclonal antibodies and affinity analysis
The procedure for generation of monoclonal antibodies is described in “Supplementary Methods”. The affinity of MAbs to antigen was determined by Scatchard assay as described previously (21).
In vivo and ex vivo near-infrared fluorescence imaging
H249 tumor bearing mice were injected with Cy5.5 labeled humanized MAb159 or control antibody. In vivo fluorescence imaging was performed using the Xenogen Lumina XR Imaging System and analyzed using the IVIS Living Imaging 3.0 software. 28 hours after injection, the tumors and organs were harvested for ex vivo fluorescence imaging. Please see “Supplementary Methods” for detailed procedure.
PTEN-null models
The prostate specific PTEN knock out model was described previously (22). Mice were treated with MAb159 and prostate tumors were monitored by luminescence imaging (xenogen). The Pten (floxed/floxed); Mx-1 cre leukemic model and the protocols for flow cytometry for analysis of leukemic blasts and peripheral blood counts have been described (11). Please see “Supplementary Methods” for detailed procedure.
Murine tumor xenograft models
The procedures for murine tumor xenograft studies and immunohistochemical analysis were as before (21) and described in “Supplementary Methods”.
Statistics
The statistical significance of differences in different samples or groups was determined using a unpaired 2-tail student T-test. Results were considered significantly different if P < 0.05.
RESULTS
Generation of monoclonal antibodies (MAbs) specific to surface GRP78
We immunized mice with a hexahistidine tagged and secreted form of human GRP78 (Figure S1A), and screened a panel of MAbs capable of binding the native form of GRP78. The criteria for the desirable antibody were high affinity binding to native cell surface GRP78 and the ability to endocytose. MAb159 fulfilled these requirements. MAb159 is highly specific for GRP78 as the binding of MAb159 to GRP78 was completely abolished by purified soluble GRP78 protein (Figure S1B). We also performed immunoprecipitation study with MAb159 and confirmed that GRP78 can be pulled down from cell lysate using mass spectrometry analysis (data not shown). MAb159 has no cross-reactivity to GRP78’s closest paralogue HSP70 (Figure S1C), but it recognizes mouse GRP78, which is 99% conserved in amino acids with human GRP78 (Figure S1D). Using scatchard analysis we have determined that MAb159 has high affinity to human GRP78 (Kd=1.7nM) (Figure S1E), hence suitable for therapeutic development. When incubated with cultured cells at 4°C, MAb159 bound the cell surface of cancer cells, but not to normal human dermal fibroblasts (Figure 1A, left). Under glucose starvation conditions which mimicked nutrient deprivation in the tumor microenvironment, greater amount of MAb159 was recruited to cancer cell surface. This is consistent with the previous findings that surface GRP78 significantly increases when the cell is under stress (14, 15). We also determined whether MAb159 preferably localizes to tumor and not normal organs by tracking biotin labeled MAb159 in HT29 xenograft tumor bearing mice in vivo. Biotin-MAb159 was only seen in the tumor, not in the normal organs including heart, liver, and kidney (Figure 1A, right).
Fig. 1. Characterization of Monoclonal Antibody Targeting Specifically to Surface GRP78.

(A) Specificity of GRP78 antibody MAb159. Left, live cells were stained with MAb159 at 4 °C. Surface GRP78 staining (green) presents on cancer cells C4-2B and MCF7, but not normal human dermal fibroblast (NHFD). Glucose starvation for 2 days significantly increased surface GRP78 on MCF7 cells (bottom right). Right, biotinylated MAb159 (50 μg) was administered I.V. to two HT29 tumor bearing mice, mice were sacrificed after six hours, tissues were harvested and MAb159 was localized with fluorochrome-conjugated streptavidin (green). MAb159 was only detected in tumor, not normal organs. Nuclei were counterstained with DAPI (blue). Scale bar, 20 μm. (B) Top, biotinylated MAb159 was incubated with glucose starved MCF7 cells for one hour at 4°C or 37°C. Localization of MAb159 is shown in green (confocal image). Endocytosis was observed only at 37°C. Bottom, MAb159 or control IgG was incubated with glucose starved MCF7 cells for 48 hours at 37°C. Surface GRP78 was detected using a polyclonal antibody (Clone H129, Santa Cruz Biotech) without fixation and permeabilization at 4°C. Scale bar, 20 μm. (C) MCF7 and HT29 cells were glucose starved and treated with control IgG or MAb159 for 5 days. Top, cell viability was determined with MTT assay. Data are presented as mean ± standard error (s.e.) (n=4). Bottom, MAb159 triggered apoptosis in glucose starved MCF7 cells determined by TUNEL assay (green). Nuclei were counterstained with DAPI (blue). Scale bar, 100 μm. (D) Glucose starved HT29 cells were treated with 50 μg/mL MAb159 for 3 days. Whole cell lysate immunoblotting shows that MAb159 decreased phosphorylated AKT (pAKT) and S6 (pS6) level. Three independent experiments were performed. Image J (NIH) was used for quantification of relative pAKT and pS6 level. Asterisks indicate P < 0.02, as determined by an unpaired 2-tail student T-test.
When incubated with cells at 37°C, MAb159 underwent endocytosis and was localized to the intracellular clathrin-coated endosomes compared to fine ring like appearance at the cell surface at 4°C (Figure 1B, top and Figure S2A). We next examined the surface GRP78 level with a second antibody and found that surface GRP78 was markedly reduced after MAb159 treatment (Figure 1B, bottom). Moreover, this reduction of GRP78 can be reverted with chlorpromazine (Figure S2B), an inhibitor specific for clathrin-mediated endocytosis (23). These data implicate that MAb159 led to internalization of surface GRP78.
MAb159 induces tumor cell apoptosis and inhibits PI3K signaling
The effect of MAb159 on tumor cells was first tested in vitro. Before incubated with MAb159, breast carcinoma cell MCF7 and colon cancer cell HT29 were cultured in glucose-free medium to enhance surface GRP78 level and thus the efficacy of the antibody. MAb159 significantly reduced the cell viability in both MTT (Figure 1C, top)and clonogenic assay (Figure S3A), correlating with increased apoptosis as determined by TUNEL assay (Figure 1C, bottom) and M30 apoptosis assay (Figure S3B). We have also measured activated caspase levels and showed that MAb159 treatment activates caspase 8 and 9 (Figure S3C), which represent the activation of extrinsic and intrinsic apoptotic pathways, respectively. This suggests that at least in part the loss of cell viability was initiated at the cell membrane.
PI3K signaling pathway regulates many biological events in the cells including cell survival and GRP78 has previously been shown to modulate its activity. Therefore, we examined whether MAb159 treated cell surface GRP78 expressing tumor cells have alterations in PI3K activity as measured by the changes in phosphorylated AKT and S6 levels. We indeed found that both phosphorylated AKT and S6 levels were reduced in antibody treated cells compared to the controls (Figure 1D). Since MAb159 induced GRP78 endocytosis led to loss of cell surface GRP78 and inhibition of pAKT signaling, we anticipate that blocking GRP78 endocytosis would antagonize MAb159’s activity. We however were unable to conduct this experiment because Chropromazine by itself reduces pAKT levels and causes cellular toxicity ((24) and data not shown).
The above results indicate that MAb159 inhibits PI3K pathway and induces cell apoptosis, which is consistent with the recent indication that cell surface GRP78 may be an upstream regulator of PI3K/AKT but not MAPK signaling (14). We thus wished to determine if there is a direct interaction between cell surface GRP78 and PI3K components. We expressed a FLAG tagged GRP78 with KDEL deletion for enhanced translocation to the cell surface (15), labeled the cell surface proteins with biotin and purified the biotin labeled proteins on monomeric avid beads. GRP78 was immunoprecipitated from this pool of surface proteins and its interacting partners were detected by Western blotting. As seen in Figure S4A, GRP78 formed complex with p85, the regulatory subunit of PI3K, but not with ERK1/2 or a cell surface specific protein EphB2. We further confirmed this interaction under endogenous expression condition. Surface GRP78 was induced with thapsigargin (15) and cell surface proteins were enriched using biotin-avidin system. Co-immunoprecipitation of surface GRP78 and p85 was achieved with either GRP78 antibody or p85 antibody (Figure S4B). These results provide the first evidence that surface GRP78 binds to PI3K component and suggest surface GRP78 regulates PI3K signaling through direct complex formation with the PI3K subunits.
MAb159 inhibits tumor growth and causes tumor regression in various xenograft tumor models
The efficacy of GRP78 antibody MAb159 in vivo was examined in various tumor xenograft models including HT29 (colon cancer), H249 (small cell lung carcinoma), and A549 (lung adenocarcinoma). These cells have relatively high (4.6–9.4%, Figure S5) surface GRP78 expression compared to normal cells (15). MAb159 treatment led to 50%, 58%, and 78% tumor growth inhibition in these models, respectively (Figure 2A).
Fig. 2. MAb159 inhibits Xenograft Tumor Growth and Attenuates PI3K Signaling.

(A) The efficacy of MAb159 was examined in HT29, H249, and A549 xenograft models. The tumors were treated with 10 mg/kg antibodies, twice a week. On the right, MAb159 enhanced the efficacy of Irinotecan (18 mg/kg, twice a week) in colon cancer colo205 xenograft model. (B) Representative immunofluorescence staining of MAb159 treated A549 tumors. MAb159 treatment reduced cell proliferation (Ki67), induced apoptosis (TUNEL), and reduced phosphorylated S6. Nuclei were counterstained with DAPI (blue). (C) Similar to (B), immunohistochemistry staining shows that MAb159 reduced phosphorylated AKT level, but not phosphorylated MAPK (ERK1/2). Scale bar, 100 μm. All quantification data above are presented as mean ± SEM. Asterisk, double asterisks and triple asterisks indicate P < 0.05, P < 0.02, and P < 0.001 respectively, as determined by an unpaired 2-tail student T-test. NS, not significant. (D) The frozen sections of A549 tumors (four tumors from each group) were lysed and the combined lysates were subjected to Western blotting. Quantification of relative phosphorylated AKT, S6, Erk1/2, and Src was performed with Odyssey (Li-Cor).
To further test if the combination of GRP78 targeted therapy and conventional chemotherapy leads to greater efficacy, we combined MAb159 and Irinotecan in a colon cancer xenograft model. Colon cancer model was chosen because GRP78 overexpression was reported to be associated with colorectal carcinogenesis (25). When administrated alone, MAb159 inhibited tumor growth by 54% compared to control group. Irinotecan mono-therapy inhibited tumor growth by 85%. The combination therapy caused tumor regression to 47% of the starting tumor volume (Figure 2A).
MAb159 reduces proliferation, induces apoptosis, impairs tumor vasculature, and inhibits PI3K signaling in tumor xenografts
At the end of the xenograft experiments, tumors were harvested for analysis. In MAb159 treated group, proliferation index (Ki67 staining) was markedly reduced, apoptosis (TUNEL assay) was significantly increased (Figure 2B), and vessel density (CD31 staining) had a modest decrease (Figure S6A). MAb159 treatment also led to a marked reduction in phosphorylated AKT, mTOR, and S6 and (Figure 2B–D and Figure S6B), indicating inhibition of PI3K signaling. Meanwhile, there was no increase in phosphorylated ERK1/2 and Src (Figure 2C and D) indicating these compensatory pathways often induced with PI3K inhibitors and resistance to therapy were not activated. In addition it is notable that the systemic administration of the antibody was well tolerated measured by the animal food intake, body weight (data not shown) and microscopic examination of vital organs (Figure S7).
MAb159 inhibits tumor metastasis
GRP78 promotes growth of blood vessels in the tumor accompanied by tumor growth and metastasis (9). Targeting surface GRP78 with a peptide conjugated to pro-apoptosis molecules inhibits metastases (19). Therefore, we tested MAb159 in an orthotopic tumor model using mouse breast adenocarcinoma cell 4T1 (26, 27). 4T1 cells were implanted into the #4 fat pads of isogenic BALB/c mice, which were treated bi-weekly with MAb159 at 10 mg/kg or saline. Within 7 days MAb159 inhibited primary tumor growth and secondary metastasis to #9 fat pads (Figure S8A, n=2). After 13 days metastatic tumors were present on the liver surface of control animals (n=3) but not in MAb159-treated mice (n=3) (Figure 3A, left). Further focal liver necrosis was seen in control but not in MAb159-treated animals (Figure 3A and data not shown). By day 14 in all control mice both primary tumor and #9 fat pad secondary tumors had become large and invaded through body wall into underlying peritoneal cavity. In comparison, MAb159-treated mice exhibited complete or near complete primary tumor regression (Figure S8B and data not shown). There was no visible contralateral metastasis in any of the MAb159-treated animals. Histological evaluation of lung metastasis showed that the majority of the lung volume of control animals was occupied by metastatic breast cancer that resulted in internal hemorrhaging in 50% of control lungs (Figure 3A, right). Lung metastasis was significantly inhibited by MAb159 treatment (Figure 3B) and there was no evidence of internal hemorrhaging in any of the M159-treated animals (Figure 3A, right). Analysis of breast tumor tissues shows that MAb159 significantly reduced pS6 level, indicative of inhibition of PI3K signaling (Figure S8C).
Fig. 3. MAb159 inhibits Xenograft Tumor Metastasis.
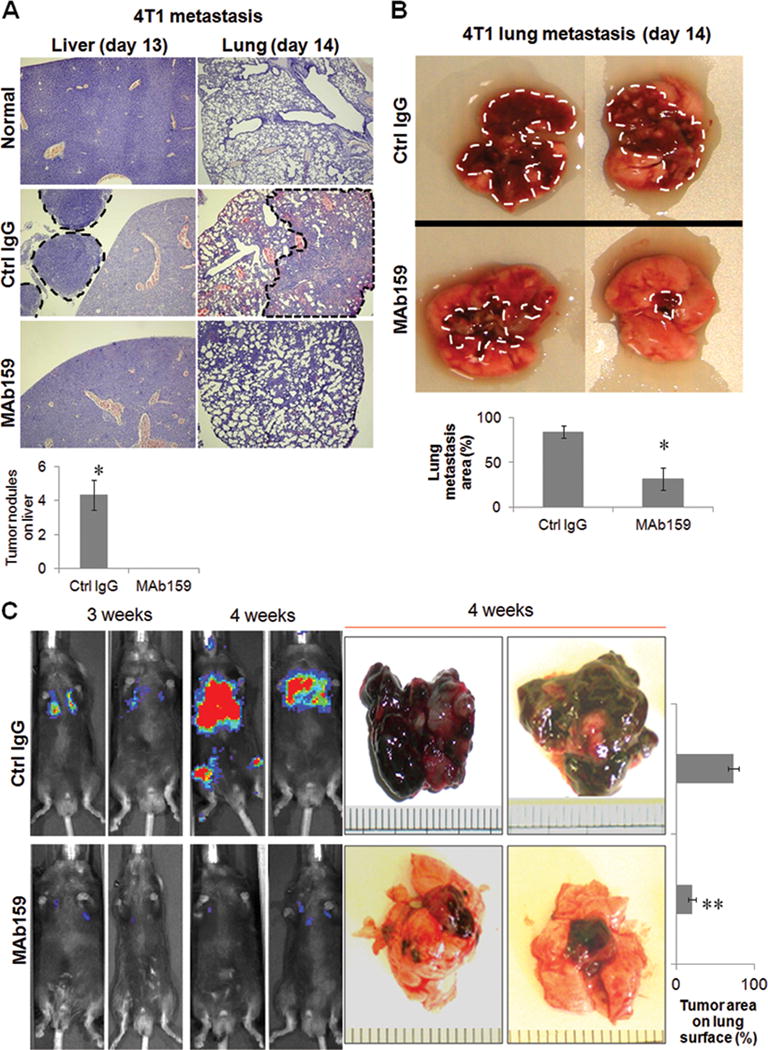
(A) MAb159 inhibited lung and liver metastasis in a 4T1 orthotopic breast cancer model. Hematoxylin and eosin (H&E) staining pictures of lung and liver from normal mice with no injection of 4T1 cells are shown on top. On day 13, no tumor was seen on the liver of MAb159 treated mice, whereas large tumors were observed on the liver of control group mice (left panel, demarcated with black dotted line). Quantification of tumor nodules per liver is shown on the bottom. On day 14, large areas of lung were infiltrated with tumor cells in the control group (demarcated with black dotted line), whereas only few tumor cells could be observed in MAb159 treated lungs (right panel). (B) Representative pictures of lung metastasis on day 14. MAb159 treatment led to significantly less metastasis compared to control IgG group. Tumor is demarcated by dashed white line. (C) MAb159 inhibits lung metastasis of B16 melanoma cell. Mice were treated with 10 mg/kg MAb159, twice a week, right after the injection of B16 cells expressing luciferase. Live mouse images taken 3 and 4 weeks after B16 cell injection are shown on the left. The representative pictures of harvested lungs at the end of the experiment (4 weeks) are shown on the right. The number of pigmented tumors on the lung surface was much less in MAb159 treated group than the control group. Quantification data are presented as mean ± SEM (n=3). Signals were quantified with Image J. Asterisk and double asterisks indicate P < 0.05 and P < 0.002 respectively, as determined by an unpaired 2-tail student T-test.
We also examined the effect of MAb159 on the metastatic growth of a syngeneic melanoma cancer cell line (9). B16-Fluc-A1 melanoma cells stably expressing luciferase were injected intravenously. Tumor metastasis and progression in the lungs was monitored live with a whole animal luminescence imaging system. MAb159 treatment significantly reduced the lung tumor formation (Figure 3C, left). At the end of the experiment, lungs were harvested and pigmented tumors were observed on the lung surface. Compared to the control group, the lungs from MAb159 treated mice had significantly fewer tumors (Figure 3C, right).
MAb159 suppresses PTEN deletion induced prostate cancer progression and leukemogenesis
We tested MAb159 in the setting of constitutively active PI3K in the PTEN knockout spontaneous tumor models including inducible PTEN knock out in prostate and hematopoietic system. In PTEN knockout spontaneous prostate cancer model, PTEN deletion is achieved with induction of Cre under probasin promoter. The luciferase expression is also induced by the same Cre so PTEN deficient prostate cells can be imaged with a luminescence imaging system (22). Tumor develops in prostate in 2–3 months. Control IgG or MAb159 was given to 2 month old PTEN null mice three times a week at 10 mg/kg dose. Tumor progression was monitored with live animal luminescence imaging. In MAb159 treated group, there was marked tumor regression (Figure 4A). In contrast, mice in control IgG treated group uniformly progressed. Histological analysis of dorsolateral prostate indicates that control IgG treated prostate had extensive adenocarcinoma (Figure 4B). In contrast, MAb159 treated prostate only had mild prostate interepithelial neoplasia (Figure 4B). Further immunohistochemical analysis shows that MAb159 significantly reduced pAKT and pS6 levels, suggesting inhibition of PI3K signaling (Figure 4B). There was an insignificant decrease in phosphorylated ERK in MAb159 treated group (Figure 4B). We also studied the efficacy of MAb159 in the xenograft model of a hormone refractory mouse prostate cancer cell line CE1 that is established from postcastration recurrent tumor of PTEN deficient mouse (20). More than 50% CE1 tumor growth inhibition was achieved with MAb159 treatment (Figure S9).
Fig. 4. MAb159 Inhibits PTEN loss Induced Spontaneous Prostate Cancer.
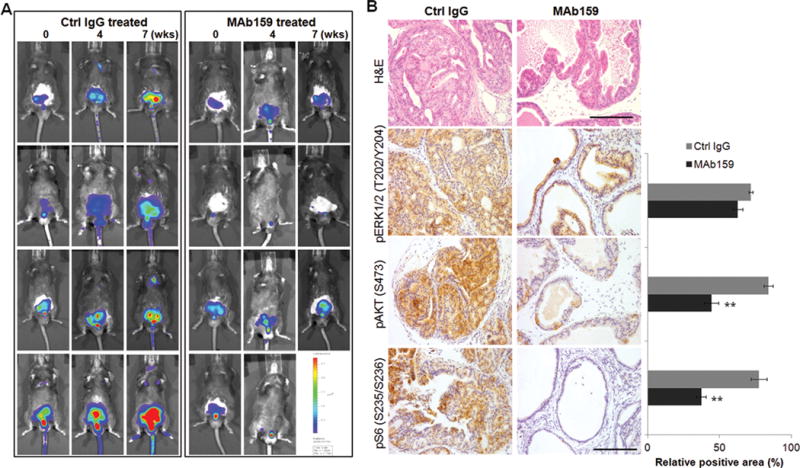
(A) Live imaging pictures of prostate specific PTEN knockout mice treated with control IgG or MAb159. MAb159 significantly inhibited prostate tumor growth. Fixed imaging setting was used throughout the study. One MAb159 treated mouse died from anesthesia at week 4. (B) H&E staining of dorsolateral prostate harvested from (A) is shown on the top. Control IgG treated prostate shows large area of prostatic adenocarcinoma, whereas MAb159 treated mice only shows mild prostate interepithelial neoplasia. Immunohistochemical stainings of pS6, pAKT, and pERK are shown on the bottom and corresponding signal quantification is shown on the right. Signal was quantified with Image J. Quantification data are presented as mean ± standard error of the mean (SEM) (n=4). Double asterisks indicate P < 0.002, as determined by an unpaired 2-tail student T-test. Scale bar, 100 μm.
As reported previously, inducible knockout of PTEN in the hematopoietic system leads to development of myeloproliferative disorders and eventual leukemia, which is suppressed by GRP78 haploinsufficiency (11, 28). These PTEN knockout mice when treated with control IgG had morbid hunched posture, while those treated with MAb159 appeared normal (Figure 5A). PTEN deficiency led to significant increase in leukemic blast cells in the bone marrow, as well as increase in spleen weight. PTEN deficient mice treated with MAb159 showed a prominent reduction in spleen size and percentage of blast cells in the bone marrow, compared to those injected with control IgG (Figure 5B), and restoration of white blood cells, lymphocytes, monocytes, and granulocytes similar to the level of wild type mice (Figure 5C). The effect on PI3K signaling was measured by the level of phosphorylated AKT with Western blotting (Figure 5D). Consistent with suppression of AKT activation following inducible heterozygous ablation of GRP78 in the same PTEN null model (11), MAb159 decreased phosphorylated AKT to normal level in PTEN deficient mice.
Fig. 5. MAb159 Suppresses PTEN Loss Induced Spontaneous Leukemia.

(A) Hunched posture of PTENf/f; MX1-Cre mice treated with IgG compared to normal posture of those treated with MAb159. (B) Quantification of the spleen weight and leukemic blast cell percentages of WT (n=4), cPf/f treated with IgG (n=5), and cPf/f treated with MAb159 (n=8). All data are presented as mean ±s.e. (C) Complete blood count with tail peripheral blood from WT (n=4), cPf/f treated with IgG (n=5), and cPf/f treated with MAb159 (n=8). All data are presented as mean ±s.e. Asterisk and double asterisks indicate P < 0.05 and P < 0.01 respectively, as determined by an unpaired 2-tail student T-test. (D) Representative Western blot results using bone marrow cell lysates for detection of the indicated protein levels. For each group, samples from two mice were used for analysis.
Affinity, Activity, and Specificity of Humanized MAb159
We have selected the antibody with the desired features suitable for therapeutic development. Next we made humanized MAb159 to avoid potential immunogenicity in humans. This antibody has affinity to GRP78 (Figure 6A) and efficacy in tumor growth inhibition (Figure 6B) comparable to the parental murine antibody.
Fig. 6. Characterization of Humanized MAb159.

(A) Comparison of the affinity and anti-tumor activity of MAb159 and humanized MAb159 using competition ELISA. Various amounts of murine and humanized MAb159 were tested for competition against a fixed concentration of biotinylated murine MAb159 for binding to GRP78-His. The binding of biotinylated MAb159 decreases with increasing amounts of both murine and humanized MAb159. Humanized MAb159 shows slightly higher affinity for GRP78 than murine MAb159. (B) A549 xenograft were treated with normal mouse IgG, murine MAb159, and humanized MAb159 (n=8 each). Antibodies were administered at 10 mg/kg, 2 times a week. Data are presented as mean ± SEM. Double asterisks indicate P < 0.005, as determined by an unpaired 2-tail student T-test. (C) Tumor bearing mice were administered with Cy5.5 conjugated humanized MAb159 or normal human IgG and fluorescent whole mouse images were taken 28 hours after injection (left panel). The mice were subsequently perfused with PBS and formalin, and the ex vivo images of the major organs were taken (right panel). MAb159, but not control IgG, shows specific localization to tumor. Quantification of the fluorescent intensity is shown on the bottom. (D) The organs in (C) were immunostained with a human Fc specific antibody, showing the preferential localization of humanized MAb159 to the tumor. Normal IgG localized to both the liver and the tumor. Scale bar, 100 μm. Quantification data are presented as mean ± SEM (n=4). Signals were quantified with Image J. Double asterisks indicate P < 0.001, as determined by an unpaired 2-tail student T-test.
To determine the tumor specific localization of humanized MAb159, we imaged tumor-bearing live mice injected with Cy5.5-labeled humanized MAb159. We found that 28 hours after injection, humanized MAb159 preferentially localized to H249 tumors but not to mouse organs (Figure 6C, left). At the end of the study, the normal organs along with tumors were harvested for an ex vivo imaging. There was a dramatic difference in the signal intensity in tumor between control IgG and humanized MAb159 (Figure 6C, right). The tissues were further subjected to immunohistochemistry analysis using a human Fc specific antibody, confirming the specific localization of humanized MAb159, but not control IgG to tumor (Figure 6D).
Pharmacokinetics and toxicology studies of humanized MAb159
We performed pharmacokinetics and toxicology studies of humanized MAb159 in mouse. A single 10 mg/kg dose of humanized Mab159 was administered i.v. and serum levels of the antibody were measured at designated time points. The mean maximum serum concentration (Cmax) of 61.5 μg/mL was achieved. The mean serum half-life was over 3 days (Table S1). At 10 mg/kg of humanized MAb159, an area under curve (AUC) of 4045 ± 1026 μg*hr/mL was achieved. Toxicology study was conducted in C57BL/7 mice treated 2 times a week for 5 weeks with 10 mg/kg humanized MAb159. Overall, there was no significant toxicity found in either the blood or vital organs of humanized MAb159 treated mice (Table S2). No histologic abnormalities were seen except for mild inflammation in the pancreas of a control and humanized MAb159 treated mouse. These results provide good safety and pharmacokinetics data to proceed to human clinical trials.
DISCUSSION
Cell surface GRP78 is a multifunctional receptor and a potential target for cancer therapy. For example, pro-apoptotic moieties or cytotoxic agents were conjugated onto peptides with high affinity for GRP78 to successfully target and kill cancer cells (12, 19). Recently an unconjugated peptidic GRP78 ligand also demonstrated toxicity to prostate cancer cell (29). A human monoclonal IgM antibody against cell surface GRP78 isolated from a cancer patient is capable of inducing lipid accumulation and apoptosis in cancer cells (30). Here we screened and identified a novel monoclonal antibody which recognizes both human and mouse GRP78 with high specificity and potently inhibits tumor growth and causes tumor regression in xenograft tumor models and spontaneous tumor models. MAb159 also suppresses tumor metastasis in multiple models. Furthermore, the humanized form of this antibody is efficacious and without toxicity.
Cell surface GRP78 is an upstream regulator of PI3K/AKT signaling. GRP78 interacts with Cripto and α2-macroglobulin on cell surface and is required for these factors to activate downstream PI3K/AKT signaling (16, 31, 32). Recently, a murine monoclonal antibody against GRP78 was reported to suppress AKT activation in a melanoma model (33). Here we show that surface GRP78 is in the same complex as the p85 subunit of PI3K, and treatment of cells with the GRP78 specific antibody MAb159 led to marked reduction in PI3K signaling in both cultured cells and in multiple tumor models. The spontaneous tumor models include PTEN deletion induced spontaneous prostate cancer and leukemia models. Furthermore, in tumor xenografts subjected to long term treatment with MAb159 for up to 30 days, inhibition of PI3K pathway was sustained and there was no evidence for induction of escape mechanisms of resistance to PI3K inhibition such as MAPK (34). Altogether, these highlight the importance of surface GRP78 in PI3K/AKT signaling and support the clinical investigation of MAb159 for PI3K driven tumors. However, cell surface GRP78 may also regulate other critical oncogenic pathways. For example, MAb159 is active to cell lines with Kras mutation (A549) and Braf mutation (HT29 and Colo205). In light of MAb159’s pro-apoptotic activity, we are also investigating the role of surface GRP78 in the extrinsic apoptosis pathway.
Recent studies reported that surface GRP78 is highly elevated in tumor associated vasculature and required for endothelial cell proliferation and survival (7, 9). These findings indicate that cell surface GRP78 targeting agents will have dual function: targeting tumor cell and tumor vasculature. Consistently, we have observed that MAb159 reduced the tumor endothelial cell density and angiogenesis dependent tumor metastasis, supporting dual targeting.
Antibody tracking and in vivo imaging studies showed that MAb159 is strictly localized to the tumor and not normal organs, indicating that antibody targeting is highly specific. In agreement with this, formal toxicology study in mice showed that the humanized antibody was well tolerated and did not induce any noticeable organ toxicity or changes in blood counts or blood chemistry.
Another potential application of MAb159 lies in its ability to be used for in vivo imaging. MAb159 specifically recognizes surface GRP78, and thus can be used to image the tumor for personalized medicine and determine whether the amount of surface GRP78 in the tumor predicts disease progression and response to therapy. Clinical trials will incorporate patient imaging as a screening process for inclusion of study subjects. This is particular important in cell surface GRP78 targeted therapy: when analyzing archival tumor samples with immunostaining, intracellular GRP78 will interfere with such analysis.
In conclusion, we have developed a novel antibody MAb159 targeting surface GRP78 expressed on tumor cells and tumor endothelial cells. This antibody disrupts PI3K signaling pathway and induces apoptosis in tumor cells, while sparing normal cells. It has the potential as both a therapeutic and diagnostic agent in cancer.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
A major obstacle in cancer therapy is the damage to normal organs by conventional chemotherapy and radiotherapy. This highlights the need for therapy that specifically target and kill cancer cells while sparing normal cells. One emerging target is GRP78, which is preferentially expressed on cancer cell surface and promotes tumor cell survival and metastasis. We have developed a monoclonal antibody MAb159 against GRP78 to target surface GRP78 and block its oncogenic functions. MAb159 effectively images tumors in vivo, suppresses PI3K/AKT signaling, induces apoptosis, and induces tumor regression in xenografts and spontaneous tumor models. The humanized MAb159 retains its GRP78 binding affinity and efficacy and is non-toxic to normal organs. We plan to initiate human clinical trial shortly.
Acknowledgments
We thank Dr. Rui Yan, Dr. Ram Subramanyan and Dr. Darryl Shibata for their technical assistance and scientific discussion in this study.
Grant Support: Research reported in this publication was supported by the L.K. Whittier Foundation (A.S.L; P.G) and the National Cancer Institute of the National Institute of Health under award number R01CA027607 (A.S.L and P.G).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
AUTHOR CONTRIBUTIONS
PSG, ASL, PC, and RL conceived the study, and participated in its design and coordination. RL, PSG, ASL, PC, and ZL drafted the manuscript. RL, XL, WG, YZ, SW, SM, VK, DD, SL, DL, GZ, SL, and ZL carried out the experiments. All authors read and approved the final manuscript.
References
- 1.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: Friend or foe? Nat Rev Cancer. 2004;4:966–77. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 2.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–18. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 4.Lee AS. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007;67:3496–9. doi: 10.1158/0008-5472.CAN-07-0325. [DOI] [PubMed] [Google Scholar]
- 5.Li Z, Li Z. Glucose regulated protein 78: A critical link between tumor microenvironment and cancer hallmarks. Biochim Biophys Acta. 2012;1826:13–22. doi: 10.1016/j.bbcan.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–16. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 7.Virrey JJ, Dong D, Stiles C, Patterson JB, Pen L, Ni M, et al. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol Cancer Res. 2008;6:1268–75. doi: 10.1158/1541-7786.MCR-08-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 9.Dong D, Stapleton C, Luo B, Xiong S, Ye W, Zhang Y, et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res. 2011;71:2848–57. doi: 10.1158/0008-5472.CAN-10-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:19444–9. doi: 10.1073/pnas.0807691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wey S, Luo B, Tseng CC, Ni M, Zhou H, Fu Y, et al. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses pten-null leukemogenesis and AKT oncogenic signaling. Blood. 2012;119:817–25. doi: 10.1182/blood-2011-06-357384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell. 2004;6:275–84. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: A multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009;11:2299–306. doi: 10.1089/ARS.2009.2568. [DOI] [PubMed] [Google Scholar]
- 14.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181–8. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem. 2010;285:15065–75. doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC. GRP78 and cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor beta signaling and enhance cell growth. Mol Cell Biol. 2008;28:666–77. doi: 10.1128/MCB.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Steiniger SC, Kim Y, Kaufmann GF, Felding-Habermann B, Janda KD. Mechanistic studies of a peptidic GRP78 ligand for cancer cell-specific drug delivery. Mol Pharm. 2007;4:435–47. doi: 10.1021/mp060122j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato M, Yao VJ, Arap W, Pasqualini R. GRP78 signaling hub a receptor for targeted tumor therapy. Adv Genet. 2010;69:97–114. doi: 10.1016/S0065-2660(10)69006-2. [DOI] [PubMed] [Google Scholar]
- 19.Miao YR, Eckhardt BL, Cao Y, Pasqualini R, Argani P, Arap W, et al. Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin Cancer Res. 2013;19:2107–16. doi: 10.1158/1078-0432.CCR-12-2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao CP, Liang M, Cohen MB, Flesken-Nikitin A, Jeong JH, Nikitin AY, et al. Mouse prostate cancer cell lines established from primary and postcastration recurrent tumors. Horm Cancer. 2010;1:44–54. doi: 10.1007/s12672-009-0005-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krasnoperov V, Kumar SR, Ley E, Li X, Scehnet J, Liu R, et al. Novel EphB4 monoclonal antibodies modulate angiogenesis and inhibit tumor growth. Am J Pathol. 2010;176:2029–38. doi: 10.2353/ajpath.2010.090755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao CP, Zhong C, Saribekyan G, Bading J, Park R, Conti PS, et al. Mouse models of prostate adenocarcinoma with the capacity to monitor spontaneous carcinogenesis by bioluminescence or fluorescence. Cancer Res. 2007;67:7525–33. doi: 10.1158/0008-5472.CAN-07-0668. [DOI] [PubMed] [Google Scholar]
- 23.Vercauteren D, Vandenbroucke RE, Jones AT, Rejman J, Demeester J, De Smedt SC, et al. The use of inhibitors to study endocytic pathways of gene carriers: Optimization and pitfalls. Mol Ther. 2010;18:561–9. doi: 10.1038/mt.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin SY, Lee KS, Choi YK, Lim HJ, Lee HG, Lim Y, et al. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis. 2013 doi: 10.1093/carcin/bgt169. Epub 2013 Jun 20. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi H, Wang JP, Zheng HC, Masuda S, Takano Y. Overexpression of GRP78 and GRP94 is involved in colorectal carcinogenesis. Histol Histopathol. 2011;26:663–71. doi: 10.14670/HH-26.663. [DOI] [PubMed] [Google Scholar]
- 26.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–405. [PubMed] [Google Scholar]
- 27.Bao L, Haque A, Jackson K, Hazari S, Moroz K, Jetly R, et al. Increased expression of P-glycoprotein is associated with doxorubicin chemoresistance in the metastatic 4T1 breast cancer model. Am J Pathol. 2011;178:838–52. doi: 10.1016/j.ajpath.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 29.Maddalo D, Neeb A, Jehle K, Schmitz K, Muhle-Goll C, Shatkina L, et al. A peptidic unconjugated GRP78/BiP ligand modulates the unfolded protein response and induces prostate cancer cell death. PLoS One. 2012;7:e45690. doi: 10.1371/journal.pone.0045690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rauschert N, Brandlein S, Holzinger E, Hensel F, Muller-Hermelink HK, Vollmers HP. A new tumor-specific variant of GRP78 as target for antibody-based therapy. Lab Invest. 2008;88:375–86. doi: 10.1038/labinvest.2008.2. [DOI] [PubMed] [Google Scholar]
- 31.Misra UK, Gonzalez-Gronow M, Gawdi G, Hart JP, Johnson CE, Pizzo SV. The role of grp 78 in alpha 2-macroglobulin-induced signal transduction evidence from RNA interference that the low density lipoprotein receptor-related protein is associated with, but not necessary for, GRP 78-mediated signal transduction. J Biol Chem. 2002;277:42082–7. doi: 10.1074/jbc.M206174200. [DOI] [PubMed] [Google Scholar]
- 32.Misra UK, Pizzo SV. Receptor-recognized alpha(2)-macroglobulin binds to cell surface-associated GRP78 and activates mTORC1 and mTORC2 signaling in prostate cancer cells. PLoS One. 2012;7:e51735. doi: 10.1371/journal.pone.0051735. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.de Ridder GG, Ray R, Pizzo SV. A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma Res. 2012;22:225–35. doi: 10.1097/CMR.0b013e32835312fd. [DOI] [PubMed] [Google Scholar]
- 34.Camp ER, Summy J, Bauer TW, Liu W, Gallick GE, Ellis LM. Molecular mechanisms of resistance to therapies targeting the epidermal growth factor receptor. Clin Cancer Res. 2005;11:397–405. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.