

Abstract
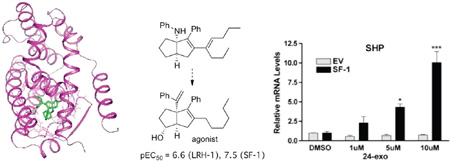
The crystal structure of LRH-1 ligand binding domain bound to our previously reported agonist 3-(E-oct-4-en-4-yl)-1-phenylamino-2-phenyl-cis-bicyclo[3.3.0]oct-2-ene 5 is described. Two new classes of agonists in which the bridgehead anilino group from our first series was replaced with an alkoxy or 1-ethenyl group were designed, synthesized, and tested for activity in a peptide recruitment assay. Both new classes gave very active compounds, particularly against SF-1. Structure-activity studies led to excellent dual-LRH-1/SF-1 agonists (e.g., RJW100) as well as compounds selective for LRH-1 (RJW101) and SF-1 (RJW102 and RJW103). The series based on 1-ethenyl substitution was acid stable, overcoming a significant drawback of our original bridgehead anilino-substituted series. Initial studies on the regulation of gene expression in human cell lines showed excellent, reproducible activity at endogenous target genes.
INTRODUCTION
The nuclear receptor (NR) superfamily in mammals comprises a highly conserved group of 49 receptors that act as transcription factors to regulate development, homeostatic physiology, and cellular metabolism.1 Nearly half of all NRs are ligand regulated, responding to dietary or endocrine signals such as steroid hormones, retinoids, vitamin D, fatty acids, and thyroid hormone. As such, NRs are attractive targets for drug discovery,2 and indeed, 13% of current FDA-approved drugs regulate NRs.3 NRs without assigned natural ligands are referred to as orphan NRs.4 On the basis of structural analyses of nearly all NR subfamilies, it is predicted that most orphan receptors might accommodate ligands, thus providing novel targets for pharmaceuticals. Finding natural or synthetic regulatory ligands for these orphan receptors is key to understanding their role in physiology,5 a process termed “reverse endocrinology”.6 Attempts to “adopt” (or deorphanize) orphan NRs have been successful for NR1H4 (farnesoid X receptor, FXR)7 and peroxisome proliferator-activated receptor family (PPARs, NR1Cs)8 and have led to useful therapeutics.
Here, we have undertaken an effort to identify synthetic ligands for subfamily V members, including steroidogenic factor-1 (SF-1, NR5A1),9 and its close homologue, liver receptor homologue-1 (LRH-1, NR5A2).10 Both receptors are known to play important roles during embryonic development and in adult physiology.
SF-1 (NR5A1) is a critical factor in vertebrate endocrine organ development, including male sexual differentiation, and is an important regulator of steroidogenic enzymes.11 Numerous genetic mutations in the DNA binding domain (DBD) and ligand binding domain (LBD) of SF-1 correlate with abnormal testis development12 and premature ovarian failure.13 By contrast, amplification of SF-1 is associated with adrenocortical tumors, with inverse agonists reported to inhibit proliferation of primary cultured tumor cells.14 SF-1 has also been implicated in the development of endometriosis and endometrial cancers.15 Finally, rodent models suggest that SF-1 expressed in the hypothalamus influences anxiety, energy homeostasis, and appetite.16
LRH-1 plays a critical role in embryonic development of the endoderm17 and is highly expressed in the intestine, liver, exocrine pancreas, and ovary.18 LRH-1 regulates crucial enzymes in hepatic bile acid biosynthesis19 and cholesterol homeostasis10 and is a key controller in the hepatic acute phase response.20 Thus, this receptor may be a target for the treatment of cardiovascular disease21 and cholinostatic liver disease.22 LRH-1 and SF-1 both regulate aromatase expression,23 and LRH-1 is expressed in several human breast cancer cell lines, suggesting that antagonists in combination with traditional aromatase inhibitors might be effective in reducing local concentrations of estrogen in human breast carcinomas.24 Finally, LRH-1 is expressed in human intestinal crypt cells where it controls cell proliferation and differentiation.25 Its expression26 and potential role in gastric cancer27 suggest that LRH-1 can influence tumor formation by accelerating the cell cycle and promoting inflammation.28
Both SF-1 and LRH-1 have emerged as potential stem cell factors because both can regulate expression of Oct-4, which is one of the four factors found to induce pluripotency of mouse embryonic fibroblasts.17,29 Moreover, these two NRs can completely replace Oct-4 to reprogram murine somatic cells to induce pluripotent cells30a and are also potent inducers in the transition from an epiblast stem cell to ground state pluripotency (iPS).30b As such, identifying effective agonists to either SF-1 or LRH-1 might help in achieving a pharmaceutical approach in generating or maintaining pluripotent human stem cells.
LRH-1 and SF-1 bind to DNA as monomers and show constitutive activity when expressed in a variety of cell types.9,10 Receptor activity can be regulated by post-translational modifications including phosphorylation31 and sumoylation32 or through interaction with the atypical orphan receptors that lack a DBD, including small/short heterodimer partner (SHP, NR0B1)33 and DAX-1, Dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1) (NR0B2).34
X-ray crystallography coupled with mass spectroscopy revealed the presence of Escherichia coli-derived phospholipids, phosphatidylglycerides (1; Figure 1) and phosphatidylethanol-amines (2a) in the ligand binding pockets of human LRH-1 and SF-1.35 The majority of phospholipids bound to either SF-1 or human LRH-1 LBD were identified as having C16 or 18 acyl groups with some showing a cis-alkene in the Δ9 position. Whether these phospholipids merely serve as structural ligands or serve a regulatory role remains to be established. Support for phospholipids as regulatory ligands includes increased coactivator peptide recruitment on binding of phospholipids to SF-135c and a correlation between binding of phospholipid and LRH-1-induced activation of gene expression.35d The exchange of bacterially derived phosphatidyl glyceride (1) with phospholipids such as 335b and phosphatidycholine (2b) has been demonstrated.36 In the latter case, the exchange had little effect on binding of coactivator peptides.36 Recently, Moore has claimed in a patent that diundecanoyl (2b, R1 = R2 = nC9H19) and dilauroyl (2b, R1 = R2 = nC11H23) phosphatidylcholine (PC) act as agonists of the LRH-1 receptor and that administration of these lipids to diabetic mice reduces blood glucose levels.37 Other correlative data include the finding that SF-1 activation by a tamoxifen analogue increases cellular levels of phosphatidylinositol (3,4,5) triphosphate (3).15c Finally sphingosine (4) is reported to bind SF-1 and suppress expression of the SF-1 target gene CYP17a1.38 Derivatives of (4) such as sphingosylpho-sphorylcholine and N-Acyl-4 (ceramides) may also be natural ligands.
Figure 1.

Natural ligands for LRH-1 or SF-1.
We reported the first synthetic small molecule agonist for LRH-1 and SF-1, GSK8470 (5), and a structure–activity relationship (SAR) study on the series 6 leading to the more active analogue 7 (Figure 2).39 Another small molecule known to activate both SF-1 and LRH-1 is the herbicide atrazine,40 but the precise mechanism leading to this increased receptor activity has yet to be defined.
Figure 2.

Synthetic compounds reported to modulate SF-1 or LRH-1 activity.
Recently, p-heptyloxyphenol (8) was reported as a potent (IC50 = 7.3 µM) inverse agonist for SF-1 but with no activity against LRH-1.41 Expression of several reported SF-1 target genes was suppressed by 8 in cell-based assays. However, although 8 was reported to inhibit proliferation of adrenocortial carcinoma cell line H295R, it had the same effect with an SF-1 negative cell line (SW-13).14 Several isoquinoline-based inverse agonists of SF-1 were discovered through high-throughput screening and further SAR studies, of which 9 was most active (IC50 = 200 nM).41,42 At high levels (10 µM), 9 modestly suppressed doxycycline (DOX) induced proliferation of H295R cell line with little effect on the SF-1 negative SW-13 cell line. Closely related isoquinolines affected both cell lines in a similar way, suggesting a non-SF-1 mechanism. The SAR for these series seems complex; for example, replacing the methoxy group in 9 with ethoxy gave a compound highly active against a range of other receptors and exhibiting strong cell toxicity, suggesting transactivation assay artifacts (promiscuous inhibition of the reporter system).
Although 5 and related compounds 6 have proven to be excellent biological probes for LRH-1 and SF-1, they have a number of limitations. Most important is that they are unstable to acid, making handling difficult and raising doubts about stability during biological application. The series also shows little discrimination between LRH-1 and SF-1. The aim of this work was thus first to develop new series of compounds that were more stable and easily handled than series 6, but with at least comparable binding and efficacy, and second to find compounds that were selective for LRH-1 and/or SF-1.
COCRYSTAL STRUCTURE OF 5 WITH hLRH-1
As a basis for the design of improved analogues of 5, we solved the cocrystal structure of compound 5 bound to the human LRH-1 LBD (Figures 3 and 4). Although racemic 5 was used, the 1S,5R-bicyclo[3.3.0]oct-2-ene enantiomer cocrystallized with LRH-1 LDB. Compound 5 occupied the same binding pocket volume as the terminal 12 carbon atoms in both acyl chains of a phospholipid bound in LRH-1.35b This smaller size permitted LRH-1 to contract, narrowing the gap between the N-terminal half of helix 3 (residues 338–350, yellow in the PL-bound structure) and the β-turn/helix 6 region (residues 408–427, orange in the PL-bound structure) where the phospholipid headgroup is situated in the PL + LRH-1 structure, and completely enclose the ligand within the binding pocket. Making no polar interactions with the protein, 5 was a compact, hydrophobic mass complementing the inner surface of the binding pocket. There was π stacking between the His390 and the aniline of 5 with a centroid–centroid distance of 3.8 Å and a deviation from coplanarity of around 10°.
Figure 3.

Cα trace from the phospholipid35b (magenta, orange, and yellow) and 5 (white) bound conformations of LRH-1 were superimposed for comparison. The pocket contracts around the smaller 5, with the biggest shifts occurring for residues 338–350 in helix 3 (yellow) and residues 408–427 in the β-turn and helix 6 (orange), where the phospholipid headgroup protrudes.
Figure 4.

Compound 5 is shown in the LRH-1 ligand binding pocket. Contacting residues are indicated, along with the His390–aniline distance. The surface of the pocket is shown, colored by the nearest atom type. The region of the pocket adjacent to helix 5 and the β-turn contain polar residues and several trapped water molecules.
The structure suggested some chemical strategies for improving compound properties. The acid-labile aniline nitrogen made no polar interactions with the protein, suggesting that another more stable linker might be tolerated. We thus designed compounds with oxygen- and carbon-based linkages from the substituted bridgehead carbon. Additionally, His390, Arg393, and Gln432 were within 5 Å of the compound. Polar moieties could be incorporated to interact with these residues, which might both improve binding and increase hydrophilicity of the ligand (Figure 5).
Figure 5.

Two-dimensional compound interaction diagram depicting adjacent residues and the π stacking interaction with His390. The crystal structure is available from the RCSB with access code 3PLZ and is further described in the Experimental Section.
CHEMISTRY
Alkoxy-Substituted Series
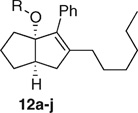
Racemic tertiary alcohol 11 was synthesized as previously reported39 from hept-6-en-1-yn-1-ylbenzene via Pauson–Khand cyclization to give the cyclopentenone 10 and cerium trichloride-assisted 1,2-addition of hexylmagnesium bromide (Scheme 1). Exposure of 11 to various alcohols in the presence of catalytic amounts of camphorsulphonic acid gave a series of racemic analogues 12 of 7 in which an alkoxy group replaced the aniline substituent. Yields for the last step were generally good (Table 1), the exception being when the alcohol carried an α-branch. The products 12 were unstable to silica, so were purified by column chromatography on basic grade III alumina.
Scheme 1.

Preparation of Bridgehead-Alkoxy Seriesa
aReagents and conditions: (a) Co2(CO)8, DMSO (10 equiv), THF, reflux, 5 h, 80%. (b) nC6H13MgBr, CeCl3, −10 °C to room temperature, 1 h, 90%. (c) ROH (10 equiv), camphorsulfonic acid (0.1 equiv), 20 °C, 3.5 h.
Table 1.
Synthesis of 1-Alkoxy-3-hexyl-2-phenyl-cis-bicyclo-[3.3.0]oct-2-enes 12a–j
| compound | R | yield (%)a |
|---|---|---|
| 12a | Me | 75 |
| 12b | Et | 69 |
| 12c | nPr | 71 |
| 12d | iPr | 7 |
| 12e | nBu | 68 |
| 12f | CH2CH(Me)(Et) | 64 |
| 12g | nPent | 62 |
| 12h | nHex | 65 |
| 12i | cHex | 12 |
| 12j | CH2Ph | 69 |
Isolated yield from 11.
"All Carbon" Series
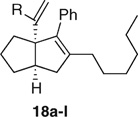
A previously reported43 tandem reaction sequence on zirconocene was used to construct “all carbon” analogues 18 of 7. Thus, intramolecular cocyclization of 1,6-enyne 13 gave the zirconacyclopentenes 14 (Scheme 2). The addition of a 1,1-dihaloalkyl compound followed by lithium diisopropylamide (LDA) or lithium 2,2,6,6-tetramethylpiperidide (LiTMP) generated a carbenoid R2X2CLi in situ, which when inserted into the alkyl-zirconium bond of 14 afforded the ring-expanded zirconacyclohexene 15. The addition of a lithiated alkyne induced a ring-closing rearrangement to the bicyclo[3.3.0]oct-1-ene 16, which rearranged further to the zirconocene alkylideneate complex 17, incorporating a second alkyne moiety. Aqueous workup gave the racemic 1-(2-alkenyl)-bicyclo[3.3.0]oct-2-enes 18a–w in generally excellent yield (Table 2) for a single-pot, three-component coupling. The yields were poorer when dichloromethane was the carbenoid precursor due to bis- and trisinsertion of the derived carbenoid into the zirconacycle.
Scheme 2.

Preparation of Bridgehead 1-Alken-2-yl-Substituted Systemsa
aReagents and conditions: (a) (i) Cp2ZrBu2, THF, −78 °C, 0.5 h; (ii) room temperature, 2 h. (b) R2 CHX2 (X = Br for R2 = nHex, nBu, nOct; X = Cl for R2 = H, SiMe2Ph), LDA or LiTMP, −78 °C, 15 min. (c) R3 C≡CLi (3 equiv), −78 to −60 °C over 0.5 h. (d) MeOH, NaHCO3, room temperature, 12–16 h.
Table 2.
Synthesis of 1-(2-Alkenyl)-bicyclo[3.3.0]oct-2-enes 18
| R1 | R2 | R3 | yield (%)a | |
|---|---|---|---|---|
| 18a | Ph | nHex | Ph | 86 |
| 18b | Ph | nHex | 3-MeOPh | 78 |
| 18c | Ph | nHex | 4-MeOPh | 72 |
| 18d | Ph | nHex | 4-MePh | 62 |
| 18e | Ph | nHex | 4-EtPh | 70 |
| 18f | Ph | nHex | 4-nBuPh | 78 |
| 18g | Ph | nHex | 4-tBuPh | 51 |
| 18h | Ph | nHex | 4-PhPh | 60 |
| 18i | Ph | nHex | nPr | 54 |
| 18j | Ph | nHex | nBu | 75 |
| 18k | Ph | nHex | nHex | 65 |
| 18l | Ph | nHex | nOct | 56 |
| 18m | 3-MeOPh | nHex | Ph | 75 |
| 18n | 4-EtPh | nHex | Ph | 70 |
| 18o | nPr | nHex | Ph | 82 |
| 18p | nBu | nHex | Ph | 88 |
| 18q | nHex | nHex | Ph | 86 |
| 18r | cHex | nHex | Ph | 80 |
| 18s | (CH2)3OH | nHex | Ph | 61 |
| 18t | Ph | H | Ph | 58 |
| 18u | Ph | nBu | Ph | 71 |
| 18v | Ph | nOct | Ph | 73 |
| 18w | Ph | SiMe2Ph | Ph | 45 |
Isolated yield of >95% pure material.
The tandem reaction sequence also worked for the formation of the racemic bicyclo[4.3.0]non-8-ene 19 and pyrrolidine fused systems 20a,b (Scheme 3). The low yield for compound 19 reflected a difficult separation from by-products —the yield estimated from the partially purified product was 46%.44
Scheme 3.

Preparation of Cyclohexyl- and Pyrrolidine-Fused Systemsa
aReagents and conditions: (a) (i) Cp2ZrBu2, THF, −78 °C, 0.5 h; (ii) room temperature, 2 h; (iii) nHexCHBr2, LDA, −78 °C, 15 min; (iv) PhC≡CLi (3 equiv), −78 to −60 °C over 0.5 h; (v) MeOH, NaHCO3, room temperature, 12–16 h.
Compounds carrying oxygen substitution on the saturated cyclopentane ring were required. The synthesis of the 6-oxygenated compounds is shown in Scheme 4.
Scheme 4.

Preparation of 6-Oxygenated Systemsa
aReagents and conditions: (a) (i) NaI, Me3SiCl, MeCN; (ii) EtOH, 0 °C to room temperature, 2 h. (b) PhC≡CLi (1.5 equiv), THF, HMPA (1.5 equiv), −78 °C, 1 h then room temperature, 14 h. (c) THF:H2O (4:1), HCl (2 M), room temperature, 3 h. (d) (i) CH2CHMgBr, −78 to −65 °C, 1 h; (ii) NH4Cl(aq). (e) tBuMe2SiOTf, imidazole, DMAP, THF, room temperature, 18 h. (f) (i) Cp2ZrBu2, THF −78 °C, 0.5 h then 3 h at room temperature; (ii) −78 °C, nHexCHBr2, LDA, 15 min; (iii) PhC≡CLi (3.0 equiv), −78 °C to −60 °C over 45 min; (iv) MeOH/NaHCO3(aq), room temperature, 5 h. (g) TBAF (5 equiv), THF, room temperature, 20 h. (h) Pyridine, Ac2O (73 equiv), DMAP (0.58 equiv), 13 h.
Using ethanol and in situ-generated Me3SiI, acrolein was converted into 1,1-diethoxy-3-iodopropane,45 which after rapid purification by chromatography on basic alumina (grade III) was reacted with lithium phenylacetylide to give (5,5-diethoxypent-1-yn-1-yl)benzene. Hydrolysis of the acetal to give an aldehyde was followed by reaction with vinylmagnesium bromide to afford the alcohol 21. Protection of the hydroxyl group with tBuMe2SiOTf gave the desired cyclization precursor 22, which underwent the zirconocene-induced cocyclization, carbenoid insertion, and phenyl acetylide addition to give a 1.6:1 mixture of the exo and endo isomers 23 after protonolysis. Tetrabutylammonium fluoride (TBAF) cleavage of the silyl group furnished the desired racemic bicyclic alcohols 24. The exo and endo isomers were separated by careful chromatography on silica, with the endo isomer eluting first. The relative stereochemistries of the exo and endo isomers were clear from coupling patterns to the proton adjacent to the hydroxyl group. In the endo isomer, it appears as a ddd, J = 9.1, 8.5, 5.5 Hz, and in the exo isomer, it appears as a broad singlet, the patterns being in accord with expectations from molecular modeling and the Karplus relationship of couplings to dihedral angles.46 The separated diastereoisomers of 24 were acylated to afford 25-endo and 25-exo. A crystal structure of 25-endo confirmed the stereochemical assignment47. The 7-oxygenated series was synthesized as shown in Scheme 5.
Scheme 5.

Preparation of 7-Oxygenated Systemsa
aReagents and conditions: (a) (i) NaH, THF, −10 °C; (ii) N-tosyl imidazole, −10 °C, 1 h. (b) PhC≡CLi (2 equiv), THF, HMPA (2 equiv), −10 to 0 °C over 1 h then room temperature, 40 h; (iii) saturated NaHCO3(aq). (c) Me2tBuSiOTf, imidazole, DMAP, THF, room temperature, 15 h. (d) (i) Cp2ZrBu2, −78 °C, 0.5 h then 3 h at room temperature; (ii) nHexCHBr2, LDA, −78 °C; (iii) PhC≡CLi (3 equiv), −78 °C to −60 °C over 45 min; (iv) MeOH/NaHCO3(aq), room temperature, 5 h. (e) TBAF (2 equiv), THF, room temperature, 20 h. (f) Pyridine, Ac2O (75 equiv), DMAP (0.58 equiv), room temperature, 17 h.
A single-pot ring-closure/ring-opening procedure was used to convert pent-4-ene-1,2-diol48 into alcohol 26.49 Thus, selective tosylation of the primary alcohol was followed by in situ ring closure to an epoxide and ring-opening with lithium phenylacetylide to afford the alcohol 26.50 The overall yield was poor despite considerable optimization. tBuMe2Si (TBDMS) protection of 26 gave 27, which underwent zirconocene-mediated cocyclization, dibromocarbenoid insertion, and phenylacetylide-driven zirconate rearrangement to give a 1:1 mixture of the exo and endo isomers 28. After TBAF cleavage of the silyl group, the racemic exo and endo isomers of 29 were separated by careful chromatography, with the endo isomer eluting first. Acylation of the separated diastereoisomers furnished 30-exo and 30-endo.
The relative stereochemistries of 29-exo and 29-endo (and hence 30-exo and 30-endo) were established by nuclear magnetic resonance (NMR) studies combined with molecular modeling (Table 3) using the MMFF94 force field as implemented in Spartan 06 (Wavefunction Inc.).51 The proton next to the hydroxyl group in one isomer appeared as a tt, J = 8.5, 5.9 Hz (due to couplings of 8.5, 8.3, 6.3, and 5.5 Hz), consistent with 29-exo or the equatorial conformer of 29-endo (29-endo eq. conf.), but as a quintet (J = 5.5 Hz) in the other diastereoisomer, not consistent with any minimum energy structure. Molecular modeling showed that 29-exo had a well-defined minimum energy conformer, but for 29-endo, the “equatorial” and “axial” hydroxy conformers (29-endo eq. and 29-endo ax.) were <1 kJ/mol different in energy. The expected coupling patterns for each conformer of 29-endo were calculated using the Altona modification46b of the Karplus relationship46a between dihedral angle and 3J as implemented in the Mspin program52 from Mestrec. The average showed a good correlation to the observed coupling constants (Table 3).
Table 3.
Correlation between Predicted and Observed Coupling Constants for 29
 | ||||||
|---|---|---|---|---|---|---|
| between | H7–H8b | H7–H8a | H7–H6b | H7–H6a | H6b–H5 | H6a–H5 |
| 29-exo | ||||||
| dihedral angle (°) | 164 | 47 | 154 | 37 | 14 | 104 |
| calcd 3J (Hz) | 10.2 | 6.1 | 8.9 | 7.6 | 9.7 | 1.9 |
| observed 3J (Hz) | 8.5 | 5.5 | 8.3 | 6.3 | 10 | 3.6 |
| 29-endo ax. conf. | ||||||
| dihedral angle (°) | 38 | 80 | 33 | 87 | 18 | 101 |
| Calcd 3J (Hz) | 5 | 1.3 | 5.9 | 1.3 | 9.4 | 1.6 |
| 29-endo eq. conf. | ||||||
| dihedral angle (°) | 44 | 159 | 46 | 165 | 35 | 155 |
| calcd 3J (Hz) | 6.6 | 9.5 | 6.3 | 10.4 | 7.3 | 10.5 |
| average calcd 29-endo eq. and –ax. conf. 3J (Hz) | 5.8 | 5.4 | 6.1 | 5.9 | 8.4 | 6.1 |
| observed 3J (Hz) | 6.2 | 4.6 | 5.6 | 5 | 9.5 | 6 |
Stability Tests
Compounds 18a, 24-exo, 24-endo, 28-exo, and 28-endo were stable after being kept in CDCl3 at room temperature in the presence of daylight for 2 weeks or being exposed to 1.0 equiv of (+)-camphorsulfonic acid in CDCl3 at room temperature for 1 week.
RESULTS AND DISCUSSION
The compounds 12a–j, 18a–w, 19, 20a,b, 24, 25, 29, and 30 were screened for activity against both hLRH-1 and hSF-1 using fluorescence resonance energy transfer (FRET)-based peptide recruitment assay. Purified bacterial-expressed LBDs35b of human LRH-1 or human SF-1 were labeled with biotin and incubated with APC-labeled streptavidin (Molecular Probes). Peptides derived from TIF-2 amino acids 737–757 (B-QEPVSPKK-KENALLRYLLDKDDTKD-CONH2) for LRH-1 or from DAX-1 amino acids 1–23 (B-MAGENHQWQGSILYNMLMSAKQT-CONH2) for SF-1 were labeled with biotin and incubated with europium-labeled streptavidin (Perkin-Elmer, Wallac). The labeled receptor and peptide were incubated in the presence of various concentrations of test compound, and the associated complexes were quantified by time-resolved fluorescence energy transfer (TR-FRET). The pEC50 values of the test compounds, which serve as a measure of the binding affinity for the receptor, were estimated from a plot using the ratio of fluorescence values collected at 671 nm to fluorescence values collected 618 nm versus concentration of test compound added. Typically, 11 points over the concentration range 1 nm to 10 µM was used to construct each dose–response curve, and the pEC50 was calculated using ActivityBase 5.4 software. Six and three repeats were carried out for LRH-1 and SF-1, respectively. Test compounds that increased the affinity of the receptors for the peptide yielded an increase in fluorescent signal. The dose–response curves were sigmoidal with a clear plateau at high concentrations of test compound, the level of which we report as the relative efficacy (RE) of peptide recruitment. In the absence of a known standard, the value of RE was normalized to 24-exo for both LRH-1 and SF-1. The average standard deviations of the pEC50 values for LRH-1 and SF-1 were 0.07 and 0.08, respectively, hence, the retention of the first decimal place. The average standard deviations of the RE values for LRH-1 and SF-1 were 0.034 and 0.025, respectively, hence, the retention of the second decimal place in the reported values but with an approximately ±0.03 95% confidence limit.
The screening data are presented in several tables. Table 4 shows the alkoxy-substituted series 12a–j, and Tables 5–7 show the series 18 compounds with emphasis on variation at the 1-, 2-, and 3-positions of the bicyclo[3.3.0]oct-2-ene skeleton, respectively. Table 8 contains the alternative core structures of 19 and 20. Finally, Table 9 provides results from compounds 24, 25, 29, and 30 with oxygen substitution on the cyclopentane ring.
Table 4.
LRH-1 and SF-1 Binding and Activation of Alkoxy-Substituted Series 12
 | |||
|---|---|---|---|
| compound | R | pEC50 (RE) |
|
| LRH-1 | SF-1 | ||
| 5 | 6.2 (0.89) | 6.8 (0.36) | |
| 7 | 7.5 (0.38) | 7.4 (0.70) | |
| 12a | Me | ia | ia |
| 12b | Et | 5.6 (0.17) | 6.3 (0.55) |
| 12c | nPr | 5.6 (0.21) | 6.7 (0.67) |
| 12d | iPr | 5.5 (0.19) | 6.4 (0.49) |
| 12e | nBu | 5.3 (0.39) | 6.6 (0.62) |
| 12f | CH2CH(Me)(Et) | 5.6 (0.18) | 6.7 (0.56) |
| 12g | nPent | ia | 6.4 (0.43) |
| 12h | nHex | ia | ia |
| 12i | cHex | 6.1 (0.16) | 7.1 (0.66) |
| 12j | CH2Ph | 7.0 (0.13) | ia |
Table 5.
Variation of Bridgehead Substituent in Series 18 Compounds
 | |||
|---|---|---|---|
| compound | R | pEC50 (RE) |
|
| LRH-1 | SF-1 | ||
| 18a | Ph | 6.6 (0.24) | 7.2 (0.78) |
| 18b | 3-MeOPh | 6.5 (0.25) | 7.2 (0.28) |
| 18c | 4-MeOPh | ia | ia |
| 18d | 4-MePh | 5.7 (0.25) | 6.8 (0.28) |
| 18e | 4-EtPh | ia | ia |
| 18f | 4-nBuPh | 6.0 (0.14) | ia |
| 18g | 4-tBuPh | ia | ia |
| 18h | 4-PhPh | ia | ia |
| 18i | nPr | 5.5 (0.23) | 6.8 (0.66) |
| 18j (RJW102) | nBu | 5.6 (0.19) | 6.7 (0.76) |
| 18k | nHex | ia | ia |
| 18l | nOct | ia | ia |
Table 7.
Variation of 3-Substituent in Series 18 Compounds
 | |||
|---|---|---|---|
| compound | R | pEC50 (RE) |
|
| LRH-1 | SF-1 | ||
| 18a | nHex | 6.6 (0.24) | 7.2 (0.78) |
| 18t | H | ia | ia |
| 18u | nBu | 6.7 (0.54) | 7.4 (0.86) |
| 18v | nOct | 5.8 (0.15) | 6.8 (0.67) |
| 18w | SiMe2Ph | 6.8 (0.53) | 7.2 (0.33) |
Table 8.
Effect of Variations in Skeleton on Biological Activity
 | ||
|---|---|---|
| compound | pEC50 (RE) |
|
| LRH-1 | SF-1 | |
| 18a | 6.6 (0.24) | 7.2 (0.78) |
| 19 | 6.6 (0.23) | 7.2 (0.42) |
| 20a (RJW103) | 5.9 (0.20) | 6.5 (0.49) |
| 20b | 6.5 (0.13) | ia |
Table 9.
Effect of Oxygen Substitution on the Cyclopentane Ring
 | ||
|---|---|---|
| compound | pEC50 (RE) |
|
| LRH-1 | SF-1 | |
| 18a | 6.6 (0.24) | 7.2 (0.78) |
| 24-exo (RJW100) | 6.6 (1.00) | 7.5 (1.00) |
| 24-endo | 6.4 (0.67) | 7.2 (0.77) |
| 25-exo | 6.4 (0.18) | 6.4 (0.25) |
| 25-endo | 6.6 (0.18) | 7.1 (0.24) |
| 29-exo | 6.0 (0.53) | 6.6 (0.49) |
| 29-endo | 5.8 (0.33) | 6.5 (0.95) |
| 30-exo | ia | ia |
| 30-endo | 6.3 (0.13) | 6.7 (0.34) |
We were pleased to find that many of the alkoxy-substituted analogues were active against both LRH-1 and SF-1, showing that the nitrogen in the aniline series 6 (exemplified by 5 and 7) was not necessary. All of the compounds (except 12j) bound less strongly to LRH-1 and induced less recruitment of peptide than the aniline series, suggesting that an aromatic group is preferred by LRH-1 in this region. Unfortunately, it was not possible to make the OPh-substituted system as it was highly unstable, both because phenoxide is a much better leaving group than alkoxides but also because phenol will act as an acid catalyst for decomposition. The compounds showed good selectivity for SF-1, with binding approaching and efficacy exceeding that of our currently most used biological tool 5. For both LRH-1 and SF-1, there is a clear SAR relating to the size of the R group with both small groups (Me) and large groups giving inactive compounds. The cutoff for large chains is sharp, indicating a defined pocket being filled. For LRH-1, this is above four carbons long [cyclohexyl, CH2CH(Me)(Et), and n-butyl all fit, while n-pentyl does not]. For SF-1, the pocket appears slightly larger with n-pentyl, but not n-hexyl fitting. These results are comparable to those observed with aniline series 6, compounds where 3- or 4-substitution on the NAr ring gave reduced binding or inactive compounds.39 An interesting exception was the benzyloxy derivative 12j, which displayed excellent binding to LRH-1, although poor efficacy, but was inactive against SF-1. It seems likely that π stacking with His-390 as described above for compound 5 results in the strong binding. Although the alkoxy-substituted series provided candidates for some of our key aims including the SF-1 and LRH-1 selective compounds 12g and 12j, respectively, these compounds proved as acid sensitive as the aniline series, as evidenced by their decomposition on attempted chromatography on silica or when stored in glassware that had not been washed with base.
The acid instability of series 12 prompted us to test compound 18a with a similar cis-bicyclo[3.3.0]oct-2-ene structure but which lacked the acid-sensitive bridgehead leaving group, and it was found to exhibit good binding and activation of both LRH-1 and SF-1 (Table 5).
Variation of the lithiated alkyne used in the multicomponent synthesis of 18 allowed variation of the bridgehead substituent (Table 5). As with the series 12 compounds, we observe a strict cutoff in the size of group R, which can be accommodated. Even addition of a para-methyl group to the preferred phenyl substituent decreased binding (18d), and anything larger (18e–h) gave inactive compounds. The potent binding, although poor efficacy, of 18f with LRH-1 is an anomaly and perhaps indicates an approach to developing reverse agonists of LRH-1. Interestingly, a 3-MeO group was well tolerated perhaps indicating some stabilization via dipolar interactions or weak H-bonding, whereas a 4-OMe substitution gave an inactive compound (18c). When R is an alkyl group (18i–l), LRH-1 binds only the shortest tried (R = nPr, nBu 18i,j) and then with low pEC50 and efficacy. The comparison of 18j (R = nBu) with the similarly sized but much more active 18a (R = Ph) indicates a strong preference for an aryl group correlating with aromatic stacking with His390 noted in the crystal structure of LRH-1 and 5 above. SF-1 bound the R = nBu compound 18j strongly and with good efficacy, confirming the larger pocket noted in the SAR of series 12 and providing a good SF-1 selective analogue in the FRET assay. The longer chain compounds 18k (R = n-hexyl) and 18i (R = n-octyl) were inactive.
Changing the 1,7-enyne starting material allowed the SAR due to variation of the substituent on position 2 of the bicyclo-[3.3.0]oct-2-ene to be probed (Table 6). The tight constraints and preference for aryl groups of the LRH-1 binding site were again apparent. Although a 3-methoxyphenyl group (18m) was tolerated, 4-ethylphenyl (18n) was not, and surprisingly, neither of R = nPr or nBu (18o,p), both similar sizes to a phenyl group, gave active compounds. R = cyclohexyl (18r) gave modest binding and poor efficacy but confirms the preference for cyclohexyl over n-alkyl observed in the alkoxy series above (Table 4). SF-1 proved much more accommodating, consistent with the larger ligand binding pocket, although compounds with 4-ethylphenyl (18n) or n-hexyl (18q) substituents were not active. nPr-, nBu-, and c-hexyl-substituted compounds (18o,p,r) all gave similar binding and efficacy with SF-1 but distinctly poorer than compound 18a (R = Ph). Remarkably, the 3-hydroxypropyl substituent in 18s gave good binding and activation of LRH-1 but was inactive against SF-1, providing the only example of a highly LRH-1 selective compound. The hydroxyl group may be in a position to form a hydrogen bond with a backbone carbonyl in the N-terminal region of helix 3 of LRH-1. The inactivity of 18s indicates this to be a very nonpolar area in SF-1.
Table 6.
Variation of 2-Substituent in Series 18 Compounds
 | |||
|---|---|---|---|
| compound | R | pEC50 (RE) |
|
| LRH-1 | SF-1 | ||
| 18a | Ph | 6.6 (0.24) | 7.2 (0.78) |
| 18m | 3-MeOPh | 6.6 (0.13) | 6.9 (0.42) |
| 18n | 4-EtPh | ia | ia |
| 18o | nPr | ia | 6.4 (0.56) |
| 18p | nBu | ia | 6.6 (0.59) |
| 18q | nHex | ia | ia |
| 18r | cHex | 5.9 (0.15) | 6.7 (0.59) |
| 18s (RJW101) | (CH2)3OH | 6.1 (0.38) | ia |
Using alternative dihalocarbenoids in the formation of 18 allowed the biological effect of variation in the 3-substituent (Table 7) to be studied. The case R = H gave an inactive compound against both receptors, indicating that the ligand binding pocket needs to filled for binding and/or activity. Shortening the n-hexyl (8a) substituent to n-butyl (18u) gave a substantial increase in efficacy for LRH-1 without affecting SF-1 in the FRET assay. The bulky SiMe2Ph substituent gave a substantial increase in efficacy for LRH-1 and a modest decrease in both binding and efficacy for SF-1 (compare 18w with 18a). An n-octyl chain gave some reduction in binding and efficacy, particularly for LRH-1.
We also looked at variation in the core skeleton (Table 8). The only effect of changing the fused saturated ring from cyclopentane (in 18a) to cyclohexane (in 19) was reduced efficacy against SF-1. The pyrrolidine-fused systems 20a,b offered the potential of improved solubility. The N-methyl compound 20a was active against both receptors, although with reduced binding and efficacy, particularly against LRH-1. Given the improved solubility characteristic of 20a, c.f., the hydrocarbons 18, it is a useful SF-1 selective compound. Compound 20b was LRH-1 selective but with very poor efficacy.
Finally, we examined some analogues with oxygen substitution of the cyclopentane ring. The design was driven by the crystal structure of compound 5 bound to LRH-1 described above, which indicated a polar patch (Arg-393 and His-390) in the generally hydrophobic ligand binding site (Figure 4).
We were delighted to find that introduction of a 6-exo-hydroxy substituent (24-exo) gave a substantial increase in both binding and efficacy against LRH-1 with more modest increases for SF-1. The endo-epimer of 24 showed very similar binding and efficacy against both receptors as compound 18a lacking the oxygen substitution. Examination of Figure 4 indicates that only the exo alcohol is placed to interact with Arg-393 or His-390. Acylation of the endo-alcohol (to give 25-endo) had little effect on binding to either receptor, although substantially reduced efficacy. Acylation of the exo-alcohol (to give 25-exo) substantially reduced binding to SF-1 with little effect on LRH-1, although both showed greatly reduced efficacy.
The 7-hydroxyl-substituted systems 29-exo and 29-endo showed reduced binding as compared to the unsubstituted analogue 18a, although efficacies were reasonable. Acylation of the exo-alcohol gave compound 30-exo, which was inactive against both LRH-1 and SF-1, indicating a size limitation of this part of the binding pocket. Acylation of the endo alcohol to give 30-endo was well tolerated.
Choice of Compounds as Biological Probes from the Peptide Recruitment Assay
Compound 24-exo provides an excellent replacement for compound 5 as a biological tool. It has excellent stability, reasonable solubility in polar solvents, and improved binding and efficacy against both LRH-1 and SF-1. Little is lost in this FRET peptide recruitment assay by using the 1.6:1 mixture of 24-exo and 24-endo produced directly from the zirconium-mediated reaction, thus avoiding a tricky separation. Compounds 18o and 18p provide the most selective compounds for SF-1, although the greater efficacy against SF-1, but still weak binding and efficacy against LRH-1 favors 18j. The main disadvantage of all of these is their very non-polar nature, which may cause solubility problems in polar solvents, in which case pyrrolidine 20a provides reasonable selectivity and may be preferred. Compound 18s was unique in providing LRH-1 selectivity, although the activity was modest.
Direct Binding to Purified NR5A Receptors
Several compounds were tested for direct binding using a native gel assay, which generally maintains native protein structure and binding activities.53 We purified both mSF-1 LBD complexed with dipalmitoyl phosphatidyl-inositol (4,5) bisphosphate (PIP2) and hLRH-1 LBD alone (no lipid added) to homogeneity as previously described,36 then added either dimethylsulfoxide (DMSO) vehicle (0) or the indicated doses of 24-exo, 20a, 18j, or 18s compounds (Figure 6). These binding reactions were allowed to equilibrate for 16 h, separated by native polyacrylamide gel electrophoresis (PAGE), and SF-1 protein was visualized with Coomassie stain. The lower band in SF-1/PIP2 lanes is PIP2-dependent, while the upper band represents SF-1 no longer bound by phospholipid. Compound 24-exo clearly displaces the bound PIP2 phospholipid from SF-1 almost completely at 1 µM, and similarly, 20a can also displace PIP2 from SF-1, however, with reduced potency and efficacy (Figure 6, left panel). Compounds 18j and 18s do not appear competent to displace PIP2 from SF-1 LBD in this in vitro assay, even up to 100 µM.
Figure 6.

Differential binding to NR5A receptors. Migration of purified mouse SF-1 LBD complexed with PIP2 (SF-1/PIP2) or with human LRH-1 LBD (LRH-1 alone) in native PAGE after incubation with increasing amounts of indicated compounds. The faster migrating lower band of SF-1 LBD without compound (0) is the SF-1/PIP2 complex.
Using hLRH-1 LBD alone that had not been complexed with any phospholipids (Figure 6, right panel), we observed a clear dose-dependent shift in hLRH-1 LBD native PAGE migration upon 24-exo binding, although 20a was nearly as efficacious binding to LRH-1, a similar shift was only observed at 100 µM. Compound 18j does not appear competent to shift hLRH-1 LBD migration in native gels. Although not very efficacious, 18s can consistently shift hLRH-1 LBD migration in this native gel assay, albeit with reduced potency as compared to 24-exo.
Transcriptional Activation of Endogenous NR5A Target Genes
Given this direct binding data, we then asked if 24-exo was capable of regulating the SF-1-mediated transcription of an endogenous target gene in living human cells. Human embryonic kidney 293 (HEK293) cells stably expressing either empty vector or SF-1 were treated with either DMSO vehicle or the indicated doses of 24-exo for 24 h; each sample was subjected to real-time quantitative polymerase chain reaction (RT-qPCR) to evaluate the relative mRNA expression of the endogenous SF-1 target gene SHP (Figure 7). Compound 24-exo induced a significant dose-dependent increase in SHP transcripts beginning at 5 µM; induction was dependent on the presence of SF-1 in these cells.
Figure 7.

Dose-dependent transcriptional activation of SF-1 by 24-exo in living cells. Relative mRNA levels of SHP transcript are shown in HEK293 cells stably expressing either empty vector (EV) or mouse SF-1 (SF-1) following 24 h of treatment with the indicated dosage of 24-exo (µM). Expression levels are shown relative to DMSO. *p < 0.05, and ***p < 0.001.
We next constructed a stable hLRH-1 HEK293 cell line in addition to the SF-1 line and tested each compound for activation of multiple endogenous target genes, which were previously identified using microarrays and gene profiling.54 SF-1 or LRH-1 cells were treated with 10 µM of indicated compounds for 24 h, and the relative mRNA expression of each transcript was determined by RT-qPCR. SF-1 and LRH-1 protein levels were comparable in stably expressing cell lines, as determined by Western blots (data not shown). We noted that cell viability and expression of housekeeping genes appeared unchanged following treatment with compounds when compared to DMSO treatment.
Consistent with the phospholipid displacement assay, both 24-exo and 20a significantly activate SF-1-mediated transcription of the endogenous SHP, StAR, G0S2, and MeOX1 target genes, while 18j and 18s do not (Figure 8, left panel). Also consistent with the native gel assay, selective activation of all genes by LRH-1 is observed in response to 10 µM 24-exo but not with similar amounts of 18j and 18s (Figure 8, right panel). Interestingly, while 20a activated LRH-1 transcription of the StAR and G0S2 genes, this compound failed to activate the SHP and MeOx1 genes, suggesting that when bound to 20a, LRH-1's ability to recruit needed cofactors at all genomic targets might be compromised. Although 18j binds LRH-1 well, it exhibits poor efficacy at these particular target genes in the HEK293 cell line, raising the possibility that a more relevant cellular or tissue context (such as liver cells) might be required for 18j to fully activate LRH-1.
Figure 8.

Differential transcriptional activation of endogenous NR5A targets in living cells. Relative mRNA expression of endogenous NR5A target genes in HEK293 cells stably expressing either mSF-1 or hLRH-1. For each gene assessed, expression levels are shown relative to DMSO following 24 h of treatment with indicated compound (10 µM). *p < 0.05, and **p < 0.01.
CONCLUSION
A series of 1-alkoxy- and 1-alken-2-yl-substituted bicyclo-[3.3.0]oct-2-enes and related bicycles were designed, synthesized, and evaluated as agonists for the orphan NRs LRH-1 and SF-1. The study achieved its main aims: a compound (in 24-exo, RJW100) with similar activity to the best of our previous agonists but with excellent stability; several compounds including 18j (RJW102) and 20a (RJW103) with selectivity for SF-1, although the former was inactive in in vivo studies, probably due to very poor solubility in aqueous media; and a compound 18s (RJW101) selective for LRH-1, although further work to improve potency is needed. The precision and consistency of the SARs are notable and probably due to the rigid bicyclic core used for all of the compounds. Our preliminary cell-based studies have demonstrated that the 24-exo is a consistently active agonist of SF-1 and LRH-1 target gene expression in a variety of cell lines. While the toxicity of 24-exo in animals remains to remains to be determined, we are currently assessing its utility in more complex in vivo biological model systems such as in Caenorhabditis elegans and zebrafish, both of which express NR5A receptors. Furthermore, we expect that 24-exo will be a useful chemical tool to probe the in vivo biological effects of SF-1 and LRH-1 in mammalian systems. For instance, we speculate that 24-exo might eventually replace the need to virally introduce Oct-4 in an iPS assay and would thus provide one step toward achieving chemical reprogramming.
EXPERIMENTAL SECTION
Chemistry
General
NMR spectra were recorded on Bruker AM300 or DPX400 spectrometers in CDCl3 or C6D6 and referenced to residual protonated solvent (1H NMR) or the center peak of the deuterated solvent triplet (13C NMR). Chemical shifts are reported in parts per million downfield of TMS, and the following abbreviations were used to denote coupling patterns: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet (uninterpretable or unresolved); and fs, fine splitting (resolved but unassigned small couplings). 13C NMR spectra were proton decoupled and are reported as C, CH, CH2, and CH3, depending on the number of directly attached protons, this being determined by DEPT experiments. Assignments in NMR are only given where not easily derived from the 1D data, and systematic numbering is used. Protons described as H-a and H-b are on the endo and exo faces of the bicyclic molecules, respectively. High-resolution mass spectra (HRMS) were recorded on a VG Analytical 70-250-SE double focusing mass spectrometer using electron impact ionization (EI) (at 70 eV) or on a Bruker Apex III spectrometer using positive ion electrospray ionization. Low-resolution mass spectra (LRMS) were recorded on a VG Analytical 70-250-SE double focusing mass spectrometer (EI), a ThermoQuest TraceMS gas chromatography mass spectrometry (GCMS) [EI and CI (with ammonia as the reagent gas)], or on a VG platform quadrupole spectrometer using positive ion electrospray ionization (ESI+). Values of m/z are reported in atomic mass units (a.m.u.) followed in parentheses by the peak intensity (relative to the base peak of 100%). IR spectra were obtained for all compounds but are not included. All compounds were >95% pure by gas chromatography performed on a Hewlett-Packard HP 6890 series GC system, using a HP-5 30 m column, with a film thickness of 0.25 µm and 0.32 mm internal diameter. The carrier gas was helium, and the flow rate was 2.7 mL min−1 using 100:1 split injection and an 80–275 at 25 °C/min (held at 275 °C for 4 min) temperature program with a flame ionization detector.
All organometallic reactions were carried out with dry glassware under an argon atmosphere using standard Schlenk and syringe techniques. Merck silica gel 60 (0.040–0.063 mm) was used for purification.
Unless given below, all materials were obtained from commercial sources and, if necessary, dried and distilled before use. Petrol refers to the fraction of petroleum ether that boils between 40 and 60 °C and was distilled before use. Tetrahydrofuran (THF) and diethylether used in reactions were freshly distilled from sodium/benzophenone. n-Butyllithium (nBuLi) was used as a 2.5 M solution in hexanes (Aldrich) and was stored at 4 °C.
Compounds 18b,h,k,m,o,r, 19, and 20b are reported elsewhere.44 The synthesis of the enyne starting materials, 1-ethyl-4-(hept-6-en-1-yn-1-yl)benzene, N-methyl-N-(phenylprop-2-ynyl)prop-2-en-1-amine, tert-butyldimethyl((7-phenylhept-1-en-6-yn-3-yl)oxy)silane 22, and tert-butyldimethyl((7-phenylhept-1-en-6-yn-4-yl)oxy)silane 27 as well as compounds 12a,c–j, 18c–g,i,l,n,p,q,t–w, 25, 29, and 30 are given in the Supporting Information.
rac-(1S,5R)-1-Ethoxy-3-hexyl-2-phenyl-bicyclo[3.3.0]oct-2-ene (12b)
To a stirred solution of rac-(3S,5R)-3-hexyl-2-phenyl-bicyclo-[3.3.0]oct-1-en-3-ol39 (11) (142 mg, 0.50 mmol in dry THF (4 mL at room temperature was added EtOH (0.29 mL, 5.0 mmol) followed by addition of a solution of (+)-camphorsulfonic acid (11.6 mg, 0.050 mmol) in dry THF (1 mL). The stirring was continued at the same temperature for 3.5 h after which the reaction mixture was poured onto saturated aqueous solution of NaHCO3 (100 mL), and the products were extracted with Et2O (3 × 75 mL). The combined organic phases were washed with H2O (3 × 100 mL) and brine (100 mL). Drying over anhydrous MgSO4 and filtration followed by concentration in vacuo and purification of the crude material by column chromatography on Al2O3 (basic, grade III) with 2.5% Et2O in petrol as the eluent gave the title compound as a yellow oil (0.108 g, 69%). 1H NMR (400 MHz, CDCl3): δ 7.27–7.13 (5H, m), 3.40 (1H, m), 3.26 (1H, m), 2.65 (1H, dd, J = 17.1, 8.3 Hz, H–4b), 2.45–2.37 (1H, m, H–5), 2.14–2.08 (2H, m), 2.06–1.93 (2H, m), 1.66–1.49 (3H, m), 1.44–1.29 (3H, m), 1.24–1.10 (10H, m), 0.77 (3H, t, J = 6.8 Hz, CH3. 13CNMR (100.5 MHz, CDCl3: δ 145.51 (C), 137.28 (C), 136.09 (C), 129.21 (CH), 128.10 (CH), 126.59 (CH), 103.39 (C), 58.40 (CH2), 42.99 (CH), 42.15 (CH2), 37.89 (CH2), 35.83 (CH2), 31.81 (CH2), 29.91 (CH2), 29.43 (CH2), 28.25 (CH2), 25.40 (CH2), 22.77 (CH2), 15.98 (CH3), 14.21 (CH3). LRMS (EI) m/z = 312([M]+, 93%), 283 (100%), 195 (59%). HRMS (EI) found, [M]+, 312.2458. C22H32O requires, 312.2453.
General Procedure
To a solution of Cp2ZrCl2 (0.293 g, 1.0 mmol) in dry THF (5 mL) cooled to −78 °C was added nBuLi (0.80 mL of a 2.5 M solution in hexanes, 2.0 mmol) dropwise (over ~2 min). After 25 min, a solution of the appropriate enyne (1.0 mmol) in dry THF (3 mL) was added dropwise. After 30 min at −78 °C, the reaction mixture was allowed to warm to room temperature and continued to stir for 2–3 h. After the reaction mixture was recooled to −78 °C, a solution of the appropriate 1,1-dihalo alkane (1.1 mmol) in dry THF (1 mL) was added followed by dropwise addition of LDA (0.64 mL of a 1.8 M solution, 1.15 mmol). The reaction mixture was stirred at −78 °C for 15 min before dropwise addition of the corresponding lithium acetylide. [Lithium acetylide was freshly prepared from alkyne (3.0 mmol) in dry THF (3 mL) and nBuLi (1.2 mL of a 2.5 M solution in hexanes, 3.0 mmol) at −5 °C over 15 min]. The stirring was continued for 0.5–1 h during which the reaction mixture was allowed to warm to −55 °C before addition of MeOH (10 mL) and saturated aqueous solution of NaHCO3 (10 mL). The whole mixture was allowed to warm to room temperature and left stirring for 12–16 h. The mixture then was poured onto H2O (100 mL), and the products were extracted with Et2O (3 × 75 mL). The combined organic phases were washed with H2O (3 × 100 mL) and brine (100 mL). Drying over anhydrous MgSO4 and filtration followed by concentration in vacuo gave the crude products mostly as yellow oils.
rac-(1R,5R)-3-Hexyl-2-phenyl-1-(1-phenylvinyl)-bicyclo[3.3.0]oc-t-2-ene (18a)
General procedure was used with 1-(hept-6-en-1-ynyl)benzene, 1,1-dibromoheptane, and 1-ethynylbenzene as components. Purification of the crude material by column chromatography on SiO2 (230–400 mesh) with hexanes as the eluent gave the title compound as a pale yellow oil (0.318 g, 86%). 1H NMR (300 MHz, CDCl3): δ 7.35–7.24 (10H, m), 5.04 (1H, d, J = 1.6 Hz, C═CH2), 5.02 (1H, d, J = 1.6 Hz, C═CH2), 2.43 (1H, tdd, J = 8.6, 3.2, 1.4 Hz, H–5), 2.34(1H, fs ddd, J = 16.2, 8.4, 1.0 Hz, H–4b), 2.12–1.97 (3H, m), 1.85 (1H, dtd, J = 12.2, 9.7, 6.8 Hz), 1.68 (2H, m), 1.62–1.52 (3H, m), 1.43–1.21 (8H, m), 0.88 (3H, t, J = 6.8 Hz, CH3). 13C NMR (75 MHz, CDCl3): δ 155.39 (C═CH2), 144.40 (C), 142.48 (C), 138.95 (C), 137.98 (C), 129.69 (2CH), 127.93 (2CH), 127.55 (2CH), 127.52 (2CH), 126.47 (CH), 126.33 (CH), 114.45 (C═CH2), 70.16 (C–1), 45.71 (CH–5), 43.83 (CH2), 36.33 (CH2), 35.64 (CH2), 31.70 (CH2), 29.89 (CH2), 29.41 (CH2), 27.87 (CH2), 25.60 (CH2), 22.60 (CH2), 14.06 (CH3). LRMS (EI) m/z: 370 ([M]+, 100%), 299 (35%), 267 (85%). HRMS (EI) found, [M]+, 370.2655. C26H34 requires, 370.2661.
rac-(1R,5R)-1-(Hex-1-en-2-yl)-3-hexyl-2-phenyl-bicyclo[3.3.0]oct-2-ene (18j)
General procedure was used with 1-(hept-6-en-1-ynyl)benzene, 1,1-dibromoheptane, and 1-hexyne as components. Purification of the crude material by column chromatography on SiO2 (230–400 mesh) with hexanes as the eluent gave the title compound as a pale yellow oil (0.263 g, 75%). 1H NMR (300 MHz, CDCl3): δ 7.29– 7.17 (3H, m), 7.06–7.02 (2H, m), 4.81 (1H, q, J = 1.4 Hz, C═CH2), 4.79 (1H, q, J = 1.4 Hz, C═CH2), 2.84 (1H, fs dd, J = 16.8, 8.4 Hz, H–4b), 2.43 (1H, dddd, J = 9.7, 8.4, 3.2, 1.6 Hz, H–5), 2.21–2.01 (5H, m), 1.88 (1H, dtd, J = 12.0, 9.4, 7.0 Hz, H–6), 1.70 (1H, m), 1.60–1.48 (6H, m), 1.45–1.33 (4H, m), 1.30–1.21 (6H, m), 0.95 (3H, t, J = 7.2 Hz, CH3), 0.86 (3H, t, J = 6.9 Hz, CH3). 13CNMR (75 MHz, CDCl3): δ 154.57 (C═CH2), 141.21 (C), 139.63 (C), 138.04 (C), 129.40 (2CH), 127.42 (2CH), 126.11 (CH), 107.27 (C═CH2), 71.24 (C–1), 45.53 (CH–5), 44.33 (CH2), 36.67 (CH2), 33.84 (CH2), 32.05 (CH2), 31.68 (CH2), 30.75 (CH2), 29.59 (CH2), 29.13 (CH2), 28.06 (CH2), 25.70 (CH2), 23.05 (CH2), 22.61 (CH2), 14.15 (CH3), 14.04 (CH3). LRMS (EI) m/z: 350 ([M]+, 57%), 307 (26%), 293 (100%). HRMS (EI) found, [M]+, 350.2979. C26H38 requires, 350.2974.
rac-(1R,5R)-3-Hexyl-1-(phenylvinyl)-2-(propan-3-ol)-bicyclo[3.-3.0]oct-2-ene (18s)
General procedure was used with tert-butyl(dec-9-en-4-yn-1-yloxy)dimethylsilane, 1,1-dibromoheptane, and 1-ethynylbenzene as components with the exception that the reaction was carried out on 2.0 mmol scale. Crude product from the three component coupling was purified by flash column chromatography on silica, and then, the TBDMS group was removed with TBAF [2.0 mL of a 1.0 M solution in THF (2.0 mmol) in dry THF (8.0 mL) at room temperature for 20 h]. Purification of the crude material by column chromatography on SiO2 (230–400 mesh) with hexanes as the eluent gave the title compound as a yellow oil (0.430 g, 61% over two steps). 1H NMR (400 MHz, CDCl3): δ 7.24–7.16 (5H, m), 5.15 (1H, d, J = 1.8 Hz, C═CH2), 4.98 (1H, d, J = 1.8 Hz, C═CH2), 3.65 (2H, q, J = 6.5 Hz, CH2OH), 2.34 (1H, tt, J = 9.0, 2.0 Hz, H–5), 2.24 (1H, dd, J = 16.0, 9.0 Hz, H–4b), 2.13–2.02 (4H, m), 1.87–1.61 (6H, m), 1.59–1.52 (1H, m), 1.48–1.38 (1H, m), 1.37–1.26 (10H, m), 0.91 (3H, t, J = 6.9 Hz, CH3). 13C NMR (100.5 MHz, CDC3): δ 155.95 (C═CH2), 144.36 (C), 139.80 (C), 136.21 (C), 127.83 (2CH), 127.37 (2CH), 126.33 (CH), 113.26 (C═CH2), 70.38 (C–1), 63.62 (CH2OH), 43.99 (CH–5), 43.80 (CH2–4), 36.55 (CH2), 36.05 (CH2), 33.45 (CH2), 31.84 (CH2), 29.52 (CH2), 29.27 (CH2), 27.73 (CH2), 25.34 (CH2), 22.65 (CH2), 22.54 (CH2), 14.10 (CH3). LRMS (CI) m/z: 353 ([M + H]+, 100%), 249 (37%). HRMS (EI) found, [M]+, 352.2767. C25H36O requires, 352.2766.
rac-(1S,5R)-3-Hexyl-2-phenyl-1-(phenylvinyl)-7-methylaza-bicyclo[3.3.0]oct-2-ene (20a)
General procedure was used with N-methyl-N-(phenylprop-2-ynyl)prop-2-en-1-amine, 1,1-dibromoheptane, and 1-ethynylbenzene as components. Purification of the crude material by column chromatography on Al2O3 (basic, grade III) with 2.5% of Et2O in hexanes as the eluent gave the title compound as a pale yellow oil (0.240 g, 62%). 1H NMR (400 MHz, CDCl3): δ 7.38–7.25 (10H, m), 5.10 (1H, d, J = 1.1 Hz, C═CH2), 4.93 (1H, d, J = 1.1 Hz, C═CH2), 2.69 (1H, d, J = 9.4 Hz, H–8), 2.68 (1H, dd, J = 8.4,4.0 Hz, H–6), 2.63 (1H, d, J = 9.4 Hz, H–8), 2.63 (1H, tdd, J = 7.9, 4.0, 2.0 H–5), 2.50 (1H, fs dd, J = 16.8, 8.8, Hz, H–4b), 2.45 (1H, dd, J = 8.4, 4.0 Hz, H–6), 2.31 (3H, s, N–CH3), 2.15 (1H, dd, J = 16.8, 1.7, Hz, H–4a), 2.12–2.08 (2H, m, C–3–CH2), 1.45–1.34 (2H, m), 1.32–1.22 (6H, m,), 0.89 (3H, t, J = 7.0 Hz, CH3. 13C NMR (100.5 MHz, CDCl3): δ 153.64 (C═CH2), 143.66 (C), 141.53 (C), 139.52 (C), 137.54 (C), 129.89 (2CH), 127.73 (2CH), 127.65 (2CH), 127.57 (2CH), 126.71 (CH), 126.46 (CH), 114.98 (C═CH2), 70.11 (C–1), 65.77 (2CH2–6 + 8), 46.35 (CH–5), 42.73 (N–CH3), 41.98 (CH2–4), 31.67 (CH2), 29.81 (CH2), 29.39 (C–3–CH2), 27.72 (CH2), 22.58 (CH2), 14.06 (CH3). LRMS (ESI ) m/z: 386 ([M + H]+, 100%). HRMS (ESI+) found, [M + H]+, 386.2832. [C28H36N]+ requires, 386.2842.
rac-(1R,5R,6R)-3-Hexyl-6-hydroxy-2-phenyl-1-(phenylvinyl)-bicyclo[3.3.0]oct-2-ene (24-exo) and rac-(1R,5R,6S)-3-Hexyl-6-hydroxy-2-phenyl-1-(phenylvinyl)-bicyclo[3.3.0]oct-2-ene (24-endo)
To a solution of Cp2ZrCl2 (1.465 g, 5.0 mmol) in dry THF (25 mL) cooled to −78 °C was added nBuLi (4.0 mL of a 2.5 M solution in hexanes, 10.0 mmol) dropwise. After 20 min, a solution of tert-butyl-dimethyl((7-phenylhept-1-en-6-yn-3-yl)oxy)silane (1.50 g, 5.0 mmol) in dry THF (15 mL) was added dropwise. After 30 min at −78 °C, the reaction mixture was allowed to warm to room temperature and continued to stir for 3 h. After the reaction mixture was recooled to − 78 °C, a solution of 1,1-dibromoheptane (1.42 g, 5.5 mmol) in dry THF (5 mL) was added followed by dropwise addition of LDA (3.06 mL of a 1.8 M solution, 5.5 mmol). The reaction mixture was stirred at −78 °C for 15 min before dropwise addition of lithium phenylacetylide solution [freshly prepared from phenylacetylene (1.65 mL, 15.0 mmol) in dry THF (15 mL) and nBuLi (6.0 mL of a 2.5 M solution in hexanes, 15.0 mmol) at −10 °C over 15 min]. The stirring was continued for 45 min during which the reaction mixture was allowed to warm to −55 °C before addition of MeOH (30 mL) and saturated aqueous solution of NaHCO3 (30 mL). The mixture was allowed to warm to room temperature and left stirring for 5 h before pouring onto H2O (200 mL) and extracting the products into Et2O (3 × 200 mL). The combined organic phases were washed with H2O (3 × 300 mL) and brine (300 mL). Drying over anhydrous MgSO4 and filtration followed by concentration in vacuo gave the crude product as a yellow oil. Purification by flash chromatography on SiO2 with 2.5% of Et2O in hexanes furnished the TBDMS protected alcohols 23 (1.6:1 exo:endo) as a yellow oil (2.20 g, 88%).
The TBDMS-protected alcohols 23 (2.20 g, 4.40 mmol) were dissolved in dry THF (44 mL) followed by addition of TBAF (17.60 of 1.0 M solution in THF), and the reaction mixture was stirred at room temperature for 20 h. Then, the mixture was poured onto H2O (200 mL) and extracted with Et2O (2 × 150 mL). The organic phases were washed with H2O (3 × 200 mL) and brine (1 × 200 mL) and dried over MgSO4. Concentration in vacuo, followed by separation by column chromatography on Al2O3 (basic, grade III) and 2.5% EtOAc in hexanes as the eluent gave the partly separated isomers: 0.277 g of pure 24-endo (14%), 0.650 g of mixed fractions (33%), and 0.556 g of pure 24-exo (29%), for a combined yield of 1.483 g (76%). Further chromatography of the mixed fractions allowed additional pure 24-exo and 24-endo to be isolated. Compound 24-exo: 1HNMR (300 MHz, CDCl3): δ 7.37–7.19 (10H, m), 5.08 (1H, d, J = 1.5 Hz, C═CH2), 5.00 (1H, d, J = 1.5 Hz, C═CH2), 3.96 (1H, br s, H–6), 2.39 (1H, fs dd, J = 16.2, 9.6 Hz, H–4b), 2.30 (1H, fs d, J = 9.6 Hz, H–5), 2.15–2.0 (4H, m), 1.79–1.67 (3H, m), 1.38–1.20 (9H, m), 0.87 (3H, t, J = 6.8 Hz, CH3). C NMR (75 MHz, CDCl3): δ 154.69 (C═CH2), 144.21 (C), 141.19 (C), 139.17 (C), 137.43 (C), 129.73 (2CH), 127.78 (2CH), 127.72 (2CH), 127.62 (2CH), 126.66 (CH), 126.59 (CH), 114.99 (C═CH2), 82.10 (CH–6), 69.40 (C–1), 55.92 (CH–5), 40.29 (CH2), 34.04 (CH2), 32.13 (CH2), 31.66 (CH2), 29.73 (CH2), 29.37 (CH2), 27.83 (CH2), 22.58 (CH2), 14.06 (CH3). LRMS (EI) m/z: 386 ([M]+•, 36%), 283 (21%), 283 (100%). HRMS (EI) found, [M]+, 386.2611. C28H34O requires, 386.2610. Compound 24-endo: 1H NMR (300 MHz, CDCl3): δ 7.35–7.21 (10H, m), 5.075 (1H, d, J = 1.5 Hz, C═CH2), 4.96 (1H, d, J = 1.5 Hz, C═CH2), 4.19 (1H, ddd, J = 9.1, 8.5, 5.5 Hz, H–6), 2.63 (1H, fs dd, J = 17.2, 2.1 Hz, H–4a), 2.50 (1H, fs td, J = 8.5, 2.1 Hz, H–5), 2.17–2.01 (3H, m), 1.86 (1H, ddt, J = 10.4, 5.5, 4.4 Hz, H–7b), 1.74–1.70 (2H, m), 1.57 (1H, ddd, J = 10.4, 9.1, 8.1 Hz, H–7a), 1.50–1.35 (2H, m), 1.29–1.24 (7H, m), 0.87 (3H, t, J = 6.8 Hz, CH3). 13C NMR (75 MHz, CDCl3): δ 154.86 (C═CH2), 144.00 (C), 143.28 (C), 139.37 (C), 137.06 (C), 129.82 (2CH), 127.78 (2CH), 127.70 (2CH), 127.61 (2CH), 126.70 (CH), 126.56 (CH), 114.88 (C═CH2), 74.55 (CH–6), 68.83 (C–1), 49.19 (CH–5), 33.77 (CH2), 33.49 (CH2), 31.83 (CH2), 31.66 (CH2), 29.89 (CH2), 29.47 (CH2), 27.93 (CH2), 22.60 (CH2), 14.06 (CH3). LRMS (EI) m/z: 386 ([M]+•, 12%), 368 (17%), 283 (69%). HRMS (EI) found, [M]+, 386.2600. C28H34O requires, 386.2610.
Biology Experimental
Transcriptional Assays
Stable cell lines expressing either N-terminal 3XFlag-tagged mouse SF-1 or human LRH-1 were generated with the T-Rex Flip-In system in the HEK293 T-Rex cell line (Invitrogen). Expression and tetracycline (Tet) inducibility were verified by Western blot analysis using an anti-3XFlag M2 monoclonal antibody (Sigma). For all transcriptional assays, compounds dissolved in DMSO were added for 16 h after initial addition of 100 ng/mL Tet or EtOH vehicle control. Low levels of transduced SF-1 and LRH-1 were observed in cell lines before the addition of Tet, most likely due to stochastic “leakiness” of the TET-transcriptional repressor, as often observed. Messenger RNA transcript abundance relative to the cyclophilin housekeeping gene is shown, with DMSO control set equal to 1. All transcripts were analyzed by quantitative PCR using the following sets of validated primers:
| hCyclophilinf | TTTCATCTGCACTGCCAAGA |
| hCyclophilinr | TTGCCAAACACCACATGCT |
| hSHPf | GCTTAGCCCCAAGGAATATGC |
| hSHPr | TTGGAGGCCTGGCACATC |
| hStARf | CCCATGGAGAGGCTCTATGAA |
| hStARr | GTTCCACTCCCCCATTGCT |
| hMEOX1f | GGCGGAGAAAGGAGAGTTCA |
| hMEOX1r | TCCTTGCGGGCTTTGCT |
| hG0S2f | CAGAGAAACCGCTGACATCTAGAA |
| hG0S2r | CAGCAAAACTCAATCCCAAACTC |
| hStARr | GTTCCACTCCCCCATTGCT |
| hMEOX1f | GGCGGAGAAAGGAGAGTTCA |
| hMEOX1r | TCCTTGCGGGCTTTGCT |
| hG0S2f | CAGAGAAACCGCTGACATCTAGAA |
| hG0S2r | CAGCAAAACTCAATCCCAAACTC |
Protein Purification and PI(4,5)P2 Displacement
Details of the SF-1 LBD construct, purification, and PI(4,5)P2 exchange have been previously described.35b,36 For compound displacement assays, 2.0 µL of serially diluted compounds in DMSO were dosed into 48.0 µL of crystallography pure 1.0 µM mSF-1 LBD/PI(4,5)P2 complexes, in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (8.0) and 5 mM MgCl2, for 2 h at 37 °C. This entire reaction was then separated by native PAGE on precast 4–16% gradient Bis/Tris-buffered gels (Invitrogen), which maintains SF-1LBD/PI(4,5,)P2 association. Gels were then stained with Coomassie brilliant blue to visualize compound-induced changes in the native gel migration pattern of SF-1 LBD/PI(4,5)P2.
Crystallogaphy
Purification of human LRH-1 is as described previously.35b Endogenous phospholipids were replaced with GR8470 (compound 5) using liposome-mediated exchange as described 35b with the exception that liposomes were composed of 1,2-ditetracosanoyl-sn-glycero-3-phosphocholine (PC24) (Avanti Polar Lipids). Briefly, GR8470 in DMSO was added to PC24 in water to a final concentration of 0.8 mM and was mixed with an equal volume of 8 mg/mL LRH-1, giving a final ligand:protein ratio of 3. Exchange was monitored by purifying the LRH-1/ligand complex on a PD10 size exclusion column and determining the complex molecular weight by mass spectroscopy as described.35b By this method, exchange did not improve beyond 5 days. Purified LRH-1 was then mixed with TIF2 peptide in a 1:2 molar ratio and concentrated to 6–8 mg/mL for crystallization trials.
Crystals of the complex were obtained in 0.2 M ammonium sulfate, 0.1 M sodium acetate trihydrate, pH 4.6, and 25% w/v polyethylene glycol 4000 and were quickly dunked into the same buffer with 12% glycerol and 12% ethylene glycol prior to freezing in liquid nitrogen.
Data were collected at beamline 17ID (IMCA CAT) at the Advanced Photon Source at Argonne National Laboratories using an ADSC Q210. Images were integrated and scaled using HKL2000. The complex crystallized in the space group P21 with unit cell constants of A = 52.7, B = 89.0, C = 65.4, and β = 101.7, with two complexes per asymmetric unit. The structure was solved by molecular replacement using the apo LRH-1 structure (1YOK) as a search model. The model was refined against 1.75 Å data using Refmac55 and rebuilt using Coot.56 The model was refined to a final R = 18.7 and Rfree = 21.4 and contained two copies of LRH-1 LBD, two copies of TIF2 peptide, two copies GR8470, one glycerol molecule, three ethylene glycol molecules, and 305 water molecules. The bond length and angle root-mean-square deviation from ideality are 0.007 Å and 1.098°, respectively.
Supplementary Material
ACKNOWLEDGMENT
R.J.W., J.S., and S.D. thank GlaxoSmithKline for generous funding.
ABBREVIATIONS USED
- APC
allophycocyanin
- NR
nuclear receptor
- SF-1
steroidogenic factor-1 (NR5A1)
- LRH-1
liver receptor homologue-1 (NR5A2)
- DBD
DNA binding domain
- LBD
ligand binding domain
- SHP
small/short heterodimer partner (NR01B)
- DAX-1
dosage-sensitive sex reversal adrenal hypoplasia critical region on chromosome Xgene 1) (NR0B2)
- FXR
farnesoid X receptor
- PPAR
peroxisome pro-liferator-activated receptor
- PC
phosphatidylcholine
- LDA
lithium diisopropylamide
- LiTMP
lithium 2,2,6,6-tetramethylpiperidide
- TBAF
tetrabutylammonium fluoride
- TBDMS
tBu-Me2Si
- HMPA
hexamethylphosphoramide
- DMAP
4-dimethylaminopyridine
- TR
time-resolved
- FRET
fluorescence resonance energy transfer
- RT-qPCR
real-time quantitative polymerase chain reaction
- HEK293
human embryonic kidney 293
- RE
relative efficacy
- DMSO
dimethylsulfoxide
- HEPES
4-(2-hydroxyethyl)-l-piperazineethanesulfonic acid
- iPS
induced pluripotent stem cell
- PAGE
polyacrylamide gel electrophoresis
- DOX
doxycycline
- Tet
tetracycline
- PIP2
dipalmitoyl phosphatidyl-inositol (4,5) bisphosphate
- G0S2
G0/G1 switch regulatory protein 2
- MeOXl
mesenchyme homeobox protein 1
- StAR
steroidogenic acute regulatory protein
- HRMS
high-resolution mass spectra
- EI
electron impact ionization
- CI
chemical ionization
- LRMS
low-resolution mass spectra
- GC-MS
gas chromatography—mass spectrometry
- NMR
nuclear magnetic resonance
- THF
tetrahydrofuran
- PC24
l,2-ditetracosanoyl-sn-glycero-3-phosphocholine
- TIF-2
transcriptional intermediary factor 2
Footnotes
ASSOCIATED CONTENT
Supporting Information. Details of the synthesis and characterization of enyne cyclization precursors and compounds 12a,c–j, 18c–g,i,l,n,p,q,t–w, 25-exo, 25-endo, 29-exo, 29-endo, 30-exo, and 30-endo and statistical analysis of the peptide recruitment assay results. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The X-ray coordinates of compound 5 bound to LRH-1 LBD have been deposited in the Protein Data Bank with accession number 3PLZ.
REFERENCES
- 1.Russell DW, Mangelsdorf DJ. Nuclear Receptors. 1st ed. Vol. 363. London: Academic Press Inc; 2003. [Google Scholar]
- 2.(a) Schweitzer A, Knauer SK, Stauber RH. Therapeutic potential of nuclear receptors. Expert Opin. Ther. Pat. 2008;18:861–888. [Google Scholar]; (b) Moore JT, Collins JL, Pearce KH. The nuclear receptor superfamily and drug discovery. ChemMedChem. 2006;1:504–523. doi: 10.1002/cmdc.200600006. [DOI] [PubMed] [Google Scholar]; (c) Shi YH. Orphan nuclear receptors in drug discovery. Drug Discovery Today. 2007;12:440–445. doi: 10.1016/j.drudis.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mukherjee S, Mani S. Orphan nuclear receptors as targets for drug development. Pharm. Res. 2010;27:1439–1468. doi: 10.1007/s11095-010-0117-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Overington JP, Al-Lazikani B, Hopkins AL. Opinion - How many drug targets are there? Nature Rev. Drug Discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 4.Benoit G, Cooney A, Giguere V, Ingraham H, Lazar M, Muscat G, Perlmann T, Renaud JP, Schwabe J, Sladek F, Tsai MJ, Laudet V. International Union of Pharmacology LXVI. Orphan nuclear receptors. Pharmacol. Rev. 2006;58:798–836. doi: 10.1124/pr.58.4.10. [DOI] [PubMed] [Google Scholar]
- 5.(a) Willson TM, Jones SA, Moore JT, Kliewer SA. Chemical genomics: Functional analysis of orphan nuclear receptors in the regulation ofbile acid metabolism. Med. Res. Rev. 2001;21:513–522. doi: 10.1002/med.1023. [DOI] [PubMed] [Google Scholar]; (b) Hummasti S, Tontonoz P. Adopting new orphans into the family of metabolic regulators. Mol. Endocrinol. 2008;22:1743–1753. doi: 10.1210/me.2007-0566. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schulman IG, Heyman R. A The flip side: Identifying small molecule regulators of nuclear receptors. Chem. Biol. 2004;11:639–646. doi: 10.1016/j.chembiol.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 6.Kliewer SA, Lehmann JM, Willson TM. Orphan nuclear receptors: Shifting endocrinology into reverse. Science. 1999;284:757–760. doi: 10.1126/science.284.5415.757. [DOI] [PubMed] [Google Scholar]
- 7.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile acids: Natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 8.Lee CH, Olson P, Evans RM. Minireview: Lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144:2201–2207. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 9.(a) Val P, Lefrancois-Martinez A-M, Veyssiere G, Martinez A. SF-1 a key player in the development and differentiation of steroidogenic tissues. Nucl. Recept. 2003;1:8. doi: 10.1186/1478-1336-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schimmer BP, White PC. Minireview: Steroidogenic fetor 1: Its roles in differentiation development and disease. Mol. Endocrinol. 2010;24:1322–1337. doi: 10.1210/me.2009-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fayard E, Auwerx J, Schoonjans K. LRH-1: An orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004;14:250–260. doi: 10.1016/j.tcb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Hoivik EA, Lewis AE, Aumo L, Bakke M. Molecular aspects of steroidogenic factor 1 (SF-1 ) Mol. Cell. Endocrinol. 2010;315:27–39. doi: 10.1016/j.mce.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Lin L, Achermann JC. Steroidogenic Factor-1 (SF-1, Ad4BP, NR5A1) and Disorders of Testis Development. Sex. Dev. 2008;2:200–209. doi: 10.1159/000152036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Husebye ES, Lovas K. Immunology of Addison's Disease and Premature Ovarian Failure. Endocrinol. Metab. Clin. North Am. 2009;38:389–405. doi: 10.1016/j.ecl.2009.01.010. [DOI] [PubMed] [Google Scholar]; (b) Lourenco D, Brauner R, Lin L, De Perdigo A, Weryha G, Muresan M, Boudjenah R, Guerra G, Maciel-Guerra AT, Achermann JC, McElreavey K, Bashamboo A. Mutations in NR5A1 Associated with Ovarian Insufficiency. N Engl. J. Med. 2009;360:1200–1210. doi: 10.1056/NEJMoa0806228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doghman M, Cazareth J, Douguet D, Madoux F, Hodder P, Lalli E. Inhibition of Adrenocortical Carcinoma Cell Proliferation by Steroidogenic Factor-1 Inverse Agonists. J. Clin. Endocrinol. Metab. 2009;94:2178–2183. doi: 10.1210/jc.2008-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Bulun SE, Utsunomiya H, Lin ZH, Yin P, Cheng YH, Pavone ME, Tokunaga H, Trukhacheva E, Attar E, Gurates B, Milad MP, Confino E, Su E, Reierstad S, Xue Q. Steroidogenic factor-1 and endometriosis. Mol. Cell. Endocrinol. 2009;300:104–108. doi: 10.1016/j.mce.2008.12.012. [DOI] [PubMed] [Google Scholar]; (b) Dube C, Bergeron F, Vaillant MJ, Robert NM, Brousseau C, Tremblay JJ. The nuclear receptors SF1 and LRH1 are expressed in endometrial cancer cells and regulate steroidogenic gene transcription by cooperating with AP-1 factors. Cancer Lett. 2009;275:127–138. doi: 10.1016/j.canlet.2008.10.008. [DOI] [PubMed] [Google Scholar]; (c) Lin BC, Suzawa M, Blind RD, Tobias SC, Bulun SE, Scanlan TS, Ingraham HA. Stimulating the GPR30 Estrogen Receptor with a Novel Tamoxifen Analogue Activates SF-1 and Promotes Endometrial Cell Proliferation. Cancer Res. 2009;69:5415–5423. doi: 10.1158/0008-5472.CAN-08-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Kim KW, Zhao LP, Parker KL. Central nervous system-specific knockout of steroidogenic factor 1. Mol. Cell. Endocrinol. 2009;300:132–136. doi: 10.1016/j.mce.2008.09.026. [DOI] [PubMed] [Google Scholar]; (b) Zhao L, Kim KW, Ikeda Y, Anderson KK, Beck L, Chase S, Tobet SA, Parker KL. Central nervous system-specific knockout of steroidogenic factor 1 results in increased anxiety-like behavior. Mol. Endocrinol. 2008;22:1403–1415. doi: 10.1210/me.2008-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu P, Goodwin B, Chung ACK, Xu X, Wheeler DA, Price RR, Galardi C, Li P, Latour AM, Roller BH, Gossen J, Kliewer SA, Cooney AJ. Orphan nuclear receptor LRH-1 is required to maintain Oct4 expression at the epiblast stage of embryonic development. Mol. Cell. Biol. 2005;25:3492–3505. doi: 10.1128/MCB.25.9.3492-3505.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee Y-K, Moore DD. Liver receptor homolog-1, an emerging metabolic modulator. Front. Biosci. 2008;13:5950–5958. doi: 10.2741/3128. [DOI] [PubMed] [Google Scholar]
- 19.Lee YK, Schmidt DR, Cummins CL, Choi M, Peng L, Zhang Y, Goodwin B, Hammer RE, Mangelsdorf DJ, Kliewer SA. Liver receptor homolog-1 regulates bile acid homeostasis but is not essential for feedback regulation of bile acid synthesis. Mol. Endocrinol. 2008;22:1345–1356. doi: 10.1210/me.2007-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venteclef N, Smith JC, Goodwin B, Delerive P. Liver receptor homolog 1 is a negative regulator of the hepatic acute-phase response. Mol. Cell. Biol. 2006;26:6799–6807. doi: 10.1128/MCB.00579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodwin BJ, Stewart EL, Brown PJ, Delerive P. Liver receptor homolog-1 (LRHl) activators as medicaments for diseases or conditions caused by low plasma apoA-1 levels. WO 2005082344. 2005 [Google Scholar]
- 22.Zollner G, Trauner M. Nuclear receptors as therapeutic targets in cholestatic liver diseases. Br. J. Pharmacol. 2009;156:7–27. doi: 10.1111/j.1476-5381.2008.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santen RJ, Brodie H, Simpson ER, Siiteri PK, Brodie A. History of Aromatase: Saga of an Important Biological Mediator and Therapeutic Target. Endocr. Rev. 2009;30:343–375. doi: 10.1210/er.2008-0016. [DOI] [PubMed] [Google Scholar]
- 24.(a) Zhou J, Suzuki T, Kovacic A, Saito R, Miki Y, Ishida T, Moriya T, Simpson ER, Sasano H, Clyne CD. Interactions between prostaglandin E-2, liver receptor homologue-1, and aromatase in breast cancer. Cancer Res. 2005;65:657–663. [PubMed] [Google Scholar]; (b) Annicotte JS, Chavey C, Servant N, Teyssier J, Bardin A, Licznar A, Badia E, Pujol P, Vignon F, Maudelonde T, Lazennec G, Cavailles V, Fajas L. The nuclear receptor liver receptor homolog-1 is an estrogen receptor target gene. Oncogene. 2005;24:8167–8175. doi: 10.1038/sj.onc.1208950. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen D, Reierstad S, Lu ML, Lin ZH, Ishikawa H, Bulun SE. Regulation of breast cancer-associated aromatase promoters. Cancer Lett. 2009;273:15–27. doi: 10.1016/j.canlet.2008.05.038. [DOI] [PubMed] [Google Scholar]; (d) Simpson ER, McDonnell DP, Kovacic A, Clyne CD, Sail R. Regulation of aromatase expression in the breast by LRH-1: A new potential target for breast cancer therapy. Breast Cancer Res. Treat. 2005;94:S236–S236. [Google Scholar]; (e) Miki Y, Clyne CD, Suzuki T, Moriya T, Shibuya R, Nakamura Y, Ishida T, Yabuki N, Kitada K, Hayashi S-i, Sasano H. Immunolocalization of liver receptor homologue-1 (LRH-l) in human breast carcinoma: Possible regulator of in situ steroidogenesis. Cancer Lett. 2006;244:24–33. doi: 10.1016/j.canlet.2005.11.038. [DOI] [PubMed] [Google Scholar]; (f) Safi R, Kovacic A, Gaillard S, Murata Y, Simpson ER, McDonnell DP, Clyne CD. Coactivation of Liver Receptor Homologue-1 by Peroxisome Proliferator-Activated Receptor gamma Coactivator-1.alpha, on Aromatase Promoter II and Its Inhibition by Activated Retinoid X Receptor Suggest a Novel Target for Breast-Specific Antiestrogen Therapy. Cancer Res. 2005;65:11762–11770. doi: 10.1158/0008-5472.CAN-05-2792. [DOI] [PubMed] [Google Scholar]; (g) Thiruchelvam PT, Lai CF, Hua H, Thomas RS, Hurtado A, Hudson W, Bayly AR, Kyle FJ, Periyasamy M, Photiou A, Spivey AC, Ortlund EA, Whitby RJ, Carroll JS, Coombes RC, Buluwela L, Ali S. The liver receptor homolog-1 regulates estrogen receptor expression in breast cancer cells. Breast Cancer Res. Treat. 2010 doi: 10.1007/s10549-010-0994-9. [DOI] [PubMed] [Google Scholar]
- 25.Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, Schoonjans K. Synergy between LRH-1 and beta-catenin induces G(1) cyclin-mediated cell proliferation. Mol. Cell. 2004;15:499–509. doi: 10.1016/j.molcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 26.Wang S-L, Zheng D-Z, Lan F-H, Deng X-J, Zeng J, Li C-J, Wang R, Zhu Z-Y. Increased expression of hLRH-1 in human gastric cancer and its implication in tumorigenesis. Mol. Cell. Biochem. 2008;308:93–100. doi: 10.1007/s11010-007-9616-1. [DOI] [PubMed] [Google Scholar]
- 27.D'Errico I, Moschetta A. Nuclear receptors, intestinal architecture and colon cancer: An intriguing link. Cell. Mol. Life Sci. 2008;65:1523–1543. doi: 10.1007/s00018-008-7552-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schoonjans K, Dubuquoy L, Mebis J, Fayard E, Wendling O, Haby C, Geboes K, Auwerx J. Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proc. Natl. Acad. Sci. U.S.A. 2005;102:2058–2062. doi: 10.1073/pnas.0409756102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Mullen EM, Gu P, Cooney AJ. Nuclear receptors in regulation of mouse ES cell pluripotency and differentiation. PPAR Res. 2007;2007 doi: 10.1155/2007/61563. article ID 61563. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang H-M, Do H-J, Kim D-K, Park J-K, Chang W-K, Chung H-M, Choi S-Y, Kim J-H. Transcriptional regulation of human Oct4 by steroidogenic factor-1. J. Cell. Biochem. 2007;101:1198–1209. doi: 10.1002/jcb.21244. [DOI] [PubMed] [Google Scholar]
- 30.(a) Heng JCD, Feng B, Han JY, Jiang JM, Kraus P, Ng JH, Orlov YL, Huss M, Yang L, Lufkin T, Lim B, Ng HH. The Nuclear Receptor NR5A2 Can Replace Oct4 in the Reprogramming of Murine Somatic Cells to Pluripotent Cells. Cell Stem Cell. 2010;6:167–174. doi: 10.1016/j.stem.2009.12.009. [DOI] [PubMed] [Google Scholar]; (b) Guo G, Smith A. A genome-wide screen in EpiSCs identifies NRSA nuclear receptors as potent inducers of ground state pluripotency. Development. 2010;137:3185–3192. doi: 10.1242/dev.052753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee YK, Choi YH, Chua S, Park YJ, Moore DM. Phosphorylation of the hinge domain of the nuclear hormone receptor LRH-1 stimulates transactivation. J. Biol. Chem. 2006;281:7850–7855. doi: 10.1074/jbc.M509115200. [DOI] [PubMed] [Google Scholar]
- 32.(a) Chalkiadaki A, Talianidis I. SUMO-dependent compartmentalization in promyelocytic leukemia protein nuclear bodies prevents the access of LRH-1 to chromatin. Mol. Cell. Biol. 2005;25:5095–5105. doi: 10.1128/MCB.25.12.5095-5105.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee MB, Lebedeva LA, Suzawa M, Wadekar SA, Desclozeaux M, Ingraham HA. The DEAD-Box protein DP 103 (Ddx20 or gemin-3) represses orphan nuclear receptor activity via SUMO modification. Mol. Cell. Biol. 2005;25:1879–1890. doi: 10.1128/MCB.25.5.1879-1890.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Bavner A, Matthews J, Sanyal S, Gustafsson JA, Treuter E. EID3 is a novel EID family member and an inhibitor of CBP-dependent co-activation. Nucleic Acids Res. 2005;33:3561–3569. doi: 10.1093/nar/gki667. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bavner A, Sanyal S, Gustafsson JA, Treuter E. Transcriptional corepression by SHP: Molecular mechanisms and physiological consequences. Trends Endocrinol. Metab. 2005;16:478–488. doi: 10.1016/j.tem.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 34.(a) Iyer AK, McCabe ERB. Molecular mechanisms of DAX1 action. Mol. Genet. Metab. 2004;83:60–73. doi: 10.1016/j.ymgme.2004.07.018. [DOI] [PubMed] [Google Scholar]; (b) Sablin EP, Woods A, Krylova IN, Hwang P, Ingraham HA, Fletterick RJ. The structure of corepressor Dax-1 bound to its target nuclear receptor LRH-1. Proc. Natl. Acad. Sci. U.S.A. 2008;105:18390–18395. doi: 10.1073/pnas.0808936105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Ingraham HA, Redinbo MR. Orphan nuclear receptors adopted by crystallography. Curr. Opin. Struct. Biol. 2005;15:708–715. doi: 10.1016/j.sbi.2005.10.009. [DOI] [PubMed] [Google Scholar]; (b) Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, Lebedeva L, Suzawa M, Williams JD, Williams SP, Guy RK, Thornton JW, Fletterick RJ, Willson TM, Ingraham HA. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120:343–355. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]; (c) Li Y, Choi M, Cavey G, Daugherty J, Suino K, Kovach A, Bingham NC, Kliewer SA, Xu HE. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Mol. Cell. 2005;17:491–502. doi: 10.1016/j.molcel.2005.02.002. [DOI] [PubMed] [Google Scholar]; (d) Ortlund EA, Lee Y, Solomon IH, Hager JM, Safi R, Choi Y, Guan ZQ, Tripathy A, Raetz CRH, McDonnell DP, Moore DD, Redinbo M. R Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat. Struct. Mol. Biol. 2005;12:357–363. doi: 10.1038/nsmb910. [DOI] [PubMed] [Google Scholar]
- 36.Sablin EP, Blind RD, Krylova IN, Ingraham JG, Cai F, Williams JD, Fletterick RJ, Ingraham H. A Structure of SF-1 Bound by Different Phospholipids: Evidence for Regulatory Ligands. Mol. Endocrinol. 2009;23:25–34. doi: 10.1210/me.2007-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore DD, Lee JM. Phospholipid LRH-1 receptor agonist compositions and use for the treatment of metabolic disorders and inflammatory bowel disease and lowering of blood glucose levels. WO 2009067182. 2009 [Google Scholar]
- 38.(a) Urs AN, Dammer E, Kelly S, Wang E, Merrill AH, Sewer MB. Steroidogenic factor-1 is a sphingolipid binding protein. Mol. Cell. Endocrinol. 2007;265:174–178. doi: 10.1016/j.mce.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Urs AN, Dammer E, Sewer MB. Sphingosine regulates the transcription of CYP17 by binding to steroidogenic factor-l. Endocrinology. 2006;147:5249–5258. doi: 10.1210/en.2006-0355. [DOI] [PubMed] [Google Scholar]; (c) Dammer EB, Leon A, Sewer MB. Coregulator exchange and sphingosine-sensitive cooperativity of steroidogenic factor-1, general control nonderepressed 5, p54, and p160 coactivators regulate cyclic adenosine 3′,5′-monophosphate-dependent cytochrome P450cl7 transcription rate. Mol. Endocrinol. 2007;21:415–438. doi: 10.1210/me.2006-0361. [DOI] [PubMed] [Google Scholar]
- 39.Whitby RJ, Dixon S, Maloney PR, Delerive P, Goodwin BJ, Parks DJ, Willson TM. Identification of Small Molecule Agonists of the Orphan Nuclear Receptors Liver Receptor Homolog-1 and Steroidogenic Factor-1. J. Med. Chem. 2006;49:6652–6655. doi: 10.1021/jm060990k. [DOI] [PubMed] [Google Scholar]
- 40.(a) Fan W, Yanase T, Morinaga H, Gondo S, Okabe T, Nomura M, Hayes TB, Takayanagi R, Nawata H. Herbicide atrazine activates SF-1 by direct affinity and concomitant co-activators recruitments to induce aromatase expression via promoter II. Biochem. Biophys. Res. Commun. 2007;355:1012–1018. doi: 10.1016/j.bbrc.2007.02.062. [DOI] [PubMed] [Google Scholar]; (b) Fan WQ, Yanase T, Morinaga H, Ondo S, Okabe T, Nomura M, Komatsu T, Morohashi KI, Hayes TB, Takayanagi R, Nawata H. Atrazine-induced aromatase expression is SF-1 dependent: Implications for endocrine disruption in wildlife and reproductive cancers in humans. Environ. Health Perspect. 2007;115:720–727. doi: 10.1289/ehp.9758. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Suzawa M, Ingraham H. A The herbicide atrazine activates endocrine gene networks via non-steroidal NR5A nuclear receptors in fish and mammalian cells. PLoS One. 2008;3:e2117. doi: 10.1371/journal.pone.0002117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Madoux F, Li X, Chase P, Zastrow G, Cameron MD, Conkright JJ, Griffin PR, Thacher S, Hodder P. Potent, selective and cell penetrant inhibitors of SF-1 by functional ultra-high-throughput screening. Mol. Pharmacol. 2008;73:1776–1784. doi: 10.1124/mol.108.045963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth J, Madoux F, Hodder P, Roush W. R Synthesis of small molecule inhibitors of the orphan nuclear receptor steroidogenic factor-1 (NR5A1) based on isoquinolinone scaffolds. Bioorg. Med. Chem. Lett. 2008;18:2628–2632. doi: 10.1016/j.bmcl.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas E, Dixon S, Whitby RJ. A rearrangement to a zirconium-alkenylidene in the insertion of dihalocarbenoids and acetylides into zirconacycles. Ange. Chem. Int. Ed. 2006;45:7070–7072. doi: 10.1002/anie.200602822. [DOI] [PubMed] [Google Scholar]
- 44.Stec J, Dixon S, Thomas EV, Whitby RJ. Tandem insertion of halocarbenoids and lithium acetylides into zirconacycles. A novel rearrangement to zirconium-alkenylidenates via β-addition to an alkynyl-zirconocene. Chem.—Eur. J. 2011 doi: 10.1002/chem.201002962. in press. [DOI] [PubMed] [Google Scholar]
- 45.(a) Abad A, Agullo C, Arno M, Cantin A, Cunat AC, Meseguer B, Zaragoza RJ. Stereoselective synthesis of (–)-metase-quoic acid. B.J. Chem. Soc. Perkin Trans. 1. 1997:1837–1843. [Google Scholar]; (b) Larson GL, Klesse R. An Improved Synthesis of the Ethylene Acetal of 3-Iodopropanal and the Ethylene Ketal of 4-Iodo-2-Butanone. J. Org. Chem. 1985;50:3627–3627. [Google Scholar]
- 46.(a) Karplus M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963;85:2870–2871. [Google Scholar]; (b) Haasnoot CAG, Deleeuw FAAM, Altona C. The Relationship between Proton-Proton Nmr Coupling-Constants and Substituent Electronegativities. 1. An Empirical Generalization of the Karplus Equation. Tetrahedron. 1980;36:2783–2792. [Google Scholar]
- 47.Light ME, Stec J, Whitby RJ. Private communication to the Cambridge Structural Database, deposition number CCDC 746929. 2009 [Google Scholar]
- 48.Diaz Y, Bravo F, Castillon S. Synthesis of purine and pyrimidine isodideoxynucleosides from (S)-glycydol using iodoetherification as key step Synthesis of (S,S)-iso-ddA. J. Org. Chem. 1999;64:6508–6511. [Google Scholar]
- 49.Cink RD, Forsyth CJ. Facile one-pot epoxidation-nucleophilic opening sequence for vicinal diols. J. Org. Chem. 1995;60:8122–8123. [Google Scholar]
- 50.Wang L, Maddess ML, Lautens M. Convenient access to functionalized vinylcyclopentenols from alkynyloxiranes. J. Org. Chem. 2007;72:1822–1825. doi: 10.1021/jo062107w. [DOI] [PubMed] [Google Scholar]
- 51.Halgren TA. Merck molecular force field. 1. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996;17:490–519. [Google Scholar]
- 52.Navarro-Vazquez A, Cobas JC, Sardina FJ, Casanueva J, Diez E. A graphical tool for the prediction of vicinal proton-proton (3)J(HH) coupling constants. J. Chem. Inf. Comput. Sci. 2004;44:1680–1685. doi: 10.1021/ci049913t. [DOI] [PubMed] [Google Scholar]
- 53.(a) Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat. Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]; (b) Wittig I, Schagger H. Advantages and limitation of clear-native PAGE. Proteomics. 2005;5:4338–4346. doi: 10.1002/pmic.200500081. [DOI] [PubMed] [Google Scholar]
- 54.Ingraham HA. Unpublished results. [Google Scholar]
- 55.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.