

SUMMARY
In multicellular organisms, specialized functions are delegated to distinct cell types whose identity and functional integrity is maintained upon challenge. However, little is known about the mechanisms enabling lineage inheritance and their biological implications. Regulatory T (Treg) cells, which express the transcription factor Foxp3, suppress fatal autoimmunity throughout the lifespan of animals. Here, we show that a dedicated Foxp3 intronic element CNS2 maintains Treg cell lineage identity by acting as a sensor of the essential Treg cell growth factor IL-2 and its downstream target STAT5. CNS2 sustains Foxp3 expression during division of mature Treg cells when IL-2 is limiting and counteracts pro-inflammatory cytokine signaling that leads to the loss of Foxp3. CNS2 mediated stable inheritance of Foxp3 expression is critical for adequate suppression of diverse types of chronic inflammation by Treg cells and prevents their differentiation into inflammatory effector cells. The described mechanism may represent a general principle of the inheritance of differentiated cell states.
INTRODUCTION
Differentiated somatic cells exhibit distinct functions and behaviors that are specified by their developmental programs. In the past two decades, tremendous progress has been achieved in elucidating genetic and epigenetic mechanisms underlying differentiation of specialized cell lineages and organ development. However, little is known about how, and to what degree, the differentiated cells maintain their fate or lose their identity in response to changing environment or upon cell division, the two conditions that may disturb the inheritance of lineage specifying factors (Sanchez Alvarado and Yamanaka, 2014). Consequently, factors that affect identity and function of a given cell type and molecular basis of their robustness upon environmental perturbations and its biological significance remain poorly understood.
The adaptive immune system with its somatic diversification of antigen receptors of essentially unlimited specificity affords vertebrates with an effective means of defense against previously encountered and new infectious agents. Potentially deleterious self-reactivity and “collateral” damage resulting in an impairment or loss of tissue function has been a trade-off for the emergence of adaptive immunity. Central to limiting excessive immune responses and associated inflammation is their suppression mediated by regulatory T (Treg) cells, a subset of CD4+ T cells expressing X-chromosome encoded transcription factor Foxp3. Foxp3 is specifically expressed in Treg cells and plays a key role in their differentiation and function (Josefowicz et al., 2012). During the differentiation of Treg cells, Foxp3 is induced in response to TCR and IL-2 signaling (Josefowicz et al., 2012; Sekiya et al., 2013) and Foxp3 protein expression is required for Treg cell function (Gavin et al., 2007; Lin et al., 2007). In addition to conferring cellular identity and functional competence during differentiation of Treg cells, Foxp3 plays an essential role in their maintenance because deletion of a conditional Foxp3 allele in differentiated Treg cells results in a loss of their function (Williams and Rudensky, 2007). Genetic fate mapping using inducible and constitutive Cre revealed heritable and stable Foxp3 expression in the Treg cell population in unchallenged mice as well as in the context of infection and autoimmune inflammation (Miyao et al., 2012; Rubtsov et al., 2010). In contrast, almost half of newly generated extrathymic Treg cells lose Foxp3 expression (Josefowicz et al., 2012). Thus, Treg cells represent a distinct cell lineage and that Foxp3 is its late acting specification factor, whose stable expression is a requisite for preserving Treg cell identity and functional integrity.
These findings also implied the existence of a distinct mechanism that ensures Treg cell lineage stability. A conserved intronic regulatory element CNS2 is required for the maintenance of Foxp3 expression in the progeny of dividing Treg cells, but does not affect Foxp3 induction and its amount on a per cell basis (Zheng et al., 2010). CNS2 can be bound by numerous transcription factors including STAT5, STAT3, and Foxp3, but how these factors regulate Foxp3 expression during cell division remains unknown (Samstein et al., 2012; Xu et al., 2010; Yao et al., 2007; Zheng et al., 2010).
CNS2 contains a stretch of CpG bases that are fully methylated in precursor cells, but undergo de-methylation upon Foxp3 expression (Floess et al., 2007; Kim and Leonard, 2007; Polansky et al., 2008; Toker et al., 2013). Previous studies suggested a correlation between the methylated state of CNS2 and unstable Foxp3 expression (Bailey-Bucktrout et al., 2013; Floess et al., 2007; Polansky et al., 2008). Genetic targeting of the pivotal DNA methyltransferase Dnmt1 or pharmacological inhibition of DNA methyltransferase activity results in a sharp increase in Foxp3 induction efficiency upon activation of naïve T cells (Floess et al., 2007; Josefowicz et al., 2009; Kim and Leonard, 2007).
Despite a considerable body of work, the biological role of CNS2 in the regulation of Foxp3 expression has not been elucidated and a mechanistic understanding of CNS2 function and its biological role are lacking. Here, we demonstrated that CNS2 serves as a sensor of IL-2/STAT5 signaling that prevents Treg conversion into effector T cells upon exposure to pro-inflammatory cytokines. CNS2 conferred stable inheritance of Foxp3 expression at limiting amounts of IL-2, which was of particular significance for control of chronic inflammation in a wide range of biological contexts.
RESULTS
Heritability of Foxp3 expression and CpG methylation in the Foxp3 locus
Lymphopenic or pro-inflammatory conditions are associated with the instability of Treg cells, however, its cause and consequences have not been examined (Bailey-Bucktrout et al., 2013; Komatsu et al., 2013). We observed accumulation of Foxp3− cells (exTreg) upon Treg cell division in response to TCR stimulation in vitro. This was unlikely due to the contamination with effector T cells because the starting population contained 99.8% Foxp3+ cells (Figure 1A, and data not shown). To demonstrate that the loss of Foxp3 was due to division of Treg cells, we blocked cell cycle progression using pharmacological inhibitors targeting different stages of the cell cycle. The cell cycle blockade fully rescued Foxp3 expression (Figure 1B) suggesting that its loss was indeed cell cycle dependent.
Figure 1. CNS2 opposes cell cycle-dependent loss of Foxp3 expression in mature Treg cells.

(A) Loss of Foxp3 expression during cell division. Highly purified Foxp3gfp Treg cells were labeled with CellTrace Violet and activated by CD3 and CD28 antibody-coated beads in the presence of 200U/ml IL-2 for 5 days. The data represent one of 3 independent experiments. (B) Blockage of cell division prevents loss of Foxp3 expression. Flow cytometric analysis of Foxp3 expression in Treg cells treated with indicated inhibitors in comparison to control non-dividing and dividing Treg cells. The data represent one of 3 independent experiments. (C) CpG methylation (mCpG) levels in the Foxp3 locus during Treg cell division in vitro and after loss of Foxp3 expression (exTreg) in vitro and in vivo. mCpG levels in biological replicate samples were averaged at each site of the Foxp3 locus from >5000 reads in naïve CD4+Foxp3− T cells (n=2), ex vivo isolated natural Treg (n=3), non-dividing (n=3) and dividing (n=3) Treg cells, in vitro exTreg (n=4) and in vivo exTreg (n=2) cells. Red dots represent individual CpG sites. Mean ± SEM; t-test comparisons of mCpG in non-dividing and dividing Treg cells are shown (see also Figure S1A–C). (D) CNS2 sustains heritable Foxp3 expression in dividing mature Treg cells. Highly purified Treg cells from Foxp3gfp and Foxp3ΔCNS2-gfp mice were co-cultured in the presence of CD3 and CD28 antibody-coated beads and 500U/ml IL-2 for 4–5 days. The data represent one of >4 independent experiments. Gating on CellTrace intensity is similar to (A). Mean ± SEM. (E) Similar gene expression profiles of CNS2-deficient and -sufficient resting Treg cells (rTreg). Poly-(A) RNA libraries were generated using FACS sorted CD44lowCD62high rTreg cells from male Foxp3gfp and Foxp3ΔCNS2-gfp littermates. Relative gene expression levels (cumulative fraction of genes) in CNS2-sufficient and -deficient rTreg cells were compared to those up- and -down regulated in GFP+ cells (“Treg wannabe’s”) from Foxp3gfpko/wt heterozygous females and wild type rTreg cells (n=3 each). The numbers of genes in each comparison group are indicated in parenthesis. (F) Acute deletion of CNS2 in mature Treg cells was induced upon tamoxifen treatment of Foxp3CNS2fl-gfp UBCCre-ERT2 R26Y male mice 7 days before the adoptive cell transfer. Treg (GFP+) cells were sorted by FACS and transferred together with allelically marked naïve CD4+Foxp3− T cells into T cell deficient hosts (Tcrb−/− Tcrd−/−). 7 weeks after transfer Foxp3 expression was analyzed and the remaining Foxp3+ (GFP+) Treg cells were purified by FACS, co-transferred with newly isolated naïve CD4+Foxp3− T cells into secondary Tcrb−/− Tcrd−/− recipients and Foxp3 expression was assessed 5 weeks later. (G) Flow cytometric analysis of Foxp3 expression before cell transfer and after recovery. Transferred YFP+ and YFP− Treg cells were originally isolated from tamoxifen treated Foxp3CNS2fl-gfp UBCCre-ERT2 R26Y and Foxp3CNS2fl-gfp R26Y mice respectively. The data represent one of >3 independent experiments. See also Figure S1.
To investigate whether CpG methylation (mCpG) plays a role in the regulation of heritable Foxp3 expression, we examined the mCpG levels across the Foxp3 locus by bisulfite sequencing in resting and dividing Treg cells as compared to non-Treg cells that do not express Foxp3 or “exTreg” cells that have lost Foxp3 expression either in vitro or in vivo. In agreement with previous reports −1.5 kb, CNS2 and exon -1 were methylated in non-Treg cells and largely or fully demethylated in Treg cells (Floess et al., 2007; Ohkura et al., 2012), but were re-methylated in “exTreg” cells that have lost Foxp3 expression either in vivo or in vitro. Importantly, we found a statistically significant increase in mCpG at sites including −1.5 kb region, CNS2 and exon-1 in dividing Foxp3+ Treg cells (Figure 1C and S1A–C). Accordingly, low dose of a DNA methyltransferase inhibitor 5-aza-deoxycitidine (5-aza-dC) prevented loss of Foxp3 expression in Treg cells that underwent comparable number of cell divisions in the presence or absence of the drug (Fig. S1D). These findings suggest that CpG methylation might be actively regulated when Treg cells divide and may affect the heritability of Foxp3 expression.
CNS2 opposes cell cycle-dependent loss of Foxp3 expression in mature Treg cells
Studies of Foxp3ΔCNS2-gfp mice on a mixed genetic background showed that CNS2 maintains Foxp3 expression in Treg cells (Zheng et al., 2010). To explore the biological role of CNS2 and the molecular mechanisms of its function in depth we backcrossed Foxp3ΔCNS2-gfp mice onto a C57BL/6 (B6) background (n>10) and bred them to CNS2-sufficient Foxp3gfp B6 mice expressing the exact same Foxp3 reporter allele, which serves as an ideal control when segregated into male littermates (Bettini et al., 2012; Darce et al., 2012). In agreement with previous results on a mixed genetic background (Zheng et al., 2010), significantly more Treg cells isolated from Foxp3ΔCNS2-gfp B6 mice with the germ-line CNS2 deficiency lost Foxp3 expression upon division in comparison to CNS2-sufficient Foxp3gfp B6 littermates (Figure 1D). To exclude a possibility that CNS2 may have a direct impact on expression of genes other than Foxp3 through long-range chromatin interactions that could contribute to impaired Foxp3 maintenance, we performed RNA-seq gene expression analysis of CNS2-sufficient and -deficient resting Treg cells. Comparable gene expression observed in these CNS2-sufficient and -deficient Treg cell populations (Figure 1E) indicated that CNS2 has a direct and specific role in the maintenance of Foxp3 expression and Treg cell fate in dividing activated Treg cells.
These findings raised the question, at which stage of Treg cell differentiation CNS2 exerts its function. To address this question, we deleted a conditional CNS2fl allele in mature Treg cells. Treg cells expressing Foxp3gfp reporter that underwent tamoxifen-induced Cre-ER (UBCCre-ERT2) mediated CNS2 deletion were tagged by YFP through the concomitant activation of R26Y recombination reporter allele (Figure 1F). YFP expression highly correlated with CNS2 deletion with only 4.7±0.6% YFP tagged cells retaining the unrecombined allele as measured by quantitative PCR (qPCR) (data not shown). To test the role of CNS2 in the maintenance of Foxp3 expression in mature Treg cells in vivo we transferred highly purified YFP+GFP+ Treg cells (>99.8% purity) from Foxp3CNS2fl-gfp UBCCre-ERT2 R26Y mice into T cell-deficient Tcrb−/−Tcrd−/− recipients together with allelically marked naïve Ly5.1+ CD4+Foxp3− T cells and assessed Foxp3 expression seven wk after transfer. Since ~50% GFP+YFP−Treg cells in tamoxifen-treated Foxp3CNS2fl-gfp UBCCre-ERT2 R26Y mice lost CNS2 and the R26Y underreported recombination at the Foxp3 locus (data not shown), we used YFP−GFP+ Treg cells sorted from untreated Foxp3CNS2fl-gfp UBCCre-ERT2 R26Y mice or from tamoxifen-treated Foxp3CNS2fl-gfp R26Y mice as negative controls. Consistent with our previous findings, only a minor portion of control Treg cells lost Foxp3 expression when analyzed 7 wk after transfer (Figure 1G, S1E and S1F) (Rubtsov et al., 2010). In contrast, over 55% of the progeny of Treg cells subjected to acute CNS2 ablation lost Foxp3 expression. Because we transferred the majority of Treg cells with unrecombined CNS2fl and the minority with ablated CNS2fl, or control Foxp3GFP cells the experimental and control group of recipient mice remained healthy. Thus, CNS2 maintains Foxp3 expression in mature Treg cells.
The observation that only some mature Treg cells lost Foxp3 expression raised a question whether the Treg cell population is heterogeneous in its reliance on CNS2 for Foxp3 maintenance with subsets of cells being dependent or independent of CNS2. It was possible, for example, that the loss of CNS2 affected only CD25low Treg cells prone to Foxp3 loss (Komatsu et al., 2009; Komatsu et al., 2013). Such a scenario implied that CNS2 acted in a deterministic manner to maintain Foxp3 expression in a CNS2-dependent Treg cell subset and that CNS2-deficient Treg cells that retained Foxp3 expression after homeostatic expansion were independent of CNS2 for Foxp3 maintenance. Alternatively, it was possible that CNS2 was required for the maintenance of Foxp3 expression in the entire population of dividing Treg cells, but Foxp3 was lost in a stochastic manner. To explore these alternatives we isolated CNS2-sufficient and -deficient YFP+GFP+ Treg cells 7 wk after the first transfer and transferred them again into T cell-deficient recipients. 5 wk after secondary transfer, we observed an essentially identical pattern to that of the primary transfer, i.e. ~60% loss of Foxp3 expression in the absence of CNS2 (Figure 1F and G). These results suggested that the loss of Foxp3 expression in mature CNS2-deficient Treg cells was an apparently stochastic event associated with their division.
CNS2 sustains heritable Foxp3 expression in mature Treg cells at limiting amounts of IL-2
We reasoned that the observed loss of Foxp3 by some Treg cells was either due to the deprivation of a factor(s) that is required for the maintenance of Foxp3 expression and is present in limiting amounts, or due to the exposure to a limiting factor(s) that represses Foxp3 expression. IL-2 and STAT5 signaling plays an important role in regulation of Foxp3 expression (Fontenot et al., 2005; Sakaguchi et al., 1995). Our recent cell fate mapping studies showed that IL-2 deprivation caused by administration of IL-2 neutralizing antibody results in a loss of Foxp3 expression by only a minor subset (~15%) of YFP tagged Treg cells (Rubtsov et al., 2010), although this may be an underestimate because of IL-2/STAT5 signaling facilitates Treg cell viability. Thus, IL-2 could be a factor that “positively” acts via CNS2 and whose deprivation leads to a pronounced loss of Foxp3 expression in the absence of CNS2. Indeed, CNS2 contains multiple STAT5 binding motifs and can be bound by STAT5 (Figure S1A) (Ogawa et al., 2013; Yao et al., 2007).
Previous studies suggested that Treg cells expressing low amounts of CD25, a subunit of high affinity IL-2 receptor, is unstable, whereas CD25high Treg cells faithfully maintain Foxp3 expression. Thus, we sought to examine whether CNS2 is required for the maintenance of Foxp3 expression only in CD25low or both CD25low and CD25high Treg cells in vivo that are less or more efficient in IL-2 capture and signaling respectively as assessed by STAT5 phosphorylation (Figure S2A). Upon transfer into lymphopenic host, both CD25high and CD25low CNS2-deficient Treg cells showed markedly reduced ability to maintain Foxp3 expression upon cell division, compared to their wild type counterparts with CD25low CNS2-deficient Treg cells exhibiting the most pronounced Foxp3 loss (Figure 2A and S2B). These results raised the possibility that increased IL-2 signaling may rescue, at least in part, loss of Foxp3 expression.
Figure 2. CNS2 sustains heritable Foxp3 expression in mature Treg cells in the presence of limiting amounts of IL-2.
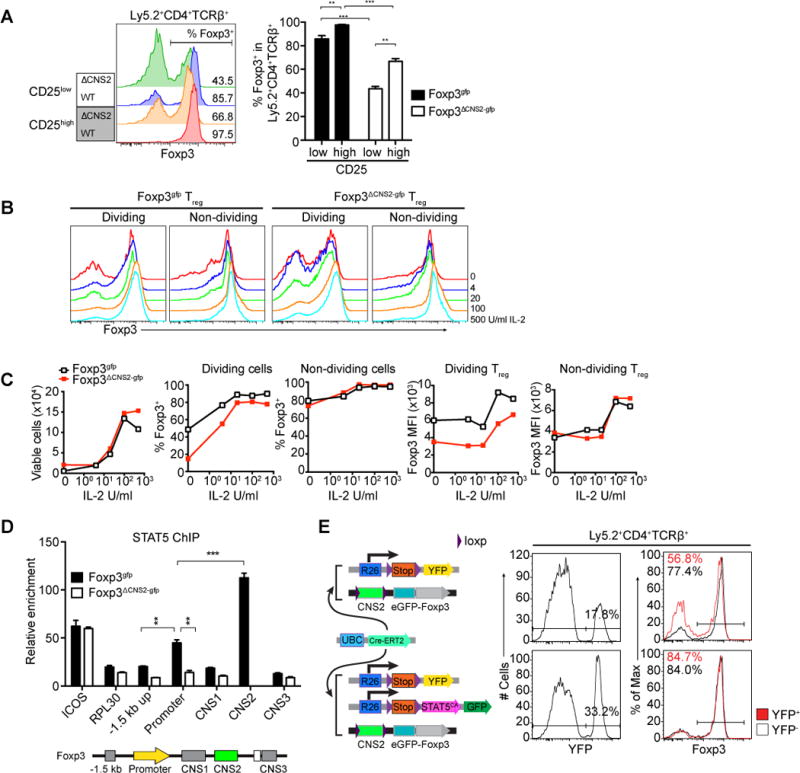
(A) The stability of Foxp3 expression is more sensitive to low levels of CD25 expression in CNS2-deficient Treg cells. Doubly sorted Ly5.2 CNS2-sufficient (Foxp3gfp) and -deficient (Foxp3ΔCNS2-gfp) Treg cells were cotransferred with Ly5.1 naïve CD4+Foxp3− T cells into T cell deficient Tcrb−/− Tcrd−/− mice and recovered for analysis 5 weeks later. CNS2-sufficient (n=5 per group), CNS2-deficient (n=3 per group). Mean ± SEM. (B, C) CNS2-deficient Treg cells are susceptible to low levels of IL-2. Treg cells were doubly sorted by FACS and activated with plate coated CD3 and CD28 antibodies in the presence of titrated amounts of IL-2 for 4 days. Foxp3 expression in the dividing (≥3 cell divisions) and non-dividing (<3 cell divisions) cells was monitored by flow cytometry. The data represent one of >3 independent experiments. (D) ChIP-qPCR analysis of STAT5 binding to the Foxp3 locus in CNS2-sufficient and -deficient Treg cells. In vitro expanded FACS purified Treg cells from Foxp3gfp or Foxp3ΔCNS2-gfp male mice were stimulated with 500U/ml IL-2 for 30 minutes. Relative enrichment was calculated by normalizing to background STAT5 binding to control region (GM5069). The data are shown as means ± SEMs of triplicates and represent one of three independent experiments. (E) Expression of hypermorphic STAT5 (STAT5CA) rescues unstable Foxp3 expression in CNS2-deficient Treg cells in vivo. Treg cells from Ly5.2 UBCCre-ERT2 R26Y Foxp3CNS2fl-gfp and UBCCre-ERT2 R26Y/ R26-Stopfl-STAT5CA Foxp3CNS2fl-gfp were doubly sorted by FACS and cotransferred with naive Ly5.1 CD4+Foxp3− T cells into Tcrb−/− Tcrd−/− mice. 4 weeks after tamoxifen gavage YFP+ or YFP− Ly5.2+ CD4+TCRβ+ cells were isolated and analyzed for Foxp3 expression (6 mice per group). The data represent one of >3 independent experiments. See also Figure S2.
We directly tested this idea by asking whether provision of high amounts of IL-2 can rescue, at least in part, loss of Foxp3 in CNS2-deficient Treg cells. Upon TCR engagement, CNS2-deficient Treg cells that divided multiple times (≥3, denoted as “dividing”) largely maintained Foxp3 expression in the presence of high amounts of IL-2, whereas at lower IL-2 levels a marked reduction in Foxp3 expression was observed on a per cell basis as well as in the percentage of Foxp3+ cells (Figure 2B and 2C). Expectedly, CNS2-deficient Treg cells that did not divide or divided less than 3 times (“non-dividing”) did not significantly change Foxp3 expression in comparison to control Treg cells. Reduced Foxp3 amounts on a per cell basis observed in CNS2-deficient Treg cells in vivo and upon their division in vitro were likely due to an incomplete Foxp3 protein turnover in Treg cells with the silenced Foxp3 locus (Figure 1D, 2B and 2C). Importantly, similar analysis of mature Treg cells subjected to tamoxifen-induced deletion of a “floxed” CNS2 allele showed similar sensitivity of heritable Foxp3 expression to low levels of IL-2 (Figure S2C). The observed effect was unlikely due to a differential effect of IL-2 on survival of CNS2-sufficient vs. -deficient Treg cells because provision of IL-2 similarly increased the viability of both CNS2-sufficient and -deficient Treg cells in vitro (Figure 2C). These results suggested that CNS2 confers the ability of dividing Treg cells to sustain Foxp3 expression upon IL-2 deprivation.
These findings raised the possibility that the observed rescue of CNS2 deficiency was due to increased STAT5 activation in response to high amounts of IL-2. Previous studies showed binding of STAT5 to CNS2 (Ogawa et al., 2013; Yao et al., 2007). In agreement with these data, we found that within the Foxp3 locus STAT5 binds predominantly to CNS2 and to a markedly lesser extent to the Foxp3 promoter (Figure 2D). Interestingly, in the absence of CNS2, binding of STAT5 was noticeably reduced in the Foxp3 promoter and its −1.5 kb upstream region, suggesting potential interaction between CNS2 and these sites. This observation was consistent with the idea that the partial rescue of the Foxp3 loss associated with the CNS2 deficiency in the presence of high amounts of IL-2 was due to heightened STAT5 activation. To address this possibility we took advantage of a conditional R26-STOPfl-STAT5CA/gfp transgene encoding a constitutively activate form of STAT5 (STAT5CA) and a GFP reporter preceded by loxP site flanked STOP cassette inserted into the Rosa26 locus (T.C. and A.R., in preparation). Forced expression of STAT5CA transgene in mature Treg cells upon deletion of a conditional CNS2 allele mediated by tamoxifen-induced Cre rescued unstable Foxp3 expression and restored Treg cell stability to a level observed in wild-type Treg cells (>90 % YFP+ cells were CNS2- deficient) (Figure 2E). These results suggested that enhanced IL-2 signaling through activation of STAT5 was able to compensate for the absence of CNS2 and that CNS2 affords Treg cell with the ability to effectively sustain Treg identity at low levels of IL-2.
CNS2 stabilizes Foxp3 expression in the presence of pro-inflammatory cytokines
In contrast to the positive role of IL-2 signaling in stabilizing heritable Foxp3 expression, pro-inflammatory cytokines IL-6, IFNγ, IL-12, and IL-4 even in the presence of optimal amounts of IL-2 severely compromised Foxp3 expression in dividing CNS2-deficient Treg cells, whereas in CNS2-sufficient Treg cells only a fairly small loss of Foxp3 was observed (Figure 3A, S3A; data not shown). Since in vitro IL-4 exposure caused particularly severe loss of Foxp3 expression in dividing Treg cells, for further investigation of this phenomenon we focused on IL-4 as an example of pro-inflammatory cytokines. Notably, the loss of Foxp3 expression induced by IL-4 in CNS2-deficient Treg cells was also observed at the mRNA level and was cell cycle dependent (Figure S3A and B).
Figure 3. IL-2-STAT5-CNS2 axis stabilizes Foxp3 expression in the presence of pro-inflammatory cytokines.
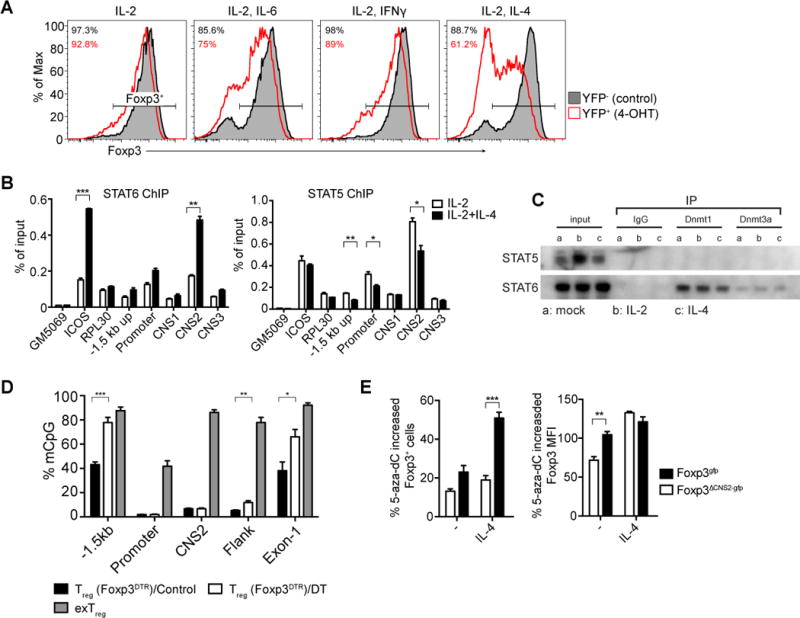
(A) Dividing Foxp3ΔCNS2-gfp Treg cells lose Foxp3 expression upon exposure to pro-inflammatory cytokines. Doubly sorted Treg cells from UBCCre-ERT2 R26Y Foxp3CNS2fl-gfp mice were activated in vitro with plate coated CD3 and CD28 antibodies in the presence of IL-2 (500U/ml) and pro-inflammatory cytokines. Deletion of CNS2 was induced upon treatment with 4-hydroxytamoxifen (4-OHT). Histograms show only the dividing cells (≥3 divisions). The data represent one of >3 independent experiments. (B) ChIP-qPCR analysis of STAT5 and STAT6 binding to the Foxp3 locus in in vitro expanded FACS purified Treg cells from Foxp3gfp mice and stimulated with IL-2 and IL-4 for 30 minutes. Relative enrichment was calculated by normalizing to the input of ChIP. The data are shown as means ± SEMs of triplicates and represent one of three independent experiments. (C) STAT6, but not STAT5 is associated with DNA methyltransferases. Dnmt1 and Dnmt3a co-precipitation with STAT5 and STAT6 was performed using nuclear extracts from in vitro expanded FACS purified Treg cells from Foxp3gfp mice as in (B). The data represent one of >3 independent experiments. (D) Elevated CpG methylation in the Foxp3 locus in Treg cells expanding under inflammatory conditions in vivo. Activated Treg cells were FACS purified from Foxp3DTR mice on day 10 after diphtheria toxin injection (see Extended Experimental Procedures). CpG methylation levels were averaged from >5000 reads in each site of the Foxp3 locus and compared to control Treg cells and ex vivo isolated exTreg cells (see also Figure S1B). The data represent 2 independent experiments. (E) Blockage of Dnmt activity with 5-aza-deoxycytidine (5-aza-dC) partially restores heritable Foxp3 expression in Treg cells exposed to IL-4. Doubly sorted Treg cells from Ly5.1 Foxp3gfp and Ly5.2 Foxp3ΔCNS2-gfp male mice were co-cultured with CD3 and CD28 antibody-coated beads for 7 days in the presence of 500 U/ml IL-2 with or without IL-4 or 0.1μM 5-aza-dC. Relative increases in the percentages of Foxp3+ cells and Foxp3 expression (MFI) on a per cell basis were calculated. The data represent one of >3 independent experiments. Mean ± SEM. See also Figure S3.
To explore whether STAT family members activated downstream of pro-inflammatory cytokine signaling bind to Foxp3 locus and possibly compete with STAT5, we employed ChIP-qPCR to examine STAT6 and STAT5 binding sites at the Foxp3 locus in Treg cells stimulated with IL-2 and IL-4 or with IL-2 alone. We found that upon combined IL-2 and IL-4 stimulation STAT6 and STAT5 bind to similar sites, namely, CNS2, the Foxp3 promoter and its −1.5 kb upstream region (Figure 3B). Concomitantly with STAT6 binding, STAT5 binding was markedly reduced at the three main sites despite the presence of the same amounts of IL-2 when it was used in combination with IL-4 or alone (Figure 3B). Of note, IL-2-dependent STAT5 phosphorylation was comparable when Treg cells were stimulated with IL-2 in the presence or absence of IL-4 (Figure S3C).
Since STAT5 and STAT6 bind to the same sites within the Foxp3 locus and promote maintenance and loss of Foxp3 expression, respectively, we asked what protein factors might associate with STAT5 and STAT6 and contribute to their opposing activity. In light of the observed connection between CpG methylation in the Foxp3 locus, cell division, and the loss of Foxp3 expression (Fig. 1C), we tested whether STAT5 or STAT6 interact with DNA methyltransferases Dnmt1 and Dnmt3a, responsible for maintenance and de novo DNA methylation, respectively (Wu and Zhang, 2011). We found that STAT6, but not STAT5 co-precipitates with Dnmt1 and to a lesser degree with Dnmt3a (Figure 3C). In agreement with a previous report (Zhang et al., 2005), we also observed Dnmt1 association with STAT3 (data not shown) and STAT3 binding to the aforementioned STAT5 binding sites in the Foxp3 locus (Figure S3D).
We previously reported that after transient depletion of Treg cells in Foxp3DTR mice recovering Treg cells rapidly divide in the presence of high amounts of Th2, Th1 and Th17 cytokines (Arvey et al., 2014; Kim et al., 2007). Under these conditions, we observed sharply increased CpG methylation at the −1.5 kb region and exon-1 in dividing activated Treg cells (Figure 3D). Furthermore, blockage of Dnmt activity with 5-aza-dC resulted in considerable, albeit partial rescue of impaired Foxp3 maintenance by dividing CNS2-deficient Treg cells in the presence of IL-4 in vitro (Figure 3E). Together, our data suggest that STAT5 competes with STAT6 for the binding to the Foxp3 locus and prevents silencing of Foxp3 expression by opposing STAT6-mediated recruitment of Dnmt1 to Foxp3 regulatory elements that are partially re-methylated during Treg cell division.
CNS2 dependent heritable Foxp3 expression and maintenance of Treg cells in the steady state
Next, we investigated the biological significance of heritable Foxp3 expression in differentiated Treg cells. In agreement with previous analysis of mice on a mixed genetic background, Foxp3ΔCNS2-gfp B6 mice displayed reduced Treg cell frequency in the peripheral lymphoid organs and heightened activation of effector T cells and pro-inflammatory cytokine production in comparison to their Foxp3gfp littermates (Figure 6C and D). Since Treg cell frequency can be altered in response to inflammation (Zheng et al., 2009), we wanted to assess the impact of CNS2 deficiency on Treg cell frequency in non-inflammatory settings. Therefore, we generated mixed bone marrow (BM) chimeras by transferring allelically marked BM cells from CNS2-deficient Ly5.2 Foxp3ΔCNS2-gfp and CNS2-sufficient Ly5.1 Foxp3gfp males mice into irradiated T-cell-deficient recipients (Figure 4A). Analyses of ratios of CNS2-deficient Ly5.2+ and -sufficient Ly5.1+ Treg cells showed that CNS2-deficient and -sufficient Treg cells were equally represented in the thymus (Ly5.2+/Ly5.1+ ratio 0.93) and that CNS2-deficient Treg cells were only moderately underrepresented in the secondary lymphoid organs (Ly5.2+/Ly5.1+ ratio 0.42–0.64) (Figure 4B). In contrast, in non-lymphoid organs of these mice we observed a pronounced skewing towards CNS2-sufficient Treg cells (Ly5.2+/Ly5.1+ ratio 0.15–0.36) and a marked decrease in the level of Foxp3 protein expression on a per cell basis (Figure 4C). Furthermore, CNS2-deficient Treg cells were underrepresented among dividing Ki67+ Treg cells, which was associated with reduced Foxp3 expression level at the single cell level (Figure 4D and S4A–C). Consistently we observed reduced numbers of CNS2-deficient Treg cells and reduced levels of Foxp3 in activated (CD44highCD62Llow) dividing CNS2-deficient Treg cells (Figure 4E, S4D and S4E). In further support for the aforementioned notion that higher IL-2 signaling in Treg cells could partially compensate for CNS2-deficiency, we noticed a marked enrichment of cells expressing higher CD25 in CNS2-deficient Treg cells and a correlation of the level of CD25 expression with the frequency and the amount of Foxp3 expression in CNS2-deficient Treg cells (Figure 4F and data not shown). These observations suggested that under non-inflammatory conditions a key function of CNS2 is to maintain Foxp3 expression in dividing Treg cells in non-lymphoid tissues.
Figure 6. CNS2 dependent inheritance of Treg cell identity prevents spontaneous age dependent autoimmunity.
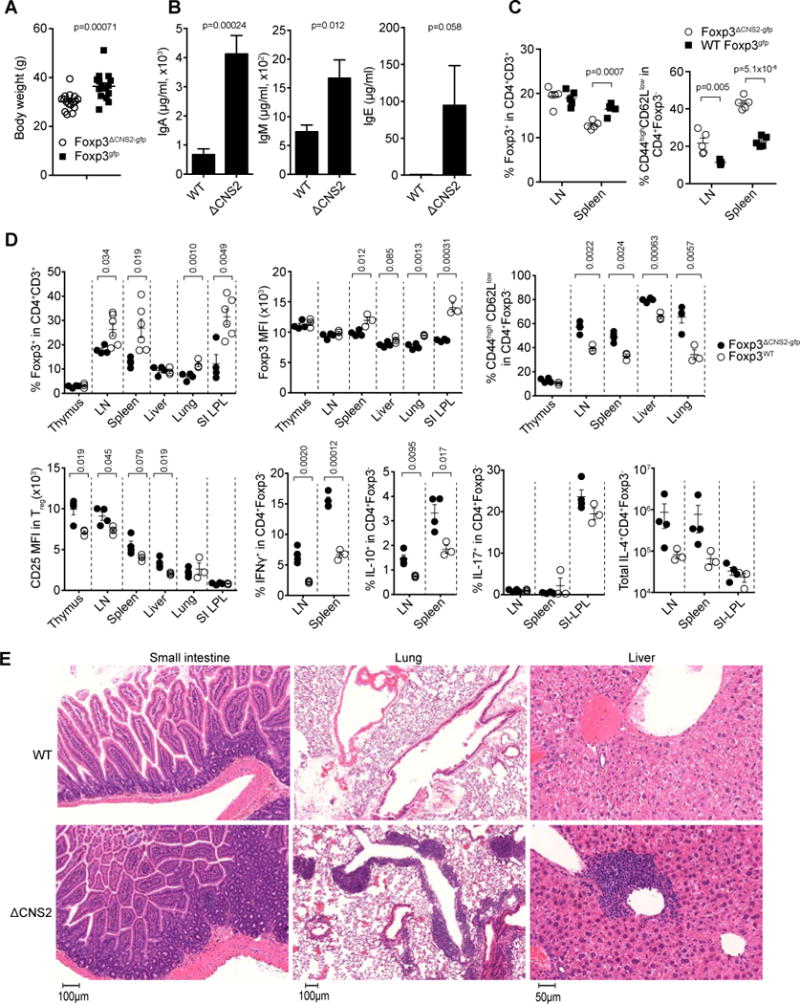
(A) Diminished body weight of Foxp3ΔCNS2-gfp mice (~12 month old) in comparison to wild type littermates. WT 36.45 ± 1.37g, ΔCNS2 30.18 ± 0.88g. n=16 per group. (B) Serum Ig levels in Foxp3ΔCNS2-gfp and littermate control mice (~12 month old) determined by ELISA. WT n=6, Foxp3ΔCNS2-gfp n=4. (C) Frequencies of Treg cells and activated CD4+Foxp3− T cells (CD44highCD62Llow) in Foxp3ΔCNS2-gfp and control mice (~3 month old; n=5 per group). (D, E) Flow cytometric analyses of Foxp3 and CD25 expression, activation of, and cytokine production by CD4+ Foxp3− T cells and tissue histopathology in 12–15 month old Foxp3ΔCNS2-gfp mice. Data represent one of 2 or more independent experiment. Mean ± SEM.
Figure 4. CNS2 dependent maintenance of heritable Foxp3 expression and Treg cell numbers in the steady state.

(A) Schematic of the generation of mixed bone marrow chimeras. (B, C) Treg cell frequency and Foxp3 expression level in Ly5.1+ Foxp3gfp and Ly5.2+ Foxp3ΔCNS2-gfp cells in the thymus, spleen, lymph nodes (LN), mesenteric lymph nodes (MLN), Peyer’s patch (PP), small intestine lamina propria (SI-LPL), liver, lungs, and visceral adipose tissue (VA) of the mixed BM chimeras. Percentages of Ly5.1+ and Ly5.2+ cells in the reconstituted Treg (CD4+GFP+Foxp3+) cells were normalized based on the reconstitution efficiency of total Ly5.1+ and Ly5.2+ lymphocytes in the thymus. Mean ± SEM. n=9 per group; the data represent one of 3 independent experiments. (D) Correlation of Ki67 expression with the difference in Foxp3 MFI between CNS2-sufficient and -deficient Treg cells (spleen). (E) Reduced frequency of, and lower Foxp3 expression level in Foxp3ΔCNS2-gfp Treg cells in activated Treg cell population (CD44highCD62Llow). (F) CD25 expression level was correlated with the frequency of CNS2-deficient Treg cells in the mixed population and anti-correlated with the difference in Foxp3 expression level between CNS2-sufficient and -deficient Treg cells (spleen). See also Figure S4.
CNS2 prevents differentiation of Treg cells into effector cells
Next, we asked whether CNS2 is required to prevent acquisition of alternative cell fates, i.e. differentiation into effector cells, by mature Treg cells under condition of autoimmune inflammation. Previous studies demonstrated a prominent capacity of Treg cells to limit inflammation in the brain and the spinal cord and limb paralysis resulting from Experimental Autoimmune Encephalomyelitis (EAE). EAE serves as an animal model for multiple sclerosis and is induced upon immunization of mice with a peptide derived from myelin oligodendrocyte glycoprotein (MOG35-55) in complete Freund’s adjuvant (CFA) (Pachner, 2011; Stromnes and Goverman, 2006; Webster et al., 2009). Furthermore, a recent study suggested that some Treg cells, in particular, those specific for MOG35-55 peptide, lose Foxp3 expression and become effector T cells in the course of EAE (Bailey-Bucktrout et al., 2013). These observations raised the question whether CNS2 was dispensable under autoimmune inflammatory conditions because its function in the maintenance of Foxp3 expression was abolished. Alternatively, it was possible that the requirement for CNS2 was heightened and that Foxp3 prevented deviation of Treg cells into pro-inflammatory effector T cells under inflammatory conditions. To answer these questions we assessed Foxp3 and pro-inflammatory cytokine expression by CNS2-deficient and -sufficient Treg cells present in the same tissue inflammatory settings during the course of EAE. To track the dynamics of cell populations in these experiments, we adoptively transferred Treg cells double sorted from Ly5.2 UBCCre-ERT2 Foxp3CNS2fl-gfp R26Y together with naïve Ly5.1+ CD4+Foxp3− T cells into lymphopenic recipients (Figure 5A). Mice were immunized with MOG peptide in CFA immediately after acute depletion of CNS2 induced with tamoxifen in ~30% of transferred Treg cells. Due to limited numbers of effector T cells and ~70% of Treg cells with the unrecombined CNS2fl allele we observed very mild disease (clinical score ~ 1.5–2) in these recipients. We found that almost all (>95%) CNS2-deficient Treg cells lost Foxp3 expression in the central nervous system (brain and spinal cord), in contrast, around a third of CNS2-sufficient Treg cells retained Foxp3 expression (Figure 5B and C). Thus, CNS2-dependent stable inheritance of Foxp3 expression plays a crucial role in maintaining Treg cell numbers under local inflammatory conditions in vivo. Loss of Foxp3 expression by a cohort of Ly5.2+ CNS2-deficient Treg cells was associated with acquisition of the production of IL-2 and pro-inflammatory cytokines IFNγ and IL-17 (Figure 5B and C). Thus, under inflammatory conditions CNS2 prevents the loss of Foxp3 by dividing Treg cells, and their differentiation into effector T cells. As a consequence of sharply reduced Foxp3 levels, CNS2-deficient Treg cells displayed lower amounts of CTLA4 and failed to effectively restrain autoimmune disease upon co-transfer into T cell-deficient hosts with effector CD4+ T cells isolated from Foxp3-deficient mice (Figure S5A and B).
Figure 5. CNS2-dependent inheritance of Foxp3 expression prevents Treg cell conversion to effector cells.
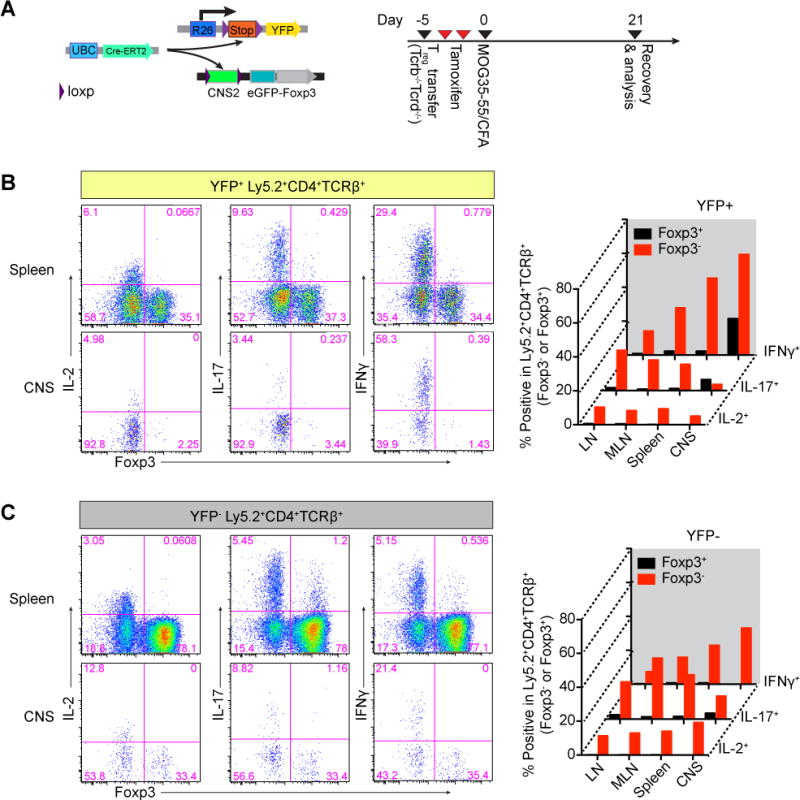
(A) Schematic of the experimental design. Treg cells doubly sorted from Ly5.2 Foxp3CNS2fl-gfp UBCCre-ERT2 R26Y male mice were cotransferred with Ly5.1 naïve CD4+Foxp3− T cells into Tcrb−/−Tcrd−/− recipients. Acute ablation of CNS2 was induced by tamoxifen gavage and 21 days after immunization with MOG35-55/CFA Ly5.2+CD4+TCRβ+ T cells were analyzed. (B, C) YFP+ and YFP− of Ly5.2+CD4+TCRβ+ T cells were separated by FACS and stimulated with PMA and ionomycin and cytokine production of the cells was analyzed by flow cytometry. The data represent one of 3 independent experiments. CNS, combined infiltrated lymphocytes from the central nervous system (brains and spinal cords). See also Figure S5.
CNS2 dependent maintenance of Foxp3 expression is essential for suppressing chronic inflammation
These results suggested that the failure to maintain the Treg cell fate in the absence of CNS2 would result in heightened inflammation and disease in a variety of settings due to a reduced suppressor function of Treg cells and their increased conversion into effector T cells (Figure 5 and S5). To test this idea we evaluated histopathology in aged 12–15 months old Foxp3ΔCNS2-gfp mice. We found that CNS2 deficiency resulted in severe inflammatory lesions in numerous non-lymphoid organs including lung, liver, stomach, small and large intestine (Figure 6E and data not shown). In addition, Foxp3ΔCNS2-gfp mice exhibited decreased body weight, elevated circulating immunoglobulin (Ig) levels and activated effector T cells (Figure 6A–C). Thus, CNS2 deficiency results in spontaneous age-dependent inflammatory lesions in non-lymphoid organs.
As the consequence of inflammation, CNS2-deficient Treg cell may lose more functional Treg cells due to cell division, pro-inflammatory cytokine exposure and/or IL-2 deprivation (Figure 3A, 2B and 2C). To direct test the impact of inflammation on CNS2-deficient Treg stability in vivo without manipulation of Treg cells in vitro, we immunized mixed bone marrow chimeras of Ly5.1 Foxp3gfp and Ly5.2 Foxp3ΔCNS2-gfp mice with MOG peptide and found that in contrast to CNS2-sufficient Treg cells CNS2-deficient Treg frequency was remarkably lower in the central nervous system, which was coupled with dramatically diminished Foxp3 protein level (Figure 7A). We also examined the course of MOG peptide-induced EAE in Foxp3ΔCNS2-gfp and Foxp3gfp mice. Although CNS2 deficiency did not impact the onset of the disease or its severity during the initial progressive phase, the disease in Foxp3ΔCNS2-gfp mice failed to remit and exhibited a chronic, relentless course after reaching its peak in contrast to the remitting-relapsing disease observed in control Foxp3gfp mice (Figure 7B and S6A). The heightened clinical manifestations including limb paralysis and weight loss correlated with a diminished Treg cell frequency among lymphocytes present in the brain and the spinal cord (Figure S6B–D and data not shown). Unlike other non-lymphoid tissues, Treg and effector T cells were barely detectable in the brain and spinal cord of unchallenged control and mutant animals (data not shown). Thus, the more severe, relapse-free disease course is unlikely due to a diminished pre-existing pool of Treg cells in CNS2-deficient mice, but rather a consequence of a loss of Foxp3 expression by CNS2-deficient Treg cells forced into the cell cycle in the setting of a localized chronic autoimmune inflammation (Figure 5 and 7A).
Figure 7. CNS2 dependent maintenance of Foxp3 expression is critical for suppressing chronic inflammation.

(A) Flow cytometric analysis of Foxp3 expression in CNS2-deficient Treg cells in the central nervous system after MOG immunization. Chimeric mice transferred with Ly5.1 Foxp3gfp and Ly5.2 Foxp3ΔCNS2-gfp BM (see also Figure 4A) were immunized with MOG35-55 in CFA and analyzed on day 14. Mean ± SEM, n=9. (B) Loss of CNS2 aggravates the chronic phase of EAE. Male Foxp3ΔCNS2-gfp and Foxp3gfp littermates were immunized with MOG35-55/CFA and their body weights and clinical disease scores were monitored. Mean + SEM. WT n=8, Foxp3ΔCNS2-gfp n=10. Data represent one of 3 independent experiments. (C) Analysis of Treg and activated effector CD4+Foxp3− T cells in Foxp3ΔCNS2-gfp and littermate control male mice on a high fat diet (HFD) and control diet (RD: regular diet). Mice were fed with HFD or RD for 10 weeks before the analysis. Data represent one of >3 independent experiments. Mean ± SEM. WT (RD) n=5, WT (HFD) n=8, Foxp3ΔCNS2-gfp (RD) n=5, Foxp3ΔCNS2-gfp (HFD) n=5. (D) Flow cytometric analysis of CD25highKi67− Treg cells in Foxp3ΔCNS2-gfp and littermate control mice on a HFD diet. (E) Body weight of Foxp3ΔCNS2-gfp and littermate control mice infected with LCMV clone 13 (p≤0.05). The data represent one of two independent experiments. Mean ± SEM. WT n=6, Foxp3ΔCNS2-gfp n=5. See also Figure S6 and S7.
In addition to spontaneous autoimmune lesions in non-lymphoid organs and autoantigen-driven focal autoimmunity, we explored a role for CNS2-dependent maintenance of Treg cell fate during sterile inflammation associated with obesity. Because metabolic perturbation induced by a high-fat diet (HFD) causes mild chronic inflammation (Feuerer et al., 2009), we first investigated Treg cell frequency and effector T cell phenotypes in wild type Foxp3gfp and Foxp3ΔCNS2-gfp mice that were fed with 60 kcal % high fat diet (>10 wk). We found that the frequency of CNS2-sufficient Treg cells was increased in spleen and liver, but decreased in visceral adipose tissue in mice on a HFD. In contrast, the frequency of CNS2-deficient Treg cells was not increased by HFD in comparison to regular diet despite a marked increase in CD25+ cells in CNS2-deficient mice (Figure 7C, 7D and S7A–C). The failure to increase Treg cell numbers was associated with a dramatic increase in the numbers of activated effector CD44highCD62Llow Foxp3−CD4+ T cells in these mice (Figure 7C, right panel). These results suggest that chronic inflammation resulting from a metabolic disorder requires continuous maintenance of Foxp3 expression when Treg cells divide in response to inflammatory cues.
Finally, we assessed the role of CNS2 during chronic viral infection. Foxp3ΔCNS2-gfp mice infected with chronic lymphocytic choriomeningitis virus (LCMV) clone 13 showed a severe progression of the wasting disease during the chronic phase of infection which was less pronounced in control Foxp3gfp mice. Interestingly, the weight loss of Foxp3ΔCNS2-gfp mice was comparable to control Foxp3gfp mice during the acute phase of infection (d7), but was severely increased during the chronic phase, when Treg cells are massively expanding (Punkosdy et al., 2011)(Figure 7E), suggesting CNS2-dependent inheritable maintenance of Treg cells is critical for limiting inflammation during chronic viral infection. Together, these data demonstrate the crucial role of CNS2-dependent inheritance of Foxp3 expression and highlights a mechanism by which a dedicated cis element serves to maintain cellular identity during the vulnerable stage of cell division, a prerequisite for Treg cell function during immune homeostasis, acute and chronic inflammation.
DISCUSSION
We found that Treg cell proliferation was a prerequisite for silencing of the Foxp3 locus and CNS2 opposed the shutdown of Foxp3 expression. Previous studies showed that CNS2 CpGs are methylated in immature and demethylated in mature Treg cells and that a demethylated state of CNS2 correlated with stable Foxp3 expression (Floess et al., 2007; Polansky et al., 2008; Toker et al., 2013). Our analysis of stage specific deletion of CNS2 revealed that its functionality is conditional upon its demethylation in mature Treg cells (data not shown). Thus, demethylated CNS2 opposed Foxp3 silencing in fully differentiated mature Treg cells in an apparently stochastic manner and this activity of CNS2 was continuously required for the maintenance of heritable Foxp3 expression.
In search of factors that converge on CNS2 to maintain Foxp3 expression during cell division we uncovered a key role for IL-2 signaling-induced STAT5 activation, which was supported by previous studies of IL-2 signaling in the differentiation and maintenance of Treg cells (Fontenot et al., 2005; Setoguchi et al., 2005). Our results suggest that at physiologic levels IL-2 signaling stabilizes heritable Foxp3 expression in dividing mature Treg cells to a large degree through CNS2. Consistent with this notion and previous work, we found STAT5 binds to CNS2 and, to a lesser degree, to the Foxp3 promoter (Ogawa et al., 2013; Yao et al., 2007). Importantly, in the absence of CNS2 STAT5 binding to the Foxp3 promoter was markedly reduced suggesting that CNS2 may interact with the promoter through a short distance loop. Consistent with these results heightened IL-2 signaling or induced expression of STAT5CA restored stable Foxp3 expression in CNS2-deficient Treg cells. On the other hand, exposure to pro-inflammatory cytokines including IL-4, IL-6, IFNγ resulted in a markedly increased loss of Foxp3 in dividing Treg cells in the absence of CNS2 despite the presence of optimal amounts of IL-2. Il-4 activated STAT6 competes with STAT5 for the binding to similar sites in the Foxp3 locus and counteracts STAT5 activity. Unlike STAT5, STAT6 interacted with Dnmt1; CpG methylation at the Foxp3 locus was increased during Treg cell division and upon loss of Foxp3 expression was fully returned to the high methylation state of effector T cells. Of note, IL-6-activated STAT3 also binds to the aforementioned sites within the Foxp3 locus and associates with Dnmt1 (data not shown) (Zhang et al., 2005). Together, these observations suggest that in the absence of CNS2 STAT proteins activated downstream of pro-inflammatory cytokine signaling may effectively compete with STAT5 for binding to the Foxp3 locus and bring DNA methyltransferases and thereby silence Foxp3 transcription. Our experiments indicate that CNS2 likely increases STAT5 occupancy at the Foxp3 promoter prior to inflammation and hence opposes pro-inflammatory cytokine-driven silencing of the Foxp3 locus. The sum of the opposing effects of IL-2 and pro-inflammatory cytokine signaling determines the inheritance of Foxp3 expression and the fate of dividing Treg cells. Although in-depth mechanistic understanding of this phenomenon would require careful quantification of active STAT proteins and their affinities for Foxp3 binding sites in Treg cells, our results suggest that CNS2 skews this balance by recruiting STAT5 to the Foxp3 promoter and enabling Treg cells to withstand diminished IL-2 and heightened inflammatory cytokine signaling. Thereby, CNS2 serves as a critical determinant of Treg cell lineage stability upon cell division in inflammatory environments where IL-2 amounts are limiting. Under physiological conditions, CNS2 maintains the Treg cell population and, thereby, prevents spontaneous age-dependent inflammation in non-lymphoid organs. Furthermore, CNS2-dependent maintenance of Foxp3 expression limits chronic, but not acute phase of organ-specific autoimmune inflammation and precludes acquisition of effector functions by Treg cells in response to inflammatory cues. Similarly, compromised inheritance of Foxp3 expression in Treg cells in the absence of CNS2 resulted in diminished health status in the course of chronic viral infection as well as increased immune cell activation during sterile metabolic inflammation.
It seems likely that the observed features of regulation of the inheritance of Treg cell identity upon integration of extracellular cues by a cis-regulatory element dedicated to cell fate maintenance may be operational in other dividing cell types. Susceptibility to a stochastic down-regulation of the expression of a lineage specification factor such as Foxp3 and the need for a dedicated regulatory element to oppose such a loss might be linked to the intrinsic inhibition of transcriptional activity, splitting and dilution of transcription factors, and histone modifications during cell division (Gottesfeld and Forbes, 1997; Mullen et al., 2001; Probst et al., 2009). Thus, the mitosis related transcriptional fluctuation could increase the probability of complete transcriptional silencing of Foxp3 expression through yet to be defined active or passive processes (Losick and Desplan, 2008). Considering the respective positive and negative roles of IL-2 and of pro-inflammatory cytokines in the heritable expression of Foxp3, it seems likely that the promotion of cell division by these factors may increase the vulnerability of Treg cells to the loss of their identity due to the stochastic silencing of Foxp3 by molecular mechanisms that remain to be explored.
In conclusion, we found that CNS2, a cis-acting regulatory element dedicated to heritable maintenance of the active state of the Foxp3 locus, enables dividing Treg cells to maintain their differentiation state upon deprivation of their essential growth factor IL-2 and upon exposure to pro-inflammatory cues driving alternative effector T cell differentiation. CNS2-dependent stability of the Treg cell lineage is critical for preventing spontaneous, age-dependent chronic inflammation in a variety of non-lymphoid organs, for restraining metabolic inflammation associated with diet-induced obesity, and for limiting organ-specific autoimmunity as well as the wasting-syndrome during chronic viral infection.
EXPERIMENTAL PROCEDURES
Animals
Foxp3ΔCNS2-gfp and Foxp3CNS2fl-gfp mice (Zheng et al., 2010) were backcrossed onto C57BL/6 background for more than 10 generations. Foxp3DTR and Foxp3gfpko were described previously (Gavin et al., 2007; Kim et al., 2007). R26-Stopfl-STAT5CA mice were generated by inserting a coding sequence of STAT5CA and a GFP reporter into the Rosa26 locus (Sasaki et al., 2006) (T. Chinen and A. Rudensky in preparation). All animals were maintained in the MSKCC animal facility under SPF conditions and the experiments were performed according to the institutional guidelines (IACUC 08-10-023).
Generation of bone marrow chimeras and adoptive T cell transfers
For the generation of bone marrow (BM) chimeras, recipient mice were irradiated (9.5 Gy) 24 hours prior to bone marrow stem cell transfer. BM cells were depleted of T cells with Dynabeads® FlowComp™ Mouse Pan T kit (Life Technologies) and injected i.v. into irradiated recipients. After BM transfer, recipient mice received neomycin with drinking water (2mg/ml) for 3 wk and were analyzed 8–12 wk later.
For adoptive cell transfers, Treg cells were purified from Foxp3gfp, Foxp3ΔCNS2-gfp or Foxp3CNS2fl-gfp males by double FACS sorting based on GFP expression after enrichment of CD4 T cells with Dynal magnetic beads (Life Technologies) and cotransferred with congenically labeled (Ly5.1) naïve CD4+Foxp3− T cells at a 1:5 ratio (total 2.5×106 cells per recipient) into T cell deficient Tcrb−/−Tcrd−/− recipients. Transferred cell subsets were analyzed by flow cytometry ~4 wk later.
Statistical analysis
For statistical analysis, unpaired two-tailed Student’s t-test was performed using Prism (GraphPad) or Excel (Microsoft). Regression analysis and plotting were performed with Excel. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 and **** p≤0.0001.
Supplementary Material
HIGHLIGHTS.
Cytokine signaling regulates heritable Foxp3 expression
Intronic element CNS2 sustains heritable Foxp3 expression in mature Treg
IL-2-STAT5 acts on CNS2 to counteract pro-inflammatory cytokine signaling
Critical roles of Treg fate heritability in physiological contexts
Acknowledgments
We thank MSKCC Genomics and Bioinformatics Core facilities for RNA sequencing, Flow Cytometry Core facility for assistance with cell sorting. W. Hu for assistance with the analysis of bisulfite sequencing data; P. DeRoos, S. Hemmers, Q. Li, G. Loeb and S. Dikiy for technical assistance. MSKCC core facilities are supported by Cancer Center Support Grant CCSG P30 CA008748. Y.F. was supported by a Postdoctoral Fellowship of the Cancer Research Institute. This study was supported by an NIH grant and the Howard Hughes Medical Institute (A.Y.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers
The GEO accession number of the RNA sequencing data reported in this study is GSE58905.
See the Extended Experimental Procedures for further information
AUTHOR CONTRIBUTIONS
Y.F. and A.Y.R. conceived of and designed the experiments and interpreted the results. A.A. analyzed RNA sequencing data. T.C. generated STAT5CA mice. G.G. performed LCMV experiments. J.v.d.V. performed pilot ChIP experiments and interpreted the results. Y.F. and A.Y.R. wrote the manuscript.
References
- Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, Rudensky AY. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol. 2014;15:580–587. doi: 10.1038/ni.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, Fehling HJ, Bluestone JA. Self-antigen-Driven Activation Induces Instability of Regulatory T Cells during an Inflammatory Autoimmune Response. Immunity. 2013;39:949–962. doi: 10.1016/j.immuni.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, Bell BD, Ziegler SF, Huehn J, Pardoll DM, et al. Loss of epigenetic modification driven by the foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36:717–730. doi: 10.1016/j.immuni.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darce J, Rudra D, Li L, Nishio J, Cipolletta D, Rudensky AY, Mathis D, Benoist C. An N-Terminal Mutation of the Foxp3 Transcription Factor Alleviates Arthritis but Exacerbates Diabetes. Immunity. 2012 doi: 10.1016/j.immuni.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- Gottesfeld JM, Forbes DJ. Mitotic repression of the transcriptional machinery. Trends in biochemical sciences. 1997;22:197–202. doi: 10.1016/s0968-0004(97)01045-1. [DOI] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Wilson CB, Rudensky AY. Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J Immunol. 2009;182:6648–6652. doi: 10.4049/jimmunol.0803320. [DOI] [PubMed] [Google Scholar]
- Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. 2007;204:1543–1551. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci U S A. 2009;106:1903–1908. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-Hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H. Pathogenic conversion of Foxp3 T cells into T17 cells in autoimmune arthritis. Nat Med. 2013 doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- Losick R, Desplan C. Stochasticity and cell fate. Science. 2008;320:65–68. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, Huehn J, Hori S. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–275. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Mullen AC, Hutchins AS, Villarino AV, Lee HW, High FA, Cereb N, Yang SY, Hua X, Reiner SL. Cell cycle controlling the silencing and functioning of mammalian activators. Curr Biol. 2001;11:1695–1699. doi: 10.1016/s0960-9822(01)00533-4. [DOI] [PubMed] [Google Scholar]
- Ogawa C, Tone Y, Tsuda M, Peter C, Waldmann H, Tone M. TGF-beta-Mediated Foxp3 Gene Expression Is Cooperatively Regulated by Stat5, Creb, and AP-1 through CNS2. J Immunol. 2013 doi: 10.4049/jimmunol.1301892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, Osaki M, Tanaka Y, Yamashita R, Nakano N, et al. T Cell Receptor Stimulation-Induced Epigenetic Changes and Foxp3 Expression Are Independent and Complementary Events Required for Treg Cell Development. Immunity. 2012 doi: 10.1016/j.immuni.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Pachner AR. Experimental models of multiple sclerosis. Current opinion in neurology. 2011;24:291–299. doi: 10.1097/WCO.0b013e328346c226. [DOI] [PubMed] [Google Scholar]
- Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–1663. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- Punkosdy GA, Blain M, Glass DD, Lozano MM, O’Mara L, Dudley JP, Ahmed R, Shevach EM. Regulatory T-cell expansion during chronic viral infection is dependent on endogenous retroviral superantigens. Proc Natl Acad Sci U S A. 2011;108:3677–3682. doi: 10.1073/pnas.1100213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability of the regulatory T cell lineage in vivo. Science. 2010;329:1667–1671. doi: 10.1126/science.1191996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151:153–166. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Alvarado A, Yamanaka S. Rethinking Differentiation: Stem Cells, Regeneration, and Plasticity. Cell. 2014;157:110–119. doi: 10.1016/j.cell.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, Schmidt-Supprian M. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity. 2006;24:729–739. doi: 10.1016/j.immuni.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Sekiya T, Kashiwagi I, Yoshida R, Fukaya T, Morita R, Kimura A, Ichinose H, Metzger D, Chambon P, Yoshimura A. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat Immunol. 2013 doi: 10.1038/ni.2520. [DOI] [PubMed] [Google Scholar]
- Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1810–1819. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- Toker A, Engelbert D, Garg G, Polansky JK, Floess S, Miyao T, Baron U, Duber S, Geffers R, Giehr P, et al. Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. J Immunol. 2013;190:3180–3188. doi: 10.4049/jimmunol.1203473. [DOI] [PubMed] [Google Scholar]
- Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, Grey ST, Sprent J. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med. 2009;206:751–760. doi: 10.1084/jem.20082824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2011;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity. 2010;33:313–325. doi: 10.1016/j.immuni.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, Laurence A, Robinson GW, Shevach EM, Moriggl R, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3-and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:6948–6953. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, Corcoran L, Treuting P, Klein U, Rudensky AY. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.