

Abstract
Designing of drug nanocarriers to aid delivery of therapeutics is an expanding field that can improve medical treatments. Nanocarriers are often functionalized with elements that recognize cell-surface molecules involved in subcellular transport to improve targeting and endocytosis of therapeutics. Combination-targeting using several affinity elements further modulates this outcome. The most studied example is endothelial targeting via multiple cell adhesion molecules (CAMs), which mimics the strategy of leukocytes to adhere and traverse the vascular endothelium. Yet, the implications of this strategy on intracellular transport and in vivo biodistribution remain uncharacterized. We examined this using nanocarriers functionalized for dual- or triple- targeting to intercellular, platelet-endothelial, and/or vascular CAMs (ICAM-1, PECAM-1, VCAM-1). These molecules differ in expression level, location, pathological stimulation, and/or endocytic pathway. In endothelial cells, binding of PECAM-1/VCAM-1-targeted nanocarriers was intermediate to single-targeted counterparts and enhanced in disease-like conditions. ICAM-1/PECAM-1-targeted nanocarriers surpassed PECAM-1/VCAM-1 in control, but showed lower selectivity toward disease-like conditions. Triple-targeting resulted in binding similar to ICAM-1/PECAM-1 combination and displayed the highest selectivity in disease-like conditions. All combinations were effectively internalized by cells, with slightly better performance when targeting receptors of different endocytic pathways. In vivo, ICAM-1/PECAM-1-targeted nanocarriers outperformed PECAM-1/VCAM-1 in control and disease-like conditions, and triple-targeted counterparts slightly enhanced this outcome in some organs. As a result, delivery of a model therapeutic cargo (acid sphingomyelinase, deficient in Niemann-Pick disease A-B) was enhanced to all affected organs by triple-targeted nanocarriers, particularly in disease-like conditions. Therefore, multi-CAM targeting may aid optimization of some therapeutic nanocarriers, where the combination and multiplicity of the affinity moieties utilized allow modulation of targeting performance.
Keywords: targeted nanocarriers, endothelial cell adhesion molecules, combination targeting, endocytosis, in vivo biodistribution, lysosomal enzyme delivery
1. Introduction
A major focus in the design of drug delivery carriers is surface functionalization with affinity moieties to enhance site-specificity and/or intracellular transport of therapeutics [1-7]. Derived from this strategy, combination-targeting to multiple cell-surface molecules is a relatively new approach that holds promise to further control drug delivery. Combination-targeting has been shown to modify in vivo biodistribution of drugs and/or their carriers [8], improve sensitivity of probes of endothelial dysfunction or angiogenesis [9, 10], enhance transport to tumor areas and delivery to multiple cell types in cases of cancer applications [11-14], and minimize off-target effects [15, 16].
One of the most studied examples of combination-targeting focuses on cell adhesion molecules (CAMs) expressed on the vascular endothelium [9, 15, 17-24]. This is the case for microparticles carrying iron oxide or fluorescent probes [9, 22], perfluorocarbon-filled microbubbles [18, 24], or gold nanorods used in imaging [21], or that of polymersomes [23], immunoliposomes [19, 20], or PLGA microspheres [15] intended for drug delivery. These examples emulate adhesive properties of leukocytes by combining targeting to an endothelial selectin and an immunoglobulin (Ig)-like CAM [9, 15, 17-24]. Selectins are involved in low-affinity interactions of leukocytes on endothelial surfaces, while Ig-like CAMs impact firm adhesion and extravasation of leukocytes [25]. Simultaneous targeting to these molecules has been shown to be beneficial. For instance, at particular flow shear rates, microspheres functionalized at certain ligand-receptor ratios with sialyl Lewis(X) and anti-ICAM bind to cells only through interaction with both receptors, enhancing selectivity [15]. Targeting polymersomes to P-selectin and ICAM-1 also enhances binding over single-targeted counterparts and improves selectivity toward inflammation [23], and combined P-selectin/VCAM-1 targeting improves binding of microbubbles in receptor-coated flow chambers [18]. These “leukomimetic” approaches are being explored for treatment and/or diagnosis of inflammation [9, 15, 19, 21-24, 26], atherosclerosis [18, 20, 22, 26], and cancer [27].
Translation of these strategies requires a good understanding of their impact in vivo. Such data are scarce, yet promising: targeting iron oxide microparticles to P-selectin/VCAM-1 enhances access to atherosclerotic sites in mouse models [26], and targeting fluorescent microspheres to P-selectin/ICAM-1 improves detection of inflammation in the choroidal or retinal microvasculature [9]. However, the overall organ biodistribution of carriers targeting multiple CAMs remains unknown, particularly for submicrometer carriers, a size more amenable for intracellular drug delivery [4]. Most studies on multi-CAM targeting have focused on micro- vs. nano-carriers and no studies have explored intracellular transport of these carriers. Combination-targeting to CAMs involved solely in leukocyte firm adhesion (vs. rolling and firm adhesion) has never been tested. The comparative impact of dual- vs. triple-targeting to CAMs is also an open question which holds relevance since it better reflects the multiple interactions between leukocytes and the endothelium [25].
We have explored these aspects using model polymer nanocarriers targeted to multiple Ig-like CAMs, including intercellular adhesion molecule-1 (ICAM-1) [28], platelet endothelial cell adhesion molecule-1 (PECAM-1) [29, 30], and/or vascular cell adhesion molecule 1 (VCAM-1) [31]. These molecules are involved in leukocyte firm adhesion and transmigration [25, 32], yet they differ in several parameters [25]. ICAM-1 and VCAM-1 are predominantly located on the luminal surface of endothelial cells, while PECAM-1 is enriched at the cell-cell border [25]. In healthy states VCAM-1 expression is low, followed by ICAM-1, vs. relatively high PECAM-1 expression [25]. VCAM-1 and ICAM-1 expression are enhanced by cytokines and other pathological stimuli, while PECAM-1 remains relatively stable [25]. ICAM-1 and PECAM-1 mediate endocytosis of nano- and micro-carriers via CAM-mediated endocytosis [33-35], and VCAM-1 mediates uptake by clathrin-mediated endocytosis [36] which is more size-restrictive (≤200-nm) [37, 38]. Given these differential properties, combination-targeting to these CAMs is interesting from both a fundamental perspective and to optimize endothelial drug delivery. Our results shed light into the potential of this strategy based on the observed efficacy of targeting and internalization in cell culture, as well as biodistibution and delivery of a model drug in mice studied in both control and disease-like conditions induced by inflammatory stimuli.
2. Materials and Methods
2.1. Antibodies and reagents
Antibodies against mouse ICAM-1, PECAM-1, or VCAM-1 were clone YN1 (ATCC; Manassas, VA), MEC13 (BD Biosciences; San Jose, CA), and MK2 (Santa Cruz Biotechnology; Dallas, TX). Non-specific IgG was from Jackson Immunoresearch (Pike West Grove, PA). Recombinant human acid sphingomyelinase (ASM) was provided by Dr. Edward Schuchman (Mount Sinai School of Medicine) [39]. Green Fluoresbrite Polystyrene particles were from Polysciences (Warrington, PA). Unless otherwise stated, all other reagents were from Sigma Chemical (St. Louis, MO).
2.2. Preparation and characterization of targeted nanocarriers
As previously described [8], model polymer nanocarriers were prepared by surface adsorption of targeting antibodies on polymer particles, which leads to random antibody orientation on the particle coat. While this is not optimal for clinical applications, it is advantageous for this study because such random orientation is similar for all formulations tested (all antibodies used are rat immunoglobulin G (IgG) type). This minimizes the contribution of antibody orientation associated to conjugation techniques, where the site and number of linkages and their final orientation need to be controlled and matched for each antibody and reaction.
Briefly, 100-nm green Fluoresbrite polystyrene nanospheres were coated with monoclonal antibodies by surface adsorption for 1 h at room temperature, including: (a) IgG (non-targeted); (b) anti-ICAM, anti-PECAM, or anti-VCAM (single-targeted; full valency) or anti-ICAM/IgG, anti-PECAM/IgG, or anti-VCAM/IgG (single targeted; one half or one third valency); (c) anti-ICAM/PECAM or anti-PECAM/VCAM (1:1 dual-targeted); (d) anti-ICAM-1/PECAM/VCAM (1:1:1 triple-targeted); or (e) antibody and ASM (1:1 mass ratio), where the antibody component contained anti-ICAM/PECAM/VCAM (1:1:1) and ASM was used as a model therapeutic cargo. For in vivo experiments, 125I-IgG or 125I-ASM were used as tracers. Uncoated molecules were removed by centrifugation at 13.8g for 3 min and coated nanocarriers were resuspended in phosphate buffer saline containing 0.3% bovine serum albumin, then sonicated to avoid aggregation with 20-30 short pulses (0.4-0.9g) with a probe sonicator. The antibody coating density was assessed using as many different 125I-labeled antibodies in independent carrier samples as antibody types on the coat. For instance, a preparation containing 125I-anti-ICAM + anti-PECAM and an independent preparation containing anti-ICAM + 125I-anti-PECAM were used to determine the density of anti-ICAM/PECAM NCs, which was done by measuring the 125I content of the coated nanocarrier suspension after centrifugation of non-coated counterparts in a gamma counter (PerkinElmer Wizard2, Waltham, MA). Potential antibody release from the coat over time was similarly assessed, as described [40]. The size, polydispersity, and zeta potential of the formulations were estimated by dynamic and electrophoretic light scattering (Malvern Zetasizer, Worcestershire, UK). Nanocarrier avidity was tested as described below (section 2.5). Table 1 summarizes the characterization of the different formulations used.
Table 1. Characterization of nanocarriers targeting ICAM-1, PECAM-1, and/or VCAM-1.
| Nanocarrier | Size (um) | PDI | Zeta potential (mv) | Antibodies per nanocarrier | Enzyme per nanocarrier | Binding avidity | |||
|---|---|---|---|---|---|---|---|---|---|
| Bmax (NCs/cell) | Kd (pM) | ||||||||
| Uncoated NCs | 137±11 | 0.04±0.03 | −44.9±9.0 | -- | -- | -- | -- | ||
| Single Targeted: | |||||||||
| Full Valency | |||||||||
| IgG NCs | 238±8.3 | 0.18±0.02 | −27.2±17 | 240±25 | -- | -- | -- | ||
| Anti-ICAM NCs | 225±6.6 | 0.15±0.01 | −32.5 ±3.3 | 273±37 | -- | 182.4 | 29.7 | ||
| Anti-PECAM NCs | 324±17 | 0.25±0.03 | −31.0±2.3 | 250±4.0 | -- | 213.8 | 18.8 | ||
| Anti-VCAM NCs | 240±18 | 0.16±0.02 | −30.3±0.07 | 239±22 | -- | 21.0 | 44.2 | ||
| One Half Valency | |||||||||
| Anti-ICAM/IgG | 235±3.2 | 0.14±0.03 | −27.3±1.1 | ICAM: 127 | IgG: 112 | -- | -- | -- | |
| Anti-PECAM/IgG | 274±0.7 | 0.15±0.04 | −31.3±0.2 | PECAM: 120 | IgG:98 | -- | -- | -- | |
| Anti-VCAM/IgG | 342±3.5 | 0.22±0.01 | −29.2±0.3 | VCAM:109 | IgG:115 | -- | -- | -- | |
| One Third Valency | |||||||||
| Anti-ICAM/IgG | 226±2.2 | 0.14±0.01 | −30.0±0.6 | ICAM: 87 | IgG: 162 | -- | -- | -- | |
| Anti-PECAM/IgG | 310±2.9 | 0.20±0.03 | −31.3±0.4 | PECAM: 69 | IgG: 154 | -- | -- | -- | |
| Anti-VCAM/IgG | 304±3.1 | 0.21±0.01 | −30.5±0.5 | VCAM: 81 | IgG: 142 | -- | -- | -- | |
| Dual Targeted: | |||||||||
| Anti-PECAM/VCAM NCs | 262±14 | 0.20±0.0l | −32.2±1.4 | PECAM: 101±26 | VCAM: 120±0.3 | -- | 37.7 | 104.4 | |
| Anti-ICAM/PECAM NCs | 272±17 | 0.18±0.03 | −32.6±1.6 | ICAM:132±6.7 | ‘ECAM: 123±6.4 | -- | 108.7 | 74.5 | |
| Triple Targeted: | |||||||||
| Anti-ICAM/PECAM/VCAM NCs | 267±12 | 0.20±0.02 | −32.6±4.9 | ICAM:92±0.2 | VCAM: 77±I8 | PECAM: 67±21 | -- | 78.0 | 48.7 |
| Anti-ICAM/PECAM/VCAM/ASM NCs | 235±6.8 | 0.16±0.01 | −35.3±0.8 | ICAM:50±4 | VCAM: 45±1.5 | PECAM:32±6 | ASM: 75±12 | -- | -- |
Data are Mean ± S.E.M; NC = nanocarrier.
2.3. Cell culture
H5V murine heart endothelial cells were cultured on gelatin-coated coverslips at 37°C, 5% CO2, and 95% relative humidity in DMEM medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 μg/ml penicillin, and 100 μg/ml streptomycin. When indicated, disease- (inflammatory)-like condition was mimicked by incubating cells for 16-20 h with 10 ng/ml tumor necrosis factor alpha (TNFα).
2.4. Endothelial expression of CAMs
The basal cell surface expression and expression level subsequent to TNFα stimulation for ICAM-1, PECAM-1, and VCAM-1 were assessed by immunostaining fixed H5V cells with 50 nM anti-ICAM-1, anti-PECAM-1, or anti-VCAM-1, respectively, followed by removal of the primary antibody and incubation with 25 nM FITC-labeled secondary antibody to allow analysis by fluorescence microscopy. The expression level was calculated as the specific staining over that of control non-specific IgG antibody, after subtraction of background fluorescence in each microscopy image.
2.5. Avidity and specificity of targeted nanocarriers toward endothelial cells
We tested nanocarrier avidity and specificity using fixed endothelial cells to avoid concomitant endocytosis that may confound the results. Binding of each nanocarrier type on fixed cells was verified to be similar to binding to live cells (see below) using an intermediate nanocarrier concentration (113 pM). Then, fixed cells were incubated for 1 h at 37°C with different concentrations (from 3.125 pM to 500 pM) of green Fluoresbrite nanocarriers single-, dual-, or triple-targeted to ICAM-1, VCAM-1 and/or PECAM-1. After washing off nanocarriers that did not bind to cells, fluorescence microscopy was employed to quantify the number of particles bound per cell. Ligand-binding saturation regression was performed to obtain the maximal binding (Bmax) and binding constant (Kd) of the different formulations. Regression curves fitted the experimental data with R2≥0.9 in all cases.
Alternatively, cells were incubated with dual or triple mixtures of nanocarriers single-targeted to these receptors (133 pM total concentration; equal split of carriers in the mix): e.g. dual-targeted anti-ICAM/PECAM NCs were compared to a mix of 66.5 pM single-targeted anti-ICAM NCs + 66.5 pM anti-PECAM NCs, where anti-ICAM NCs and anti-PECAM NCs displayed either full valency or valencies matching the dual-targeted anti-ICAM/PECAM NCs, etc. Incubations were done in the presence of control cell medium, medium containing 200 nM single, dual, or triple combination of anti-ICAM, anti-PECAM, and/or anti-VCAM antibodies to compete for specific interaction of targeted nanocarriers with cells, or medium containing 220 nM non-specific IgG as a negative control for competition assays. Fluorescence microscopy was used to quantify the number of particles bound per cell.
2.6. Binding and internalization of targeted carriers
For binding and endocytosis in live endothelial cells, cells were incubated at 37°C for 1 h in control or disease-like conditions with green Fluoresbrite nanocarriers (113 pM) single-, dual-, or triple-targeted to ICAM-1, VCAM-1 and/or PECAM-1. After washing off unbound materials, cells were fixed and surface-bound carriers were counter-stained with Texas-Red-labeled goat anti-rat IgG, as described [35]. Fluorescence microscopy was used to distinguish internalized nanocarriers (green single-labeled) from surface-bound counterparts (Texas-Red + green double-labeled). Binding was quantified as the total number of cell-associated objects and internalization was calculated as the fraction of internalized nanocarriers from the total number of cell-associated particles, as described [35]. This was done using Image-Pro 6.3 software (Media Cybernetics, Inc., Bethesda, MD) and a macro that counts the number of ∼200-nm fluorescent objects over a threshold background (not object intensity). We have verified that this technique is not influenced by potential changes in Fluoresbrite-particle intensity due to nanocarrier transit through different subcellular compartment (e.g. cell surface vs. intracellular endosomal compartments).
2.7. Biodistribution of targeted nanocarriers or their therapeutic cargo
C57BL/6J mice (untreated or injected intraperitoneally with 1 mg/kg bacterial lipopolysaccharide (LPS) ∼24 h prior to experiments, to mimic inflammation) were anesthetized and injected as follows. Single-, dual-, or triple-targeted nanocarriers containing 125I-IgG were injected intravenously (∼1.3 mg/Kg antibody and ∼1.8 × 1013 particles/Kg). Another set of mice was injected with free 125I-ASM or 125I-ASM co-coated on triple-targeted nanocarriers (∼0.7 mg/Kg ASM and ∼1.8 × 1013 particles/Kg). Blood and organs were harvested following euthanasia at 30 min post-injection. 125I and weight were determined to estimate the organ-to-blood localization ratio (LR) and specificity index (SI). LR represents the percent of injected dose accumulated per gram of tissue (%ID/g) divided by the %ID/g in blood, to account for differences in organ size and circulating nanocarriers [40]. The SI is calculated as the LR of targeted formulations divided by the LR of non-targeted counterparts (either IgG NCs or free ASM, as it corresponds) and represents the specific organ targeting [40]. These studies complied with IACUC, University of Maryland regulations, and the Guide for Care and Use of Laboratory Animals of the U.S National Institutes of Health.
2.8. Statistics
Data were calculated as mean ± standard error of the mean (S.E.M.). Cell experiments contained duplicated coverslips and were repeated ≥ 2 times (total coverslips ≥ 4), and involved individual (not bulk) analysis of ≥ 20 cells per coverslip (≥ 80 individual cells) randomly selected through the entire cell sample, which renders statistical significance. For in vivo experiments 5-6 mice were used per condition, except for groups which represented repeats under a different condition of results already observed to render statistical differences (see Table 2). Statistical significance was determined as p<0.05 by Student's t-test (for comparison between two groups) or ANOVA with post hoc Tukey's test (for comparison among > 2 groups).
Table 2. Circulation of antibodies or antibody-coated nanocarriers injected in mice.
| Ab or NC | n | 1 min | 15 min | 30 min |
|---|---|---|---|---|
| Antibody | ||||
| IgG | 6 | 91±5.7 | 79±3.7 | 76±3.7 |
| Anti-ICAM | 6 | 60±1.8 | 52±2.2 | 47±2.7 |
| Anti-PECAM | 5 | 33±7.2 | 20±5.0 | 18±4.3 |
| Anti-VCAM | 3 | 76±0.9 | 63±2.1 | 61±0.4 |
| Single-targeted nanocarrier | ||||
| Control | ||||
| IgG NCs | 4 | 26±2.3 | 4.5±1.2 | 6.8±0.3 |
| Anti-ICAM NCs | 5 | 12±3 | 6.1±0.3 | 6.1±1.3 |
| Anti-PECAM NCs | 5 | 39±8.1 | 7.2±0.7 | 5.8±0.6 |
| Anti-VCAM NCs | 8 | 31±6.0 | 4.1±0.9 | 3.5±0.6 |
| LPS | ||||
| IgG NCs | 4 | 24±6.3 | 2.1±0.3 | 3.4±0.7 |
| Anti-ICAM NCs | 5 | 33±5.2 | 2.1±0.3 | 3.3±0.6 |
| Anti-PECAM NCs | 3 | 17±2.3 | 2.8±0.3 | 3.5±0.3 |
| Anti-VCAM NCs | 6 | 28±5.2 | 2.1±0.5 | 2.0±0.2 |
| Dual-targeted nanocarriers | ||||
| Control | ||||
| Anti-PECAM/VCAM NCs | 6 | 36±5.3 | 6.8±1.9 | 6.2±0.5 |
| Anti-ICAM/PECAM NCs | 5 | 30±7.8 | 3.1±0.2 | 4.1±0.9 |
| LPS | ||||
| Anti-PECAM/VCAM NCs | 3 | 20±1.8 | 2.2±0.1 | 3.6±0.1 |
| Anti-ICAM/PECAM NCs | 3 | 8.1±0.5 | 1.8±0.3 | 2.2±0.3 |
| Triple-targeted nanocarriers | ||||
| Control | ||||
| Anti-ICAM/PECAM/VCAM NCs | 4 | 22±8.9 | 3.3±0.6 | 4.5±0.8 |
| LPS | ||||
| Anti-ICAM/PECAM/VCAM NCs | 3 | 37±3.1 | 1.7±1.1 | 2.4±0.8 |
| ASM delivery | ||||
| Control | ||||
| ASM | 11 | 60±6.9 | 33±4.4 | 30±4.0 |
| Anti-ICAM/PECAM/VCAM/ASM NCs | 3 | 19±2.7 | 2.8±0.6 | 5.1±0.4 |
| LPS | ||||
| ASM | 2 | 64±18 | 41±7.3 | 39±12 |
| Anti-ICAM/PECAM/VCAM/ASM NCs | 3 | 24±11 | 2.7±0.9 | 2.0±0.2 |
Data shown is percent injected dose (% ID), expressed as Mean ± S.E.M.
Ab = antibody; NC = nanocarrier; LPS = lipopolysaccharide.
3. Results
3.1. Expression of ICAM-1, PECAM-1, and VCAM-1 by mouse endothelial cells
The expression level and regulation by inflammatory cytokines differs for ICAM-1, PECAM-1, and VCAM-1 [25]. Using antibodies with comparable affinities (on the range of hundreds pM, as per the vendors), we first verified this in endothelial cells derived from mouse vasculature (H5V cells), which may better reflect subsequent experiments in vivo. All three CAMs were expressed by these endothelial cells (Figure 1A), although at different levels (Figure 1B): PECAM-1 had the greatest expression under control conditions (∼21-fold over IgG), followed by low expression of ICAM-1 and VCAM-1 (2.2- and 1.7-fold over IgG), as expected [25]. Also as described [25], after activation with TNFα to mimic inflammation present in many diseases (Figure 1C), ICAM-1 and VCAM-1 expression were upregulated (2.7- and 3.1-fold over control conditions) while PECAM-1's was not (0.8-fold over control).
Figure 1. Endothelial expression of ICAM-1, PECAM-1, and VCAM-1.

Control and TNFα-activated H5V cells were fixed and cell surface expression was detected using antibodies against ICAM-1, PECAM-1, or VCAM-1 (vs. non-specific IgG), followed by a FITC-secondary antibody and fluorescence microscopy. (A) Activated cells are shown. Dashed lines mark the cell border (for IgG). Scale bar = 10-μm. (B) The level of fluorescence over non-specific IgG (horizontal line = 1) is shown for control cell. (C) Enhanced expression for TNFα-activated cells over control cells (Δ expression) is shown. Data are mean ± SEM. *Compares specific antibodies vs. non-specific IgG. #Compares TNFα vs. control conditions.
3.2. Binding and endocytosis of nanocarriers single-targeted to endothelial ICAM-1, PECAM-1 or VCAM-1
Since binding of nanocarriers to their cell receptors and endocytosis by cells impact targeting and intracellular transport [2], we examined these parameters. Model polymer nanocarriers were coated with non-specific IgG antibody (IgG NCs) or antibodies targeted to ICAM-1 (anti-ICAM NCs), PECAM-1 (anti-PECAM NCs), or VCAM-1 (anti-VCAM NCs). All carriers had similar valency (∼240-270 antibodies/NC), polydispersity (∼0.2), and zeta potential (∼-30 mV), with size within ∼230-320 nm range (Table 1). Antibodies remained stably coated on particles for up to 24 h (maximum time tested; 77-80% of the initial coating density) and no changes in size, polydispersity, or zeta potential were observed.
Incubation of varying concentrations of nanocarriers with fixed, activated endothelial cells revealed their cell-surface avidity (Table 1 and Supplemental Figure 1). Anti-PECAM NCs had the greatest cell-surface avidity (Kd ∼19 pM) and maximal extent of binding (Bmax ∼214 NCs/cell), followed by anti-ICAM NCs (Kd ∼30 pM; Bmax ∼180 NCs/cell), and anti-VCAM NCs (Kd ∼44 pM; Bmax ∼21 NCs/cell), which reflected the relative level of expression of these CAMs on activated endothelial cells. This binding was specific since it was inhibited by competing antibodies (anti-ICAM, anti-PECAM, or anti-VCAM; 75-90% inhibition; data not shown) but not by control non-specific IgG antibody (0-14% inhibition).
Binding to live cells was also specific and reflected the expected expression changes in disease conditions (Figure 2A-B). After 1 h incubation with control cells, IgG NCs displayed little cell association (1.4 NCs/cell), while anti-PECAM NCs had the highest binding (128 NCs/cell; 91-fold over IgG NCs), followed by anti-ICAM NCs (50 NCs/cell; 36-fold over IgG NCs) and anti-VCAM NCs (5 NCs/cell; 3.6-fold over IgG NCs). In activated cells, binding of anti-ICAM NCs and anti-VCAM NCs increased by 2.3- and 3.9-fold over control conditions, contrary to anti-PECAM NCs (1.1-fold).
Figure 2. Binding and endocytosis of nanocarriers targeted to ICAM-1, PECAM-1, or VCAM-1.

(A) Green Fluoresbrite nanocarriers incubated for 1 h at 37°C with control or TNFα-activated H5V cells, where surface-bound (non-internalized) carriers were immunostained with a Texas-Red secondary antibody (see Methods). Total cell-associated carriers and surface-bound counterparts were imaged by fluorescence microscopy. As an example, images show anti-ICAM NCs, anti-PECAM NCs and anti-VCAM NCs compared to non-specific IgG NCs in TNFα-activated H5V cells. Dashed lines mark cell borders. Scale bar is = 10-μm. Arrows = internalized carriers. Arrowheads = surface-bound carriers. (B) Absolute number of carriers associated to cells. (C) Percentage of internalized nanocarriers. (D) Absolute number of internalized nanocarriers. Data are mean ± SEM. *Compares control vs. TNFα; #compares to anti-ICAM NCs; !compares between anti-PECAM NCs and anti-VCAM NCs.
As per uptake, this could not be detected for non-specific IgG NCs (Figure 2A). Nanocarriers targeted to ICAM-1, PECAM-1, or VCAM-1 were efficiently internalized by endothelial cells within 1 h (Figure 2A and 2C). The fraction of carriers endocytosed relative to the total number of cell-associated carriers (which reflects the endocytosis rate) was similar for anti-ICAM NCs and anti-PECAM NCs (72% and 76%), and somewhat higher for anti-VCAM NCs (84%). Uptake increased upon cell activation for anti-ICAM NCs (1.3-fold) but not anti-PECAM NCs or anti-VCAM NCs (1.0- and 1.1-fold). In terms of the absolute number of internalized carriers (Figure 2D), a parameter influenced more strongly by the absolute binding level, uptake via PECAM-1 was greatest (98 NCs/cell) compared to ICAM-1 or VCAM-1 (37 and 4.6 NCs/cells, respectively) in control cells, while in activated cells uptake via PECAM-1 or ICAM-1 was similarly high (111 and 103 NCs/cell) and greater than that of VCAM-1 (19 NCs/cell).
3.3. Binding and endocytosis of nanocarriers dual-targeted to endothelial ICAM-1 and PECAM-1 or PECAM-1 and VCAM-1
We then pursued combination targeting. We examined dual targeting to ICAM-1 and PECAM-1 (anti-ICAM/PECAM NCs), and to PECAM-1 and VCAM-1 (anti-PECAM/VCAM NCs) as these are combinations where one receptor is expressed at low level in control conditions and upregulated by inflammation (ICAM-1 and VCAM-1), while the other is expressed at high level in control condition and is not upregulated by inflammation (PECAM-1) (Figure 1 and [25]). This may facilitate detection of changes in the binding pattern of dual-targeted carriers vs. single-targeted counterparts. Also, one combination implies receptors involved in the same pathway (ICAM-1 and PECAM-1 associated with CAM endocytosis), while the other involves receptors of different routes (PECAM-1's CAM endocytosis and VCAM-1's clathrin-mediated uptake) [35, 36], to help discern differences of targeting similar vs. different pathways.
Both dual-targeted nanocarriers had equal split of each targeting antibody on the carrier surface, similar total added valency, size, polydispersity, and zeta potential compared to single-targeted counterparts or non-specific IgG NCs (Table 1). Dual-targeted carriers had lower cell-surface avidity than single-targeted counterparts (Kd was ∼75 pM for anti-ICAM/PECAM NCs and ∼104 pM for anti-PECAM/VCAM NCs; Table 1 and Supplemental Figure 1). The Bmax was greater for anti-ICAM/PECAM NCs (∼109 NCs/cell) vs. anti- PECAM/VCAM NCs (38 NCs/cell; Table 1). Binding of dual-targeted carriers to cells was specific (not shown): for anti-PECAM/VCAM NCs and anti-ICAM/PECAM NCs, respectively, binding was 33-fold and 52-fold over IgG NCs in control cells, and 48-fold and 52-fold in activated cells, which was inhibited by the corresponding competing antibodies (∼70-75% inhibition), not by non-specific IgG (∼0-30% inhibition).
Binding of dual-targeted anti-PECAM/VCAM NCs was intermediate compared to single-targeted anti-PECAM NCs or anti-VCAM NCs for control and disease-like conditions (Supplemental Figure 2A and Figure 3A). This dual-targeted formulation displayed properties of both receptors: binding was increased relative to anti-VCAM NCs (anti-PECAM NCs had also bound at a higher extent than anti-VCAM NCs) and slightly enhanced in disease conditions (a feature observed for anti-VCAM NCs). Binding of anti-PECAM/VCAM NCs was lower than that of a 1:1 mixture of parent anti-PECAM NCs + anti-VCAM NCs (double valency toward each individual receptor compared to anti-PECAM/VCAM NCs; Supplementary Figure 3A), and lower for valency-matched carriers (same valency toward each receptor; Supplementary Figure 3B). Also, the percentage of nanocarriers internalized per control cell was statistically similar compared to single-targeted counterparts (∼80%; Supplemental Figure 2B and Figure 3B) and did not change in disease (Figure 3B). The absolute number of nanocarriers internalized per cell was intermediate for dual-targeted carriers vs. single-targeted counterparts (Supplemental Figure 2C and Figure 3C) and enhanced in disease-like conditions (Figure 3C). This set of results suggest that anti-PECAM/VCAM NCs can bind to both involved receptors.
Figure 3. Endothelial binding and endocytosis of nanocarriers dual- or triple-targeted to ICAM-1, PECAM-1, and/or VCAM-1.
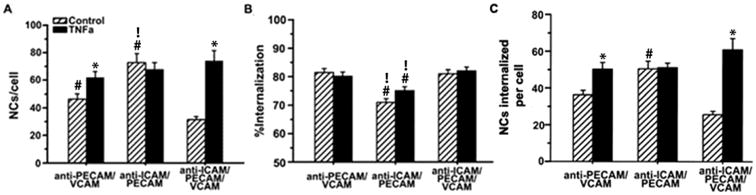
(A) Binding of FITC-labeled nanocarriers to control vs. TNFα-activated H5V cells (1 h, 37°C), quantified by fluorescence microscopy, as in Figure 2. (B) Percentage of internalized nanocarriers, assessed by fluorescence microscopy after staining surface-bound nanocarriers with a Texas-Red secondary antibody, as in Figure 2. (C) Absolute number of endocytosed nanocarriers. Data are mean ± SEM. *Compares control vs. TNFα; #compares dual-targeted to triple-targeted counterparts; !compares the two dual-targeted formulations.
For the second dual-targeted formulation (anti-ICAM/PECAM NCs), binding was similar to single-targeted anti-ICAM NCs in control cells and it did not change in activated cells, resulting in lower binding than single-targeted counterparts in disease-like conditions (Supplemental Figure 4A and Figure 3A). This combination had higher binding than that of anti-PECAM/VCAM NCs in control condition (1.6-fold). Binding of anti-ICAM/PECAM NCs was similar than that of a 1:1 mixture of parent anti-ICAM NCs + anti-PECAM NCs (Supplementary Figure 3A) and a mix of single-targeted samples with matching valency toward each independent receptor (Supplementary Figure 3B). The rate of endocytosis of anti-ICAM/PECAM NCs was relatively high and unaffected by disease (70-75%; Supplemental Figure 4B and Figure 3B), and somewhat lower than that of anti-PECAM/VCAM NCs. The absolute number of carriers internalized per cell was similar to that of single-targeted anti-ICAM NCs in control conditions and somewhat higher than anti-PECAM/VCAM NCs, while it was unchanged and similar to that of anti-PECAM/VCAM NCs in disease condition (Supplemental Figure 4C and Figure 3C). Hence, these dual-targeted formulations behaved differently from each other and their single-targeted counterparts.
3.4. Binding and endocytosis of nanocarriers triple-targeted to endothelial ICAM-1, PECAM-1 and VCAM-1
We then tested a triple-targeted formulation (anti-ICAM/PECAM/VCAM NCs), which reflects better the multiplicity of targeting of the natural ligands of these CAMs: leukocytes [25]. This is, to the best of our knowledge, the first time that triple-CAM targeting is examined. Triple-targeted nanocarriers had equal split of each targeting antibody on the carrier surface, with a similar total added valency, size, polydispersity, and zeta potential compared to single- or dual-targeted counterparts or non-specific IgG NCs (Table 1). Triple-targeted carriers had greater cell surface avidity than dual-targeted formulations and similar to single-targeted anti-VCAM NCs (Kd ∼49 pM; Table 1). With regard to the Bmax, this was lower than that of anti-ICAM/PECAM NCs and higher than that of anti-PECAM/VCAM NCs (78 NCs/cell; Table 1). Binding of anti-ICAM/PECAM/VCAM NCs to endothelial cells was specific (not shown): 23-fold and 57-fold over IgG NCs in control cells and activated cells, which was inhibited by a mixture of the three competing antibodies (∼95% inhibition), not by non-specific IgG (0% inhibition).
Under control conditions, binding of triple-targeted carriers to endothelial cells was significantly lower compared to dual-targeted carriers: 2.3- and 1.5-fold lower than anti-ICAM/PECAM NCs and anti-PECAM/VCAM NCs, respectively (Figure 3A). Binding of triple-targeted carriers was similar to a 1:1:1 mixture of parent anti-ICAM NCs + anti-PECAM NCs + anti-VCAM NCs (fully coated and possessing triple valency for each receptor; Supplementary Figure 3A) and significantly higher than valency-matched controls (5.5-fold higher; Supplementary Figure 3B). This indicates synergy of triple targeted carriers. In addition, binding was markedly increased in disease, reaching a level comparable to that of anti-ICAM/PECAM NCs and higher than anti-PECAM/VCAM NCs (Figure 3A). Thus, triple-targeted carriers showed the greatest binding difference between control and disease conditions: 2.3-fold (compared to 1.3-fold for anti-PECAM/VCAM NCs or 0.9-fold for anti-ICAM/PECAM NCs), providing the best disease-site selectivity without compromising absolute targeting level.
The endocytosis rate (percentage) of anti-ICAM/PECAM/VCAM NCs was relatively high and not affected in disease-like conditions (∼81%; Figure 3B). This was comparable to anti-PECAM/VCAM NCs and slightly greater than anti-ICAM/PECAM NCs under both conditions. Hence, the slight decrease in endocytosis rate observed for the ICAM-1/PECAM-1 combination vs. PECAM-1/VCAM-1 was abolished by triple-targeting. With regard to the absolute level of nanocarriers internalized per cell, this was higher under disease vs. control conditions, lower than anti-ICAM/PECAM NCs in control condition, and similar to both dual targeted formulations in disease (Figure 3C).
3.5. Comparative in vivo biodistribution of antibodies addressed to Ig-like CAMs
Previous studies on multi-CAM-targeting in vivo examined imaging applications of microbubbles targeted to αVβ3-integrin/P-selectin/vascular endothelial growth factor receptor 2 [41], P-selectin/ICAM-1 iron-oxide microparticles [26], and P-selectin/ICAM-1-targeted fluorescent microparticles [9].Yet, no previous in vivo studies have focused on combination-targeting to multiple Ig-like CAMs (vs. selectins with Ig-like CAMs) involving nanocarriers (vs micro-scale counterparts). For simplicity, we focused on the lungs, brain, and liver (shown in Figures) as examples of a peripheral organ, central nervous system tissue, and a clearance organ, respectively. Yet, data on other organs and circulation are also provided in Tables. Since the lungs contain 1/4-1/3 of the total body vasculature and receive low-shear total cardiac output, this peripheral organ is expected to be the main specific target with regard to these endothelial markers.
We first compared antibodies against ICAM-1, PECAM-1, or VCAM-1 after i.v. injection in mice, in part reflective of the level of expression and accessibility of their respective targets in vivo. We examined the localization ratio (LR) and specificity index (SI), which represent the organ-to-blood ratio and the specific accumulation compared to non-specific IgG (see Methods). Anti-PECAM displayed the lowest percentage of the injected dose in blood, followed by anti-ICAM, anti-VCAM, and non-specific IgG (Table 2). As shown in Figure 4 and Table 3, these blood levels inversely correlated with the absolute level of accumulation in organs and specificity of targeting over IgG control (SI > 1). For instance, anti-PECAM had both the highest accumulation (Figure 4A) and specificity (Figure 4B) in brain, lung, liver, and all other organs; and anti-ICAM displayed higher accumulation and specificity than anti-VCAM in the lungs, liver, heart, and kidneys.
Figure 4. Biodistribution of antibodies targeted to ICAM-1, PECAM-1, or VCAM-1.

Mice were injected i.v. with 125I-labeled anti-ICAM, anti-PECAM, anti-VCAM, or non-specific IgG and blood and organs were collected 30 min after injection. (A) The organ-to-blood localization ratio (LR) and (B) the targeted-to-untargeted specificity index (SI) over control IgG are shown for the brain, lungs, and liver. SI = 1 is non-specific IgG (horizontal line). Data are mean ± S.E.M. #Compares against anti-ICAM; !compares anti-PECAM to anti-VCAM.
Table 3. Biodistribution and specificity of antibodies or antibody-coated nanocarriers injected in mice.
| Ab or NC | Brain | Heart | Kidney | Liver | Lungs | Spleen | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LR | SI | LR | SI | LR | SI | LR | SI | LR | SI | LR | SI | |
| Antibody | ||||||||||||
| – IgG | 0.02±0.001 | - | 0.1±0.01 | - | 0.3±0.02 | - | 0.2±0.01 | - | 0.2±0.02 | - | 0.2±0.01 | - |
| – Anti-ICAM | 0.02±0.002 | 1.2±0.1 | 0.2±0.02 | 2.0±0.2 | 0.6±0.02 | 2.2±0.1 | 0.5±0.03 | 2.3±0.1 | 1.8±0.1 | 12±0.6 | 0.6±0.04 | 3.3±0.3 |
| – Anti-PECAM | 0.09±0.040 | 4.6±1.8 | 0.9±0.48 | 8.5±4.5 | 1.5±0.5 | 5.6±1.8 | 1.7±0.17 | 7.6±0.7 | 3.6±2.0 | 24±13 | 1.9±0.86 | 13±5.7 |
| – Anti-VCAM | 0.02±0.002 | 1.0±0.1 | 0.1±0.01 | 1.2±0.1 | 0.3±0.01 | 1.3±0.04 | 0.3±0.01 | 1.2±0.1 | 0.2±0.02 | 1.1±0.2 | 0.7±0.02 | 3.5±0.2 |
| Single-targeted nanocarriers | ||||||||||||
| Control | ||||||||||||
| – IgG NCs | 0.05±0.009 | - | 0.4±0.08 | - | 0.5±0.05 | - | 11±0.9 | - | 1.9±0.5 | - | 15±2.0 | - |
| – Anti-ICAM NCs | 0.16±0.04 | 3.2±0.9 | 0.8±0.2 | 2.1±0.4 | 1.3±0.2 | 2.7±0.5 | 11±1.38 | 1.0±0.1 | 32±8.4 | 18±4.6 | 20±4.5 | 1.3±0.3 |
| – Anti-PECAM NCs | 0.09±0.02 | 1.7±0.3 | 0.7±0.2 | 1.8±0.6 | 0.8±0.1 | 1.7±0.3 | 14±4.66 | 1.2±0.4 | 18±2.0 | 9.5±1.1 | 24±3.0 | 1.6±0.2 |
| – Anti-VCAM NCs | 0.23±0.02 | 4.6±1.1 | 0.7±0.1 | 2.0±0.4 | 1.4±0.3 | 3.0±0.5 | 24±1.61 | 2.2±0.4 | 3.8±0.3 | 2.1±0.4 | 40±2.2 | 2.6±0.3 |
| LPS | ||||||||||||
| – IgG NCs | 0.14±0.001 | - | 0.5±0.06 | - | 0.8±0.07 | - | 23±0.6 | - | 4.5±02.2 | - | 34±15 | - |
| – Anti-ICAM NCs | 0.20±0.03 | 1.4±0.2 | 1.7±0.4 | 3.5±0.8 | 1.9±0.5 | 2.3±0.6 | 16±2.80 | 0.7±0.1 | 68±17 | 15±3.9 | 48±11 | 1.4±0.3 |
| – Anti-PECAM NCs | 0.24±0.05 | 1.7±0.3 | 1.5±0.3 | 3.1±0.6 | 2.0±0.3 | 2.4±0.4 | 15±1.05 | 0.7±0.03 | 38±3.5 | 8.4±0.6 | 29±9.6 | 0.9±0.2 |
| – Anti-VCAM NCs | 0.20±0.03 | 1.4±0.2 | 1.1±0.2 | 2.1±0.3 | 1.7±0.3 | 2.0±0.3 | 48±7.24 | 2.1±0.6 | 11±2.6 | 2.6±0.6 | 71±12 | 2.1±0.3 |
| Dual-targeted nanocarriers | ||||||||||||
| Control | ||||||||||||
| – Anti-PECAM/VCAM NCs | 0.09±0.01 | 1.8±0.3 | 0.5±0.1 | 1.4±0.3 | 0.8±0.1 | 1.7±0.2 | 10±1.4 | 0.9±0.1 | 8.1±1.8 | 4.4±1.0 | 21±4.5 | 1.4±0.3 |
| – Anti-ICAM/PECAM NCs | 0.16±0.04 | 3.2±0.9 | 1.3±0.3 | 3.7±0.9 | 1.1±0.2 | 2.4±0.3 | 21±7.9 | 1.9±0.7 | 82±17 | 44±9.4 | 28±8.2 | 1.8±0.5 |
| LPS | ||||||||||||
| – Anti-PECAM/VCAM NCs | 0.20±0.02 | 1.4±0.1 | 0.8±0.0 | 1.8±0.1 | 1.2±0.1 | 1.5±0.1 | 16±1.3 | 0.7±0.1 | 22±1.9 | 4.9±0.4 | 26±3.1 | 0.8±0.1 |
| – Anti-ICAM/PECAM NCs | 0.80±0.12 | 5.6±0.9 | 4.5±0.2 | 9.4±0.4 | 4.0±0.8 | 4.8±1.0 | 8.7±3.0 | 0.4±0.1 | 266±16 | 59±3.6 | 16±1.8 | 0.5±0.1 |
| Triple-targeted nanocarriers | ||||||||||||
| Control | ||||||||||||
| – Anti-ICAM/PECAM/VCAM NCs | 0.20±0.03 | 4.0±0.7 | 1.8±0.2 | 4.8±0.4 | 1.6±0.5 | 3.3±1.0 | 9.3±3.2 | 0.8±0.3 | 87±34 | 47±18 | 19±3.9 | 1.2±0.2 |
| LPS | ||||||||||||
| – Anti-ICAM/PECAM/VCAM NCs | 0.80±0.29 | 5.6±2.1 | 4.1±1.8 | 8.6±3.7 | 3.7±1.4 | 4.4±1.7 | 21±7.1 | 0.9±0.3 | 102±36 | 23±7.9 | 73±34 | 2.2±1.0 |
| ASM delivery | ||||||||||||
| Control | ||||||||||||
| – ASM | 0.04±0.01 | - | 0.2±0.02 | - | 0.4±0.04 | - | 1.2±0.1 | - | 0.3±0.04 | - | 0.5±0.04 | - |
| – Anti-ICAM/PECAM/VCAM/ASM NCs | 0.24±0.05 | 6.4±1.6 | 1.1±0.0 | 6.4±0.2 | 1.5±0.3 | 3.6±0.9 | 15±2.4 | 13±2.4 | 10±2.0 | 31±7.6 | 23±3.8 | 50±10 |
| LPS | ||||||||||||
| – ASM | 0.05±0.02 | - | 0.2±0.01 | - | 0.4±0.1 | - | 0.6±0.2 | - | 0.3±0.04 | - | 0.3±0.07 | - |
| – Anti-ICAM/PECAM/VCAM/ASM NCs | 0.51±0.04 | 9.5±0.7 | 2.7±0.7 | 15±3.9 | 4.2±0.3 | 10±0.8 | 36±3.0 | 64±5.4 | 30±3.3 | 115±12 | 81±31 | 242±93 |
Data are Mean ± S.E.M. Ab = antibody; NC = nanocarrier; LPS = lipopolysaccharide; LR = localization ratio; SI = specificity index reported as LR ratio vs control non-targeted counterparts (IgG, IgG NCs, or free ASM).
3.6. Comparative in vivo biodistribution of nanocarriers single-targeted to Ig-like CAMs
We then compared the biodistribution of nanocarriers single-targeted to ICAM-1, PECAM-1, or VCAM-1 vs. that of non-specific IgG NCs. All nanocarriers disappeared more rapidly from the blood than naked antibodies (Table 2). IgG NCs only accumulated significantly in the liver and spleen (LR 11 and 15; Figure 5 and Table 3), but not the lungs (LR 2), brain (LR 0.05) or other organs. In contrast, all targeted nanocarriers showed specific accumulation in different organs. For instance, the greatest accumulation of anti-ICAM NCs was in the lungs (LR ∼32), followed by spleen (LR ∼20), and liver (LR ∼11). Anti-PECAM NCs accumulated preferentially in the spleen (LR ∼24), followed by lungs (LR ∼18), and liver (LR ∼14). Anti-VCAM NCs were highest in the spleen (LR ∼40), followed by the liver (LR ∼24), and lungs (LR ∼4). Spleen and liver accumulation of anti-ICAM NCs or anti-PECAM NCs was not specific compared to control IgG NCs (SI∼1), yet this was specific for anti-VCAM NCs (SI ∼2.6 in spleen and 2.1 in liver). All three formulations accumulated specifically in the kidneys, heart, lungs, and brain (SI >1). Anti-ICAM NCs and anti-PECAM NCs showed highest specificity for the lungs (18-fold and 10-fold over IgG NCs), while anti-VCAM NCs did in the brain (4.6-fold; Figure 5 and Table 3). In general, this specificity increased for anti-ICAM NCs and anti-VCAM NCs compared to naked anti-ICAM or anti-VCAM antibodies, while it decreased for anti-PECAM NCs compared to anti-PECAM (Supplemental Figure 5).
Figure 5. Biodistribution of nanocarriers single-targeted to ICAM-1, PECAM-1, or VCAM-1.
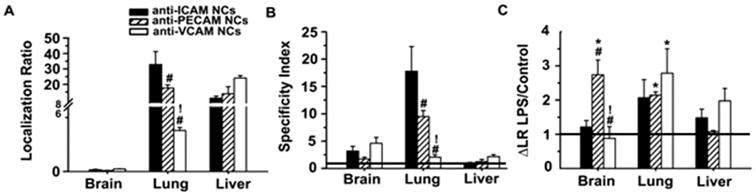
Mice were injected i.v. with 125I-labeled anti-ICAM NCs, anti-PECAM NCs, anti-VCAM NCs, or non-specific IgG NCs, and blood and organs were collected 30 min after injection. (A) The organ-to-blood localization ratio (LR) and (B) targeted-to-untargeted specificity index (SI) over IgG NCs are shown for the brain, lungs, and liver in control mice. SI = 1 is non-specific IgG NCs (horizontal line). (C) Effect of LPS challenge on nanocarrier biodistribution, assessed as the fold-change in LR (ΔLR) for LPS-treated mice over control mice. No change (Δ = 1) is shown as a horizontal line. Data are mean ± S.E.M. #Compares against anti-ICAM NCs; *compares anti-PECAM NCs to anti-VCAM NCs; *compares control vs. LPS.
With regard to mice challenged with lipopolysaccharide (LPS) to mimic inflammatory-like conditions, nanocarrier accumulation was enhanced, although differently comparing anti-ICAM NCs, anti-PECAM NCs, and anti-VCAM NCs (Figure 5C and Table 3). Accumulation of anti-ICAM NCs seemed enhanced by LPS in all organs except brain, with similar improvement in the spleen, heart, and lungs (ΔLR ∼2.4, 2.2, 2.1). For anti-PECAM NCs, all organs experienced LPS-enhanced accumulation except the spleen and liver, with highest improvements in the lungs, brain, and kidneys (ΔLR ∼2.1, 2.7, 2.5). Anti-VCAM NCs showed enhanced accumulation only in the lungs (ΔLR ∼2.8).
3.7. Biodistribution of nanocarriers dual- or triple-targeted to ICAM-1, PECAM-1, and/or VCAM-1
We next tested in vivo biodistribution and specificity of nanocarriers dual- or triple-targeted to these CAMs (Figure 6 and Table 3). In the spleen and liver, the level of accumulation (LR ∼20 and ∼10, respectively) and lack of specificity over IgG NCs (SI ∼1) for anti-PECAM/VCAM NCs or anti-ICAM/PECAM/VCAM NCs were similar to that of anti-ICAM NCs and anti-PECAM NCs, and reduced compared to anti-VCAM NCs. Hence, multi-targeting nanocarriers to ICAM-1 and/or PECAM-1 ruled biodistribution and avoided these organs.
Figure 6. Biodistribution of nanocarriers dual- or triple-targeted to ICAM-1, PECAM-1, and/or VCAM-1.
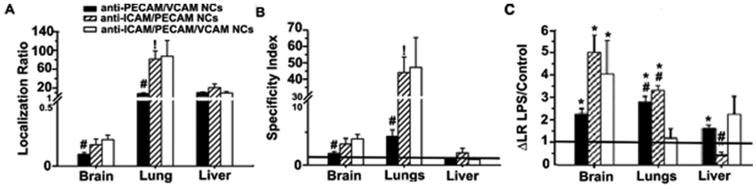
Mice were injected i.v. with 125I-labeled anti-PECAM/VCAM NCs, anti-ICAM/PECAM NCs, or anti-ICAM/PECAM/VCAM NCs and blood and organs were collected 30 min after injection. (A) The organ-to-blood localization ratio (LR) and (B) specificity index (SI) over IgG NCs are shown for the brain, lungs, and liver. SI = 1 is non-specific IgG NCs (horizontal line). (C) Effect of LPS challenge on nanocarrier biodistribution, assessed as fold-change in LR (ΔLR) for LPS-treated mice over control mice. No change (Δ = 1) is shown as a horizontal line. Data are mean ± S.E.M. #Compares against anti-ICAM/PECAM/VCAM NCs; *compares anti-ICAM/PECAM NCs vs. anti-PECAM/VCAM NCs; *compares control vs LPS.
Lung accumulation of anti-ICAM/PECAM NCs was significantly increased compared to single-targeted counterparts (Supplemental Figure 6 and Table 3): LR and SI were enhanced ∼2.6-fold over anti-ICAM NCs, and SI was enhanced 4.6-fold over anti-PECAM NCs. But this was not significant in the case for the spleen and liver. Triple-targeted nanocarriers showed similar (1.1-fold) lung accumulation and specificity to the anti-ICAM/PECAM formulation. Anti-PECAM/VCAM NCs had intermediate lung accumulation and specificity vs. single-targeted counterparts (Supplemental Figure 7 and Table 3). As per other organs, the biodistribution and specificity patterns also varied. In the heart, anti-ICAM/PECAM NCs surpassed its single-targeted counterparts and so was the case for triple-targeted formulation. Anti-PECAM/VCAM NCs showed intermediate accumulation and specificity in the lungs as compared to their single-targeted counterparts, and lower as compared to anti-ICAM/PECAM NCs. Yet, in the heart, anti-PECAM/VCAM NCs had similar accumulation and specificity than anti-PECAM NCs or anti-VCAM NCs, but this was also lower than for anti-ICAM/PECAM NCs. In the brain, anti-ICAM/PECAM NCs performed similarly to single-targeted counterparts and this was also the case for anti-PECAM/VCAM NCs compared to parent counterparts. Triple-targeted nanocarriers provided a slight improvement in some organs as compared to double-targeted formulations.
In disease, anti-ICAM/PECAM NCs and anti-ICAM/PECAM/VCAM NCs enabled specific targeting to the kidneys, heart, lung, and brain (not liver or spleen) vs. IgG NCs, while anti-PECAM/VCAM NCs did not (Figure 6C and Table 3). Compared to control mice, anti-ICAM/PECAM NCs displayed higher accumulation in disease in the kidneys, heart, lungs, and brain, with the greatest enhancement in kidneys and brain (3.5-fold and 4.9-fold). Anti-PECAM/VCAM NCs had enhanced accumulation in disease vs. control mice for the kidneys, heart, lungs, liver, and brain, with most acute improvement in the lungs and brain (2.8-fold and 2.2-fold). Triple-targeted nanocarriers had enhanced accumulation in brain in disease conditions (4.0-fold). A slight enhancement was also observed in the spleen (3.9-fold), kidneys (2.3-fold), and heart (2.3-fold).
3.8. Biodistribution of a model therapeutic cargo delivered by triple-CAM-targeted nanocarriers
The previous set of experiments indicated that, overall, dual- and triple-targeted formulations enhanced nanocarrier accumulation and specificity over non-specific carriers in those organs of the body where the targets are expressed. In disease-like conditions, triple-targeted formulations performed closely but slightly better than anti-ICAM/PECAM NCs, particularly in cell culture and with regard to endocytosis, and were clearly superior to anti-PECAM/VCAM NCs. Hence, we selected this triple-targeted formulation to investigate its potential to improve the biodistribution of a therapeutic cargo.
Since endothelial CAMs are expressed throughout the vasculature, they represent appropriate targets for applications requiring broad delivery of therapeutics or delivery to particular tissues where inflammation causes local upregulation of these molecules. As a representative example involving pathological effects through the body accompanied by inflammation, we tested delivery of recombinant acid sphingomyelinase (ASM), a lysosomal enzyme deficient in type A-B Niemann-Pick disease, explored for enzyme replacement therapy [39, 42]. ASM is an example of a cargo that is necessary throughout the entire body and predominantly in the lungs, brain, and reticulo-endothelial system (liver and spleen), severely affected in this disease [42, 43]. Our previous studies showed that targeting of ASM to ICAM-1 using anti-ICAM NCs or by combined-targeting to ICAM-1/transferrin receptor significantly enhanced ASM accumulation in the brain, lungs, and liver (vs. the free enzyme), with greatest accumulation values for ICAM-1 targeting and more uniform distribution for ICAM-1/transferrin receptor pair [8, 38, 44].
Triple-CAM-targeted nanocarriers showed ASM accumulation in all organs tested, with greatest LR in the spleen and liver, and lowest LR in the brain (Figure 7A, and Table 3). Nanocarriers very markedly enhanced delivery over the free enzyme (SI; Figure 7C and Table 3), including organs where LR was relatively low (e.g. 6.4-fold enhancement in the brain) and all other cases (3.6-fold in kidney, 49.9-fold in spleen, 6.4-fold in heart, 30.9-fold in lungs, and 12.7 in liver). ASM delivery by anti-ICAM/PECAM/VCAM NCs was further improved in all organs in disease conditions (Table 3), which surpassed the free enzyme by 10.2-fold in kidneys, 242-fold in spleen, 15.3-fold in heart, 115-fold in lungs, 63.8-fold in liver, and 9.5-fold in brain (Figure 7B and Table 3). Therefore, this triple-targeting strategy seems promising to improve broad ASM biodistribution, as required, and demonstrates the potential of combination-targeting in drug delivery.
Figure 7. ASM delivery by nanocarriers triple-targeted to ICAM-1, PECAM-1, and VCAM-1.

Control or LPS-treated mice were injected i.v. with 125I-labeled ASM either as a naked enzyme or loaded on anti-ICAM/PECAM/VCAM NCs, and blood and organs were collected 30 min after injection. (A) The organ-to-blood localization ratio (LR) under control or (B) LPS conditions, and (C) the specificity index (SI) over free ASM at these two conditions are shown for brain, lungs, and liver. SI = 1 is naked ASM (this horizontal line is adjacent to the X-axis in C and, hence, hardly visible). Data are mean ± S.E.M. *Compares free enzyme vs. enzyme coupled to anti-ICAM/PECAM/VCAM NCs; #compares control vs LPS-challenged mice.
4. Discussion
In summary, we have characterized cellular binding, internalization, and in vivo biodistribution of nanocarriers functionalized for multi-CAM targeting toward ICAM-1, PECAM-1, and/or VCAM-1. This is the first time that multi-CAM targeting is studied: (a) for combinations encompassing only Ig-like CAMs, (b) in the context of intracellular transport, (c) for nanoscale carriers in vivo, and (d) for delivery of biological therapeutics. Our results demonstrate that multi (dual and, mainly, triple) targeting to Ig-like CAMs enhanced binding and selectivity toward diseased cells, resulted in efficient endocytosis, and improved in vivo accumulation and specificity of drug carriers and therapeutic cargoes, which depended upon the combination and multiplicity of affinity moieties used.
With regard to cell culture, both the cellular avidity and level of binding of targeted nanocarriers somewhat reflected the relative level of expression of these CAMs by endothelial cells, which were greater for PECAM-1 > ICAM-1 > VCAM-1, and was enhanced in disease vs. control conditions only for the two latter molecules. Yet, the binding of nanocarriers to ICAM-1 vs. PECAM-1 was more similar than expected, and binding of nanocarriers to VCAM-1 lower than expected based on the level of expression of these molecules (also reflected by the binding level of free antibodies). While anti-ICAM clone YN1 and anti-PECAM clone MEC13 bind on membrane distal domains 1 and 2 of their respective antigens [45, 46], anti-VCAM clone MK2 binds somewhere between domains 1 and 4 [47, 48] of VCAM-1. It is possible that binding a more membrane proximal epitope on VCAM-1 (e.g. domains 3, 4) may have been hindered by steric hindrance of anti-VCAM NCs, even that this epitope was accessible to free antibodies. This phenomenon has been observed in other cases [8, 33, 49] and may be due to different location of these receptors on the plasmalemma or different location of their antigenic epitopes from the plane of the cell surface (e.g. proximal vs. distal). This may result in a different accessibility for bulkier antibody-coated nanocarriers vs. the free antibodies used for cell expression.
Binding of the PECAM-1/VCAM-1 combination acquired properties of both target molecules: enhanced binding over VCAM-targeting alone and enhanced selectivity for disease-like conditions over PECAM-targeting alone. For the ICAM-1/PECAM-1 combination binding was intermediate to either single-targeted nanocarriers and did not display disease selectivity, as for PECAM-1. Similar binding of anti-ICAM/PECAM NCs in control vs. disease conditions seems counterintuitive based on the observed ICAM-1 overexpression in disease and enhanced binding of nanocarriers in this situation. An analogous result has been observed for ICAM-1-targeted polymersomes, which showed similar binding in control and disease-like conditions [23]. This indicates that the nanocarrier targeting performance can be controlled according to the combination of Ig-like CAMs selected. Additionally, the multiplicity of affinity moieties also affected binding level and disease selectivity (as observed when comparing dual- vs. triple-targeted formulations) and, hence, this parameter can be utilized to modulate targeting performance. Triple-targeted nanocarriers displayed higher cell avidity than dual-targeted combinations in diseased cells and lower binding level to control cells, resulting in the highest disease selectivity. This pairs well with the natural selectivity of leukocytes toward diseased endothelium, which involves multi-CAM interactions [50]. Interestingly, the targeting behavior observed in cell cultures differed from that of carriers dual-targeted to selectins and Ig-like CAMs, which synergistically enhanced binding vs. single-targeted counterparts [17, 18, 20, 23, 24, 26]. But this synergy was observed for the triple-targeted formulation. Also, the targeting performance in vivo appeared synergistic in the heart and lungs in the case of ICAM-1/PECAM-1 or ICAM-1/PECAM-1/VCAM-1 combinations, particularly for LPS-treated mice, suggesting that synergy is possible when multiple Ig-like CAMs are targeted in combination as well.
With regard to endocytosis, this was highly efficient for all nanocarriers and conditions tested, implying that the potential for controlling intracellular delivery of these formulations is rather ruled by the level of carrier binding. Nevertheless, a few interesting observations arose from this study. We found an intermediate rate of endocytosis of dual-targeted anti-PECAM/VCAM NCs compared to its single-targeted counterparts. Since PECAM-1 is preferentially located in the cell-cell border (arguably less amenable for endocytosis) vs. VCAM-1 expression on the free luminal surface, this result could be due to engagement of receptors at these two different locations [25]. In addition, anti-ICAM/PECAM NCs were internalized with lower efficiency compared to anti-PECAM/VCAM NCs. This is counterintuitive since ICAM-1 and PECAM-1 associate with the same endocytic mechanism (CAM endocytosis) [35], while VCAM-1 and PECAM-1 associate with distinct routes (CAM vs. clathrin pathways [35, 36]). Yet, it is possible that simultaneous engagement of receptors that use the same cell machinery may result in a competition phenomenon, leading to decreased uptake. Supporting this, sequential stimulation with antibodies to ICAM-1 followed by PECAM-1 inhibits ICAM-1-induced RhoA activation and actin cytoskeletal rearrangement in endothelial cells [35, 51]. Additional targeting to VCAM-1 in the case of the triple-targeted formulation enhanced endocytosis. Hence, binding to CAMs associated with different vs. same endocytic route may be beneficial by relaxing the competition for the same cell signaling/machinery involved in uptake.
Most results in mice were generally in agreement with cell culture data. For instance, lung accumulation was high for most formulations. Although 100-nm “naked” polystyrene nanoparticles have been shown to accumulate non-specifically in this organ [52] this was not the case in our study since non-specific IgG NCs did not accumulate in the lungs, likely due to differences in size and surface-coat of our antibody targeted nanoparticles. Specific lung accumulation of targeted particles reflects endothelial targeting in vivo, since the pulmonary vasculature constitutes ∼20-30% of the total endothelium in the body, is exposed to low shear stress, and receives full cardiac output [53]. Lower pulmonary accumulation of anti-VCAM NCs vs. anti-ICAM NCs or anti-PECAM NCs is in agreement with lower binding of anti-VCAM NCs to control endothelial cell cultures. Yet, contrary to cell culture where binding of anti-PECAM NCs surpassed anti-ICAM NCs, in vivo pulmonary accumulation and specificity of anti-ICAM NCs were higher than anti-PECAM NCs. Yet this was opposite from the behavior of naked antibodies in vivo. Perhaps the shear stress of the blood flow and/or different accessibility of these receptors in vivo vs. cell cultures dictates this, e.g. PECAM-1 more prevalently located in the cell-cell borders in vivo [53] may account for this difference. Indeed, PECAM-1 expression is more displaced toward the cell-cell border in endothelial cells exposed to flow vs. static cultures [54]. In addition, LPS-treatment enhanced accumulation of anti-ICAM NCs in the lung, which was expected and in agreement with cell culture data for ICAM-1 targeting, but not PECAM-1 targeting [25]. Yet, it is possible that under inflammatory stimulation PECAM-1 redistributes toward luminal areas, increasing its accessibility relative to control mice. Supporting this, such an effect has been reported for PECAM-1 in cell cultures stimulated with TNFα [55].
In the case of dual-targeted formulations, as in cell culture, enhanced targeting of anti-ICAM/PECAM NCs vs. anti-PECAM/VCAM NCs was also observed in vivo (e.g. in the lungs). Yet in mice, anti-ICAM/PECAM NCs had improved accumulation in disease conditions and displayed higher selectivity vs. control conditions. This result is contrary to cell culture data, where anti-ICAM/PECAM NCs did not exert enhanced binding under pathological stimulation. This may be due to enhanced ICAM-1 expression and/or accessibility in vivo [44, 53], potential effects of the blood flow shear stress, and/or a consequence of PECAM-1 redistribution to the luminal surface, as discussed above [55]. Also following the trend observed in cell culture, anti-ICAM/PECAM/VCAM NCs outperformed double-targeted counterparts, yet selectivity toward disease vs. control conditions was more apparent in the case of cell cultures. Focusing on the lungs as an example of endothelial targeting, triple-targeted nanocarriers displayed the highest accumulation in control mice and perhaps this represents a saturating level, explaining lack of an effect in LPS-challenged mice. Indeed, when triple-targeted nanocarriers were co-coated with recombinant ASM (a model for a therapeutic cargo) which may have reduced the valency of carriers toward each particular receptor, triple-CAM targeted nanocarriers markedly enhanced delivery of this cargo, particularly comparing disease vs. control conditions. Therefore, combination-targeting shows translational potential with regard to modulating the biodistribution of therapeutic agents. This pairs well with previous studies, such as that of triple-targeting of microbubbles to αVβ3-integrin, P-selectin, and vascular endothelial growth factor receptor 2 for cancer imaging [41], or triple-targeting to tumor endothelial marker 7, folate receptor, and PECAM-1 to improve targeting to cancer cells and angiogenic blood vessels [11].
The application of multiple-CAM targeting for delivery of lysosomal ASM is relevant in that this strategy allows broad and enhanced distribution of this enzyme through the body, meeting the requirements of this particular intervention, and also this syndrome associates with inflammation, making CAMs adequate targets for said treatment [42, 43]. Indeed, comparison with single targeted anti-ICAM/ASM NCs published previously [44] shows advantages for the triple-CAM targeted ASM formulation shown here. For instance, in control animals the accumulation of triple-CAM targeted ASM NCs was reduced compared to that of anti-ICAM/ASM NCs in the heart, kidney, brain, and lungs (similar for the liver and spleen), while in disease models it was enhanced in most organs (including brain, heart, kidney, liver, and spleen). Although the disease models used in these studies are different (LPS for this work and ASM knockout mice for the previous one), both are characterized by systemic inflammation and this result is in good agreement with the concept that multi-CAM targeting improves disease-to-health targeting ratio.
In addition, nanocarriers with combined targeting to ICAM-1/PECAM-1 or ICAM-1/PECAM-1/VCAM-1 may be particularly beneficial for delivering therapeutics to the lungs and heart to develop other applications, e.g. aiming at cardiovascular and pulmonary disorders. Although beyond the scope of the present report, future studies will aim to explore this possibility and optimize the valencies of these formulations in relevant disease models.
5. Conclusions
The data shown here demonstrates that addressing nanocarriers to multiple Ig-like CAMs allows modulation of their targeting, endocytosis, and organ biodistribution, and also enhances delivery of model therapeutic cargoes, both in control and predominantly in disease-like conditions. PECAM-1/VCAM-1 targeting enabled nanocarriers to acquire properties of both target molecules, where binding to cells improved over anti-VCAM NCs and the selectivity for diseased vs. control cells improved over anti-PECAM NCs. Targeting ICAM-1/PECAM-1 provided greater binding than anti-PECAM/VCAM counterparts, yet with lack of selectivity for diseased vs. control cells. Increasing the mulitiplicity of affinity moieties from dual- to triple-CAM-targeting enabled similarly high binding to cells but with superior selectivity in disease. Internalization rate was highly efficient for all conditions tested and the absolute uptake, hence, depended on the level of binding of the functionalized nanocarriers. Interestingly, however, endocytosis may be more efficient when targeting multiple receptors associated with CAM-mediated and clathrin-mediated endocytosis, rather than targeting multiple receptors associated solely with CAM-mediated endocytosis. As in cell culture, enhanced targeting of anti-ICAM/PECAM NCs vs. anti-PECAM/VCAM NCs was also observed in vivo. Yet in mice, anti-ICAM/PECAM NCs also displayed more selectivity toward diseased conditions. Triple-targeted nanocarriers slightly outperform both double-targeted counterparts and showed greatest selectivity toward disease, mimicking the behavior of leukocyte integrins engaged in endothelial binding through multi-CAM interactions during inflammation [50]. Therefore, multi-targeting to CAMs can be controlled by the combination of targets utilized and/or by the multiplicity of affinity moieties. This strategy may allow manipulation of the targeting performance of nanocarriers, which is tailored by combining properties of multiple targets rather than optimizing features of the nanocarrier or targeting ligand, or engaging into discovery of new target molecules. Overall, multi-CAM-targeting is a promising example of the potential that combination-targeting strategies hold for the development of targeted nanocarriers for therapeutic applications.
Supplementary Material
Supplemental Fig. 1 Cell-binding avidity of nanocarriers targeted to ICAM-1, PECAM-1, and/or VCAM-1. TNFα-activated H5V cells were fixed and incubated for 1 h at 37°C with varying concentrations of Fluoresbrite-labeled nanocarriers targeted to ICAM-1, PECAM-1, and/or VCAM-1, followed by analysis by fluorescence microscopy.
Supplemental Fig. 2 Binding and endocytosis of anti-PECAM/VCAM nanocarriers vs. their single-targeted counterparts. Green Fluoresbrite nanocarriers incubated for 1 h at 37 °C with control or TNFα-activated H5V cells. Surface-bound (non-internalized) carriers were immunostained with a Texas-Red secondary antibody (see Materials and methods). Total cell-associated carriers and surface-bound counterparts were imaged by fluorescence microscopy. (A) Absolute number of carriers associated to cells. (B) Percentage of internalized nanocarriers. (C) Absolute number of internalized nanocarriers. Data are mean ± SEM. *Compares control vs. TNFα; #compares to anti-PECAM/VCAM NCs; !compares between anti-PECAM NCs and anti-VCAM NCs.
Supplemental Fig. 3 Comparative binding of nanocarriers dual- or triple-targeted to ICAM-1, PECAM-1, and/or VCAM-1 vs. their single-targeted counterparts. Anti-ICAM/PECAM NCs, anti-PECAM/VCAM NCs, and anti-ICAM/PECAM/VCAM NCs, all labeled with green Fluoresbrite, were incubated for 1 h at 37 °C with fixed, TNFα-activated H5V cells. Total cell-bound carriers were imaged by fluorescence microscopy and compared to the binding of mixtures of single-targeted carriers, including anti-ICAM NCs, anti-PECAM NCs, and/or anti-VCAM NCs mixtures carrying full valency for each of their receptors (A), or valencies matching those of dual- or triple-targeted carriers (B); (see Methods Section 2.5). Data are mean ± SEM.
Supplemental Fig. 4 Binding and endocytosis of anti-ICAM/PECAM nanocarriers vs. their single-targeted counterparts. Green Fluoresbrite nanocarriers incubated for 1 h at 37 °C with control or TNFα-activated H5V cells. Surface-bound (non-internalized) carriers were immunostained with a Texas-Red secondary antibody (see Materials and methods). Total cell-associated carriers and surface-bound counterparts were imaged by fluorescence microscopy. (A) Absolute number of carriers associated to cells. (B) Percentage of internalized nanocarriers. (C) Absolute number of internalized nanocarriers. Data are mean ± SEM.*Compares control vs. TNFα; #compares to anti-ICAM/PECAM NCs; !compares between anti-ICAM NCs and anti-PECAM NCs.
Supplemental Fig. 5 Change in the in vivo specificity index of antibody-nanocarriers vs. antibodies targeted to ICAM-1, PECAM-1, or VCAM-1. Mice were injected i.v. with nanocarriers or free antibodies targeted to ICAM-1, PECAM-1, VCAM-1, or with nonspecific IgG. Blood and organs were collected 30 min after injection. The change (Δ) in the specificity index (SI) of antibody nanocarriers over free antibody is shown for the brain, lungs, and liver. SI=1 indicates no change in specificity (horizontal line). Data are mean ± S.E.M. #Compares SI of nanocarrier to SI of antibody for a given receptor.
Supplemental Fig. 6 In vivo biodistribution of anti-ICAM/PECAM nanocarriers vs. their single-targeted counterparts. Mice were injected i.v. with anti-ICAM/PECAM NCs, anti-ICAM NCs, anti-PECAM NCs, or IgG NCs, and blood and organs were collected 30 min after injection. (A) The organ-to-blood localization ratio (LR) and (B) targeted-to-untargeted specificity index (SI) over IgG NCs are shown for the brain, lungs, and liver in control mice. SI = 1 is non-specific IgG NCs (horizontal line). (C) Effect of LPS challenge on nanocarrier bio distribution, assessed as the fold-change in LR (ΔLR) for LPS-treated mice over control mice. No change (Δ = 1) is shown as a horizontal line. Data are mean ± S.E.M. #Compares to anti-ICAM/PECAM NCs; !compares anti-PECAM NCs to anti-ICAM NCs; *compares control vs. LPS.
Supplemental Fig. 7 In vivo bio distribution of anti-PECAM/VCAM nanocarriers vs. their single-targeted counterparts. Mice were injected i.v. with anti-PECAM/VCAM NCs, anti-PECAM NCs, anti-VCAM NCs, or IgG NCs, and blood and organs were collected 30 min after injection. (A) The organ-to-blood localization ratio (LR) and (B) targeted-to-untargeted specificity index (SI) over IgG NCs are shown for the brain, lungs, and liver in control mice. SI = 1 is non-specific IgG NCs (horizontal line). (C) Effect of LPS challenge on nanocarrier bio distribution, assessed as the fold-change in LR (ΔLR) for LPS-treated mice over control mice. No change (Δ = 1) is shown as a horizontal line. Data are mean ± S.E.M. #Compares to anti-PECAM/VCAM NCs; !compares anti-PECAM NCs to anti-VCAM NCs; *compares control vs. LPS.
Acknowledgments
The authors thank Dr. Edward Schuchmann (Mount Sinai School of Medicine, New York, NY) for providing recombinant human ASM. This study was funded by NIH grant (R01 HL098416) to S. Muro.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Debbage P. Targeted drugs and nanomedicine: present and future. Curr Pharm Des. 2009;15:153–172. doi: 10.2174/138161209787002870. [DOI] [PubMed] [Google Scholar]
- 2.Duncan R, Richardson SC. Endocytosis and intracellular trafficking as gateways for nanomedicine delivery: opportunities and challenges. Mol Pharm. 2012;9:2380–2402. doi: 10.1021/mp300293n. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman AS. The origins and evolution of “controlled” drug delivery systems. J Control Release. 2008;132:153–163. doi: 10.1016/j.jconrel.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Muro S. Challenges in design and characterization of ligand-targeted drug delivery systems. J Control Release. 2012;164:125–137. doi: 10.1016/j.jconrel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 2012;8:147–166. doi: 10.1016/j.nano.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Torchilin VP. Multifunctional nanocarriers. Adv Drug Deliv Rev. 2006;58:1532–1555. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Weissleder R, Kelly K, Sun EY, Shtatland T, Josephson L. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat Biotechnol. 2005;23:1418–1423. doi: 10.1038/nbt1159. [DOI] [PubMed] [Google Scholar]
- 8.Papademetriou IT, Garnacho C, Schuchman EH, Muro S. In vivo performance of polymer nanocarriers dually-targeted to epitopes of the same or different receptors. Biomaterials. 2013;34:3459–3466. doi: 10.1016/j.biomaterials.2013.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun D, Nakao S, Xie F, Zandi S, Schering A, Hafezi-Moghadam A. Superior sensitivity of novel molecular imaging probe: simultaneously targeting two types of endothelial injury markers. FASEB J. 2010;24:1532–1540. doi: 10.1096/fj.09-148981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willmann JK, Lutz AM, Paulmurugan R, Patel MR, Chu P, Rosenberg J, Gambhir SS. Dual-targeted contrast agent for US assessment of tumor angiogenesis in vivo. Radiology. 2008;248:936–944. doi: 10.1148/radiol.2483072231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veeranarayanan S, Poulose AC, Mohamed MS, Varghese SH, Nagaoka Y, Yoshida Y, Maekawa T, Kumar DS. Synergistic targeting of cancer and associated angiogenesis using triple-targeted dual-drug silica nanoformulations for theragnostics. Small. 2012;8:3476–3489. doi: 10.1002/smll.201200874. [DOI] [PubMed] [Google Scholar]
- 12.Xu Q, Liu Y, Su S, Li W, Chen C, Wu Y. Anti-tumor activity of paclitaxel through dual-targeting carrier of cyclic RGD and transferrin conjugated hyperbranched copolymer nanoparticles. Biomaterials. 2012;33:1627–1639. doi: 10.1016/j.biomaterials.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Ying X, Wen H, Lu WL, Du J, Guo J, Tian W, Men Y, Zhang Y, Li RJ, Yang TY, Shang DW, Lou JN, Zhang LR, Zhang Q. Dual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animals. J Control Release. 2010;141:183–192. doi: 10.1016/j.jconrel.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Zhu C, Pardridge WM. Antisense gene therapy of brain cancer with an artificial virus gene delivery system. Mol Ther. 2002;6:67–72. doi: 10.1006/mthe.2002.0633. [DOI] [PubMed] [Google Scholar]
- 15.Eniola AO, Hammer DA. In vitro characterization of leukocyte mimetic for targeting therapeutics to the endothelium using two receptors. Biomaterials. 2005;26:7136–7144. doi: 10.1016/j.biomaterials.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Saul JM, Annapragada AV, Bellamkonda RV. A dual-ligand approach for enhancing targeting selectivity of therapeutic nanocarriers. J Control Release. 2006;114:277–287. doi: 10.1016/j.jconrel.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 17.Eniola AO, Willcox PJ, Hammer DA. Interplay between rolling and firm adhesion elucidated with a cell-free system engineered with two distinct receptor-ligand pairs. Biophys J. 2003;85:2720–2731. doi: 10.1016/s0006-3495(03)74695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrante EA, Pickard JE, Rychak J, Klibanov A, Ley K. Dual targeting improves microbubble contrast agent adhesion to VCAM-1 and P-selectin under flow. J Control Release. 2009;140:100–107. doi: 10.1016/j.jconrel.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gunawan RC, Almeda D, Auguste DT. Complementary targeting of liposomes to IL-1alpha and TNF-alpha activated endothelial cells via the transient expression of VCAM1 and E-selectin. Biomaterials. 2011;32:9848–9853. doi: 10.1016/j.biomaterials.2011.08.093. [DOI] [PubMed] [Google Scholar]
- 20.Gunawan RC, Auguste DT. The role of antibody synergy and membrane fluidity in the vascular targeting of immunoliposomes. Biomaterials. 2009;31:900–907. doi: 10.1016/j.biomaterials.2009.09.107. [DOI] [PubMed] [Google Scholar]
- 21.Ha SH, Carson A, Agarwal A, Kotov NA, Kim K. Detection and monitoring of the multiple inflammatory responses by photoacoustic molecular imaging using selectively targeted gold nanorods. Biomedical Optics Express. 2011;2:645–657. doi: 10.1364/BOE.2.000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McAteer MA, Mankia K, Ruparelia N, Jefferson A, Nugent HB, Stork LA, Channon KM, Schneider JE, Choudhury RP. A leukocyte-mimetic magnetic resonance imaging contrast agent homes rapidly to activated endothelium and tracks with atherosclerotic lesion macrophage content. Arterioscler Thromb Vasc Biol. 2012;32:1427–1435. doi: 10.1161/ATVBAHA.111.241844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robbins GP, Saunders RL, Haun JB, Rawson J, Therien MJ, Hammer DA. Tunable leukopolymersomes that adhere specifically to inflammatory markers. Langmuir. 2010;26:14089–14096. doi: 10.1021/la1017032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weller GE, Villanueva FS, Tom EM, Wagner WR. Targeted ultrasound contrast agents: in vitro assessment of endothelial dysfunction and multi-targeting to ICAM-1 and sialyl Lewisx. Biotechnol Bioeng. 2005;92:780–788. doi: 10.1002/bit.20625. [DOI] [PubMed] [Google Scholar]
- 25.Serrano D, Muro S. Endothelial Cell Adhesion Molecules and Drug Delivery Applications. In: Aranda-Espinoza H, editor. Mechanobiology of the Endothelium. CRC Press; Boca Raton: 2014. [Google Scholar]
- 26.McAteer MA, Schneider JE, Ali ZA, Warrick N, Bursill CA, von zur Muhlen C, Greaves DR, Neubauer S, Channon KM, Choudhury RP. Magnetic resonance imaging of endothelial adhesion molecules in mouse atherosclerosis using dual-targeted microparticles of iron oxide. Arterioscler Thromb Vasc Biol. 2008;28:77–83. doi: 10.1161/ATVBAHA.107.145466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammer DA, Robbins GP, Haun JB, Lin JJ, Qi W, Smith LA, Ghoroghchian PP, Therien MJ, Bates FS. Leuko-polymersomes. Faraday Discuss. 2008;139:129–141. doi: 10.1039/b717821b. discussion 213-128, 419-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rothlein R, Springer TA. The requirement for lymphocyte function-associated antigen 1 in homotypic leukocyte adhesion stimulated by phorbol ester. J Exp Med. 1986;163:1132–1149. doi: 10.1084/jem.163.5.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman PJ, Berndt MC, Gorski J, White GC, Lyman S, Paddock C, Muller WA. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science. 1990;247:1219–1222. doi: 10.1126/science.1690453. [DOI] [PubMed] [Google Scholar]
- 30.Phillips DR, Agin PP. Platelet plasma-membrane glycoproteins - Evidence for presence of nonequivalent disulfide bonds using non-reduced-reduced 2-dimensional gel-electrophoresis. J Biol Chem. 1977;252:2121–2126. [PubMed] [Google Scholar]
- 31.Osborn L, Hession C, Tizard R, Vassallo C, Luhowskyj S, Chi-Rosso G, Lobb R. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell. 1989;59:1203–1211. doi: 10.1016/0092-8674(89)90775-7. [DOI] [PubMed] [Google Scholar]
- 32.Lorenzon P, Vecile E, Nardon E, Ferrero E, Harlan JM, Tedesco F, Dobrina A. Endothelial cell E- and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J Cell Biol. 1998;142:1381–1391. doi: 10.1083/jcb.142.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garnacho C, Albelda SM, Muzykantov VR, Muro S. Differential intra-endothelial delivery of polymer nanocarriers targeted to distinct PECAM-1 epitopes. J Control Release. 2008;130:226–233. doi: 10.1016/j.jconrel.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muro S, Garnacho C, Champion JA, Leferovich J, Gajewski C, Schuchman EH, Mitragotri S, Muzykantov VR. Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol Ther. 2008;16:1450–1458. doi: 10.1038/mt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muro S, Wiewrodt R, Thomas A, Koniaris L, Albelda SM, Muzykantov VR, Koval M. A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J Cell Sci. 2003;116:1599–1609. doi: 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- 36.Ricard I, Payet MD, Dupuis G. VCAM-1 is internalized by a clathrin-related pathway in human endothelial cells but its alpha(4)beta(1) integrin counter-receptor remains associated with the plasma membrane in human T lymphocytes. Eur J Immunol. 1998;28:1708–1718. doi: 10.1002/(SICI)1521-4141(199805)28:05<1708::AID-IMMU1708>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 37.Hillaireau H, Couvreur P. Nanocarriers' entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66:2873–2896. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papademetriou J, Garnacho C, Serrano D, Bhowmick T, Schuchman EH, Muro S. Comparative binding, endocytosis, and biodistribution of antibodies and antibody-coated carriers for targeted delivery of lysosomal enzymes to ICAM-1 versus transferrin receptor. J Inherit Metab Dis. 2013;36:467–477. doi: 10.1007/s10545-012-9534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He X, Miranda SR, Xiong X, Dagan A, Gatt S, Schuchman EH. Characterization of human acid sphingomyelinase purified from the media of overexpressing Chinese hamster ovary cells. Biochim Biophys Acta. 1999;1432:251–264. doi: 10.1016/s0167-4838(99)00069-2. [DOI] [PubMed] [Google Scholar]
- 40.Hsu J, Serrano D, Bhowmick T, Kumar K, Shen Y, Kuo YC, Garnacho C, Muro S. Enhanced endothelial delivery and biochemical effects of alpha-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J Control Release. 2011;149:323–331. doi: 10.1016/j.jconrel.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Warram JM, Sorace AG, Saini R, Umphrey HR, Zinn KR, Hoyt K. A triple-targeted ultrasound contrast agent provides improved localization to tumor vasculature. J Ultrasound Med. 2011;30:921–931. doi: 10.7863/jum.2011.30.7.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuchman EH, Desnick RJ. Niemann-Pick Disease Types A and B: Acid Sphingomyelinase Deficiencies. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, Vogelstein B, Bunz F, Gibson KM, Mitchell G, editors. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 2001. pp. 3589–3610. [Google Scholar]
- 43.Muro S. New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2:189–204. doi: 10.1002/wnan.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garnacho C, Dhami R, Simone E, Dziubla T, Leferovich J, Schuchman EH, Muzykantov V, Muro S. Delivery of acid sphingomyelinase in normal and niemann-pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J Pharmacol Exp Ther. 2008;325:400–408. doi: 10.1124/jpet.107.133298. [DOI] [PubMed] [Google Scholar]
- 45.Liu G, Vogel SM, Gao X, Javaid K, Hu G, Danilov SM, Malik AB, Minishall RD. Scr phosphorylation of endothelial cell surface intercellular adhesion molecule-1 mediates neutrophil adhesion and contributes to the mechanism of lung inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1342–1350. doi: 10.1161/ATVBAHA.110.222208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chacko AM, Nayak M, Greineder CF, Delissier HM, Muzycantov VR. Collaborative enhancement of antibody binding to distinct PECAM-1 epitopes modulates endothelial targeting. PLoS One. 2012;7:e34958. doi: 10.1371/journal.pone.0034958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar AG, Dai XY, Kozak CA, Mims MP, Gotto AM, Ballantyne CM. Murine VCAM-1 - molecular-cloning, mapping, and analysis of a truncated form. J Immunol. 1994;153:4088–4098. [PubMed] [Google Scholar]
- 48.Lee S, Yoon IH, Yoon A, Cook-Mills JM, Park CG, Chung J. An antibody to the sixth Ig-like domain of VCAM-1 inhibits leukocyte transendothelial migration without affecting adhesion. J Immunol. 2012;189:4592–4601. doi: 10.4049/jimmunol.1103803. [DOI] [PubMed] [Google Scholar]
- 49.Frey A, Giannasca KT, Weltzin R, Giannasca PJ, Reggio H, Lencer WI, Neutra MR. Role of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting. J Exp Med. 1996;184:1045–1059. doi: 10.1084/jem.184.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 51.Couty JP, Rampon C, Leveque M, Laran-Chich MP, Bourdoulous S, Greenwood J, Couraud PO. PECAM-1 engagement counteracts ICAM-1-induced signaling in brain vascular endothelial cells. J Neurochem. 2007;103:793–801. doi: 10.1111/j.1471-4159.2007.04782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao WY, Li HJ, Chang MY, Tang AC, Hoffman AS, Hsieh PC. Comprehensive characterizations of nanoparticle biodistribution following systemic injection in mice. Nanoscale. 2013;5:11079–11086. doi: 10.1039/c3nr03954d. [DOI] [PubMed] [Google Scholar]
- 53.Muzykantov VR, Radhakrishnan R, Eckmann DM. Dynamic factors controlling targeting nanocarriers to vascular endothelium. Curr Drug Metab. 2012;13:70–81. doi: 10.2174/138920012798356916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han J, Zern BJ, Shuvaev VV, Davies PF, Muro S, Muzykantov V. Acute and chronic shear stress differently regulate endothelial internalization of nanocarriers targeted to platelet-endothelial cell adhesion molecule-1. ACS Nano. 2012;6:8824–8836. doi: 10.1021/nn302687n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romer LH, McLean NV, Yan HC, Daise M, Sun J, DeLisser HM. IFN-gamma and TNF-alpha induce redistribution of PECAM-1 (CD31) on human endothelial cells. J Immunol. 1995;154:6582–6592. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1 Cell-binding avidity of nanocarriers targeted to ICAM-1, PECAM-1, and/or VCAM-1. TNFα-activated H5V cells were fixed and incubated for 1 h at 37°C with varying concentrations of Fluoresbrite-labeled nanocarriers targeted to ICAM-1, PECAM-1, and/or VCAM-1, followed by analysis by fluorescence microscopy.
Supplemental Fig. 2 Binding and endocytosis of anti-PECAM/VCAM nanocarriers vs. their single-targeted counterparts. Green Fluoresbrite nanocarriers incubated for 1 h at 37 °C with control or TNFα-activated H5V cells. Surface-bound (non-internalized) carriers were immunostained with a Texas-Red secondary antibody (see Materials and methods). Total cell-associated carriers and surface-bound counterparts were imaged by fluorescence microscopy. (A) Absolute number of carriers associated to cells. (B) Percentage of internalized nanocarriers. (C) Absolute number of internalized nanocarriers. Data are mean ± SEM. *Compares control vs. TNFα; #compares to anti-PECAM/VCAM NCs; !compares between anti-PECAM NCs and anti-VCAM NCs.
Supplemental Fig. 3 Comparative binding of nanocarriers dual- or triple-targeted to ICAM-1, PECAM-1, and/or VCAM-1 vs. their single-targeted counterparts. Anti-ICAM/PECAM NCs, anti-PECAM/VCAM NCs, and anti-ICAM/PECAM/VCAM NCs, all labeled with green Fluoresbrite, were incubated for 1 h at 37 °C with fixed, TNFα-activated H5V cells. Total cell-bound carriers were imaged by fluorescence microscopy and compared to the binding of mixtures of single-targeted carriers, including anti-ICAM NCs, anti-PECAM NCs, and/or anti-VCAM NCs mixtures carrying full valency for each of their receptors (A), or valencies matching those of dual- or triple-targeted carriers (B); (see Methods Section 2.5). Data are mean ± SEM.
Supplemental Fig. 4 Binding and endocytosis of anti-ICAM/PECAM nanocarriers vs. their single-targeted counterparts. Green Fluoresbrite nanocarriers incubated for 1 h at 37 °C with control or TNFα-activated H5V cells. Surface-bound (non-internalized) carriers were immunostained with a Texas-Red secondary antibody (see Materials and methods). Total cell-associated carriers and surface-bound counterparts were imaged by fluorescence microscopy. (A) Absolute number of carriers associated to cells. (B) Percentage of internalized nanocarriers. (C) Absolute number of internalized nanocarriers. Data are mean ± SEM.*Compares control vs. TNFα; #compares to anti-ICAM/PECAM NCs; !compares between anti-ICAM NCs and anti-PECAM NCs.
Supplemental Fig. 5 Change in the in vivo specificity index of antibody-nanocarriers vs. antibodies targeted to ICAM-1, PECAM-1, or VCAM-1. Mice were injected i.v. with nanocarriers or free antibodies targeted to ICAM-1, PECAM-1, VCAM-1, or with nonspecific IgG. Blood and organs were collected 30 min after injection. The change (Δ) in the specificity index (SI) of antibody nanocarriers over free antibody is shown for the brain, lungs, and liver. SI=1 indicates no change in specificity (horizontal line). Data are mean ± S.E.M. #Compares SI of nanocarrier to SI of antibody for a given receptor.
Supplemental Fig. 6 In vivo biodistribution of anti-ICAM/PECAM nanocarriers vs. their single-targeted counterparts. Mice were injected i.v. with anti-ICAM/PECAM NCs, anti-ICAM NCs, anti-PECAM NCs, or IgG NCs, and blood and organs were collected 30 min after injection. (A) The organ-to-blood localization ratio (LR) and (B) targeted-to-untargeted specificity index (SI) over IgG NCs are shown for the brain, lungs, and liver in control mice. SI = 1 is non-specific IgG NCs (horizontal line). (C) Effect of LPS challenge on nanocarrier bio distribution, assessed as the fold-change in LR (ΔLR) for LPS-treated mice over control mice. No change (Δ = 1) is shown as a horizontal line. Data are mean ± S.E.M. #Compares to anti-ICAM/PECAM NCs; !compares anti-PECAM NCs to anti-ICAM NCs; *compares control vs. LPS.
Supplemental Fig. 7 In vivo bio distribution of anti-PECAM/VCAM nanocarriers vs. their single-targeted counterparts. Mice were injected i.v. with anti-PECAM/VCAM NCs, anti-PECAM NCs, anti-VCAM NCs, or IgG NCs, and blood and organs were collected 30 min after injection. (A) The organ-to-blood localization ratio (LR) and (B) targeted-to-untargeted specificity index (SI) over IgG NCs are shown for the brain, lungs, and liver in control mice. SI = 1 is non-specific IgG NCs (horizontal line). (C) Effect of LPS challenge on nanocarrier bio distribution, assessed as the fold-change in LR (ΔLR) for LPS-treated mice over control mice. No change (Δ = 1) is shown as a horizontal line. Data are mean ± S.E.M. #Compares to anti-PECAM/VCAM NCs; !compares anti-PECAM NCs to anti-VCAM NCs; *compares control vs. LPS.