

Advances in synchrotron beamlines bring opportunities with accompanying challenges for the study of soft condensed (biological) matter. This article describes improvements to the BioCAT beamline that include micro-focus, scanning and cryo-cooling of soft connective tissues yielding X-ray data from whole rat-tail tendons to better than 4 Å.
Keywords: X-ray diffraction, microfocus, amyloid, collagen, biological samples
Abstract
The small source sizes of third-generation synchrotron sources are ideal for the production of microbeams for diffraction studies of crystalline and non-crystalline materials. While several such facilities have been available around the world for some time now, few have been optimized for the handling of delicate soft-tissue specimens under cryogenic conditions. Here the development of a new X-ray micro-diffraction instrument at the Biophysics Collaborative Access Team beamline 18-ID at the Advanced Photon Source, and its use with newly developed cryo-diffraction techniques for soft-tissue studies, are described. The combination of the small beam sizes delivered by this instrument, the high delivered flux and successful cryo-freezing of rat-tail tendon has enabled us to record data to better than 4 Å resolution. The ability to quickly raster scan samples in the beam allows selection of ordered regions in fibrous samples for markedly improved data quality. Examples of results of experiments obtainable using this instrument are presented.
1. Introduction
Wide-angle fiber diffraction is the method of choice for studying filamentous viruses, most cyto-skeletal proteins, collagenous structures in connective tissues and amyloid systems because of the tendency of these biopolymers to align their long axes to form fibers (Chandrasekaran & Stubbs, 2001 ▶; Tsuruta & Irving, 2008 ▶). Such systems can rarely be studied by conventional macromolecular crystallographic techniques. In those few cases where crystals can be grown, the molecular interactions in the crystals rarely correspond to key aspects of these systems as the biologically significant interactions in the fibers are disrupted or simply not present in the crystal (Orgel et al., 2014 ▶; Orgel & Irving, 2014 ▶).
The fibrillar collagens form the structural basis of organs and connective tissues (Ayad et al., 1998 ▶; Kadler et al., 2007 ▶; Orgel et al., 2011 ▶). While collagen’s helical structure has been widely studied (Orgel et al., 2014 ▶; Rich & Crick, 1961 ▶; Bella et al., 1994 ▶; Okuyama et al., 1971 ▶; Kramer et al., 2001 ▶; Fan et al., 1993 ▶; Okuyama, 2008 ▶; Bella, 2010 ▶; Rainey & Goh, 2002 ▶), collagen’s packing structure is possibly of greater significance to collagen’s roles in nature (Orgel et al., 2006 ▶, 2011 ▶; Perumal et al., 2008 ▶; San Antonio et al., 2012 ▶). Understanding the packing structure of collagen from a variety of different source tissues, and in normal and diseased states, has the potential to provide insights into the binding of important macromolecules within the extracellular matrix (ECM) of mammalian connective tissues and to shed light on both the origins and possible treatments for numerous diseases (including arthritis, heart disease, cancer) and injuries.
Protein misfolding reactions (amyloidosis) are the source of numerous human pathologies, including Alzheimer’s disease (Malinchik et al., 1998 ▶), Parkinson’s disease, amyotrophic lateral sclerosis, cystic fibrosis and polyglutamine diseases (Soto, 2003 ▶). Protein misfolding has also been associated with cataracts, type II diabetes (Chiti & Dobson, 2006 ▶; Vestergaard et al., 2007 ▶) and a variety of diseases caused by prions [reviewed by Inouye & Kirschner (1998 ▶) and Weissmann (2004 ▶)]. The connections between the specific form of the protein folds and disease pathology have been starting to emerge in recent years, although the form of amyloid structure may vary even within the brain of a single patient, depending on the origins of the original misfolded ‘seed’ (Tycko & Wickner, 2013 ▶; Cloe et al., 2011 ▶; Tycko et al., 2009 ▶). As with collagenous systems, many more structural studies will be needed to help describe the links between structure and pathology and in order to answer fundamental questions regarding protein folding. We also expect such studies to reveal details concerning the physiology of amyloid accumulation and allow for the rational design of drugs to prevent or reverse amyloid formation or to inhibit the physiological effects.
Diffraction studies of all these systems require high-intensity X-ray beams at the sample of a size of the order of 3–20 µm. This focal spot size range allows localization of small ordered domains in tissue samples (as will be presented in this study) with low enough divergence so that diffraction spots on the detector are well separated and that diffraction signals are well resolved above the diffuse background scattering from amorphous material. Here we describe how we have adapted our existing X-ray fluorescence microscopy instrument (Barrea et al., 2010 ▶) to be a new, highly flexible, micro-diffraction instrument on the BioCAT beamline 18-ID at the Advanced Photon Source (Fischetti et al., 2004 ▶) that is optimized for the study of partially ordered biological material within tissue samples. We report the improvements of the X-ray optics and the new sample handling capabilities, including cryo-preservation of samples, essential for soft-tissue studies with microbeams. The performance of the instrument for selected biological diffraction problems of biomedical relevance are also described.
2. Instrumentation
2.1. X-ray optics
The layout of the main components of the microbeam instrument on the BioCAT undulator beamline 18ID are shown in Fig. 1 ▶. These comprise an APS undulator type ‘A’ [3.3 cm period, 10.8 µm minimum gap (Lai et al., 1993 ▶; Dejus et al., 1994 ▶)], a sagittal bending double-crystal Si(111) liquid-nitrogen-cooled monochromator, and a Kirkpatrick–Baez mirror bender system (KB mirror system) of the University of Chicago design (Eng et al., 1998 ▶). The undulator gap can be adjusted to place the fundamental peak in the energy spectrum (3.2 keV at closed gap, 14 keV at open gap) close to the desired monochromatic beam energy. Relevant source parameters at 12 keV are σx = 270.8 µm, 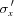 = 11.4 µrad, σy = 10.8 µm,
= 11.4 µrad, σy = 10.8 µm, 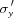 = 3.8 µrad. The KB mirror system (metallic coating being 40 nm of Rh and 10 nm Cr, slope error of <2 µrad) is mounted on a motorized table located 68 m downstream from the undulator source for a source demagnification of 166:1 (vertical) and 318:1 (horizontal). The X-ray energy used for most experiments is 12 keV (delivering a flux density of 1 × 1010 photons s−1 µm−2), although the energy can be easily changed to 8 keV to adjust the range of accessible d-spacings in the diffraction patterns. The incoming beam is defined by a set of beam-defining (‘Pre-KB’) slits located immediately upstream of the mirror assembly. The beam intensity is monitored by a small ion chamber mounted immediately downstream of the KB system. The ion chamber signal may be used as a feedback signal to a lock-in amplifier, which controls the tuning of the monochromator’s second crystal (which oscillates at 220 Hz) in order to maintain the beam intensity at its maximum value. A second set of vacuum-compatible slits located just downstream of the mirrors are used to reduce parasitic scattering from the optics. A third set of slits (guard slits) and the final guard aperture, selectable from 50 µm up to 250 µm, may be independently aligned using separate sets of motorized linear stages (see Fig. 1 ▶). Additional details of the beamline optics can be found elsewhere (Fischetti et al., 2004 ▶; Barrea et al., 2005 ▶, 2009 ▶). The great advantage of the KB mirror system for our purposes is the insensitivity of focal position to changes in X-ray beam energy and the ability to re-focus the mirrors relatively quickly and easily for different focal distances (∼20 min for a complete realignment) (see Fig. 1 ▶ and Table 1 ▶ for schematic and technical details). There are three focusing configurations typically used with this system: (a) the beam focused at the sample position for maximum spatial resolution, (b) the beam focused at the detector position for maximum resolution of features in the diffraction patterns, and (c) the beam focused at midpoint between sample and detector as a useful compromise. Focusing at the sample provides the smallest spot size on the sample, ideal for scanning X-ray fluorescence microscopy experiments, or diffraction experiments where the smallest possible focal spot is desired and the relatively high divergence can be tolerated. Focusing at the detector provides the lowest divergence under these conditions, ideal to resolve closely spaced diffraction peaks.
= 3.8 µrad. The KB mirror system (metallic coating being 40 nm of Rh and 10 nm Cr, slope error of <2 µrad) is mounted on a motorized table located 68 m downstream from the undulator source for a source demagnification of 166:1 (vertical) and 318:1 (horizontal). The X-ray energy used for most experiments is 12 keV (delivering a flux density of 1 × 1010 photons s−1 µm−2), although the energy can be easily changed to 8 keV to adjust the range of accessible d-spacings in the diffraction patterns. The incoming beam is defined by a set of beam-defining (‘Pre-KB’) slits located immediately upstream of the mirror assembly. The beam intensity is monitored by a small ion chamber mounted immediately downstream of the KB system. The ion chamber signal may be used as a feedback signal to a lock-in amplifier, which controls the tuning of the monochromator’s second crystal (which oscillates at 220 Hz) in order to maintain the beam intensity at its maximum value. A second set of vacuum-compatible slits located just downstream of the mirrors are used to reduce parasitic scattering from the optics. A third set of slits (guard slits) and the final guard aperture, selectable from 50 µm up to 250 µm, may be independently aligned using separate sets of motorized linear stages (see Fig. 1 ▶). Additional details of the beamline optics can be found elsewhere (Fischetti et al., 2004 ▶; Barrea et al., 2005 ▶, 2009 ▶). The great advantage of the KB mirror system for our purposes is the insensitivity of focal position to changes in X-ray beam energy and the ability to re-focus the mirrors relatively quickly and easily for different focal distances (∼20 min for a complete realignment) (see Fig. 1 ▶ and Table 1 ▶ for schematic and technical details). There are three focusing configurations typically used with this system: (a) the beam focused at the sample position for maximum spatial resolution, (b) the beam focused at the detector position for maximum resolution of features in the diffraction patterns, and (c) the beam focused at midpoint between sample and detector as a useful compromise. Focusing at the sample provides the smallest spot size on the sample, ideal for scanning X-ray fluorescence microscopy experiments, or diffraction experiments where the smallest possible focal spot is desired and the relatively high divergence can be tolerated. Focusing at the detector provides the lowest divergence under these conditions, ideal to resolve closely spaced diffraction peaks.
Figure 1.

Beamline schematic. (a) BioCAT beamline schematic; parts (b–(d) are possible sample mounts/options at the position labeling ‘Sample’. (b) Cryo-jet that operates without a gas shield and plunge-cooled sample. Note that there is no ice build-up at the nozzle while there is ice building up on the sample outside of the cryo-jet stream, although the sample at the beam position is cryo-preserved and ice-free in this image. (c) Cryo-stream that operates with a gas shield to prevent icing; note that the sample is dried by the very low humidity gas shield, even within part of the sample that is within the cryo-stream. (d) Example of a room-temperature sample enclosure, designed to preserve the sample against dehydration with two sections so that multiple samples can be scanned (one after the other) per sample mount change during an experimental run.
Table 1. Parameters for the KB-based micro-diffraction instrument.
KB mirrors aperture size 0.6 mm, flux 1.0 × 1012 photons s−1.
| Focus at sample | Focus at midpoint | Focus at detector | |
|---|---|---|---|
| 250 mm camera length | |||
| Beam size at sample (V × H) | 3.5 mm × 5 mm | 120 µm × 160 µm | 200 µm × 250 µm |
| Beam size at detector (V × H) | 350 µm × 500 µm | 120 µm × 160 µm | 10 µm × 10 µm |
| Divergence (V × H) | 1.2 mrad × 2.0 mrad | 0.9 mrad × 1.4 mrad | 0.8 mrad × 1.1 mrad |
| 500 mm camera length | |||
| Beam size at sample (V × H) | 3.5 µm × 5 µm | 200 µm × 270 µm | 300 µm × 350 µm |
| Beam size at detector (V × H) | 600 µm × 1000 µm | 200 µm × 270 µm | 15 µm × 15 µm |
| Divergence (V × H) | 1.2 mrad × 2.0 mrad | 0.8 mrad × 1.1 mrad | 0.6 mrad × 0.75 mrad |
2.2. Sample handling and positioning
Sample positioning is performed by a set of high-precision (0.1 µm-resolution) motorized Newport® UTM25PE and ILS50PP stages mounted in XYZ configuration. These stages allow the users to locate and to scan the samples with high precision and reproducibility. There is a set of custom-designed sample holders that provide the needed flexibility to accommodate different requirements from the users. Visualization and alignment of the samples is performed using an optical system mounted at 90° from the beam direction using a prism to view the X-ray flight path, redirecting the view to the optical equipment outside of the beam. The prism is mounted on a motorized stage between 20 and 50 mm upstream of the sample position (depending on the specific experiment-dependent set-up the viewer can be set up to visualize downstream of the sample also) and it is moved out of the beam path during data collection. Its main use is to view sample features and for alignment for diffraction experiments. The optical view is calibrated against an image of the X-ray beam captured on X-ray-sensitive paper. The system consists of a small-size prism (5 mm × 5 mm) mounted on a manual XY positioner for independent alignment and a CCD Hitachi HV-C20 color camera with an Infinity optical system with a set of CFV-2/3/4 lenses.
Biological samples are quite sensitive to damage from exposure to the X-ray beam; therefore they require special conditions to avoid or minimize the effects of radiation exposure. Unlike crystallography experiments with crystals usually on the microscopic scale and rarely measuring more then a millimeter in any dimension, fiber diffraction samples generally need to be on the millimeter scale. This is due to their semi or non-crystalline nature requiring more sample area to capture sufficient signal in the X-ray diffraction experiment, and/or because cutting or extracting elements out of a biological specimen indirectly damages the organization of the sample distant from the site of a cut. Since these samples are composed of fibers much longer then they are wide, this is not surprising, since cutting in one site may disturb the fiber organization along the remaining part of the cut fibers. Hence our specimens might be up to 5 mm in diameter and several tens of millimeters in length. Even though on average an extracted collagen sub-tendon fiber is around 0.05–0.5 mm in diameter, its relatively large size requires specific attention to vitreously freeze and it is possible that it is not completely vitreously frozen throughout (for instance, its center for the thicker samples), requiring a microbeam to probe the outer edges of the sample to find the best preserved sample parts and longer exposure time to the X-rays to collect signal from weakly diffracting peaks. The extended survival time in the beam that the cryo-cooling enables appears to more then compensate for the reduced sample size and beam area.
To maintain fibrous samples at liquid-nitrogen temperature we have a customized cold nitrogen stream system (CRYOCOOL-PC, Cryoindustries of America Inc, Manchester, NH, USA); as designed by the manufacturer, it lacks the warm shield gas layer used in conventional crystallography cryo-jets which can dry out our relatively large samples, some of the length of which may extend beyond the cryo-jet’s footprint which is ∼8 mm in diameter (prevention of icing of the cryo-jet is achieved with the manufacturers supplied nozzle heater; we have added a small additional heater to the tip for prolonged use, on the hour range at low temperatures). So-called ‘flash-freezing’ can be achieved by rapidly inserting samples in cryo-protectant into the nitrogen stream although this method does not give highly reproducible results presumably due to the size of the samples. For reproducibility, we have used a Leica CPC (Buffalo Grove, IL, USA) cryo-workstation with liquefied propane for plunge-freezing of samples before being placed in the cryo-jet stream. Samples plunged in liquefied propane are pre-mounted on pins that fit into a customized Hampton haemostat where the inner surfaces are widened to accept larger sample holders than the mounts typically used for macromolecular crystallography, and transferred to the sample position in the beam in the cryo-jet using the normal cryo-crystallgraphy transfer technique with a haemostat. Both flash-freezing and plunge-freezing techniques have been explained elsewhere (Vera & Stura, 2014 ▶; Warkentin et al., 2006 ▶), but for our larger fibrous samples flash-freezing requires particular care and speed in handling to prevent warming of the sample during transfer. Using either flash-freezing or plunge-freezing, we have been successful in preserving connective tissue samples in the X-ray beam for extended periods of time, up to 60 min whilst frozen, and without any obvious signs of degradation (as determined from comparison of diffraction patterns from the start, middle and end of data collection, changes in intensity and intensity peak positions being clear indicators of damage). Although more difficult to freeze because of their relatively large size, once frozen it may be their larger size and lower water content relative to protein crystals that allows such excellent extended preservation time in the cryo-jet.
2.3. Detectors
For micro-diffraction applications we use one of two detectors. The first is a Mar165 CCD detector (Rayonix Inc. Evanston, IL, USA) that has been modified for fiber diffraction applications with a thinner (25 µm versus 40 µm FWHM) phosphor to provide a ∼65 µm point spread function (down from ∼100 µm FWHM at the expense of a reduction in DQE from ∼88% to ∼78%) and software modifications to allow it to be read out in 1 × 1 unbinned mode (39.9 µm × 39.9 µm pixel size) for the full resolution of 4k × 4k pixels. The large active area (165 mm-diameter circle active area) and the large number of resolution elements is a benefit for systems that provide detailed patterns with closely spaced diffraction peaks. We also have a Pilatus 100k pixel array detector that can be read out at speeds up to 200 Hz (1 ms detection, 4 ms readout) for fast scanning applications. A limitation is the very small active area of only 33.5 mm × 84 mm. This, combined with its relatively large pixel size of 172 µm × 172 µm, limits the angular resolution of the detector. Another limitation is that this version of the detector starts becoming inefficient at energies above 10–12 keV (51% efficient at 15 keV with 320 µm-thick sensor thickness). The high speed of readout, however, can be very useful for scanning diffraction mapping applications where high spatial resolution in the detector plane is not required and the diffraction features of interest are localized to a relatively small region.
2.4. Scanning micro-diffraction
Two-dimensional micro-diffraction mapping experiments are performed by conventional step-scanning. The rate-limiting step for scanning X-ray diffraction mapping experiments is either (i) exposure time (typically 1–20 s depending on sample type and thickness) or (ii) readout time of the detector (10 s for the Mar165, 4 ms for the Pilatus 100k). Since in either case the dwell time per pixel is likely to be longer than 1 s, step-scanning is appropriate. The scan software controls the sample positioning and the triggering of the CCD detector and the shutters. The sample is visualized on a video monitor screen, via the prism/camera system described above, in order to select the area to be scanned. During the scans, the sample is moved to the desired position, and then the trigger signal is sent to the detector to start the exposure time and for the opening of the shutter. Once the exposure time is completed, the system closes the shutter and waits for the image to be read out and stored in file before moving the sample to the next position. The program continues with the exposure of the next spot until the whole predefined area has been scanned. The system collects the image information and sample positioning simultaneously. Currently the images are stored in separate tiff file format and the sample position is saved in HDF5 file format. Efforts are continuing to integrate all the information into a single file in HDF5 format for easy handling and archiving of the collected data.
3. Results and discussion
Table 1 ▶ summarizes the X-ray beam sizes at the sample and the detector, as well as the beam divergences for various configurations of the micro-diffraction instrument. Much of our efforts over the last few years have gone into reducing the parasitic scattering background and improving the ergonomics and robustness of the system. The useful d-space range from the micro-diffraction instrument is 1/200 to 1/3.4 Å−1 at 12 keV and 1/900 to 1/20 Å−1 at 8 keV with the Mar165 detector, and 1/200 to 6 Å −1 at 12 keV and 1/900 to 40 Å−1 at 8 keV with the Pilatus detector (along the long dimension). We have demonstrated that it is possible to collect very useful wide-angle fiber diffraction patterns with a relatively high flux of ∼1 × 1012 photons s−1 and ∼5 µm × 8 µm beams when focused at the sample, and ∼60 µm to 100 µm beams when focused at the detector. These small beams allow selection of small ordered domains in the sample to yield particularly high quality patterns for detailed structural analysis. By scanning the sample to find the more ordered domains, it is possible to concentrate on these areas in order to collect higher quality data then would be possible with larger beam sizes because of inhomogeneity in the sample. Antipova & Orgel (2010 ▶) used this capability to find the part of lamprey notochord that gave the best diffraction patterns (Fig. 2 ▶) allowing them to solve the one-dimensional structure of collagen type II. In another study, Antipova & Orgel (2012 ▶), examining the tissue disruptive effects of various substances including antibodies related to rheumatoid arthritis, were able to find small domains where the fibrillated fibril-bundles still maintained a degree of alignment and hence obtain diffraction data from these small regions in the disrupted tissues. In another study, Landahl et al. (2011 ▶) used the micro-diffraction instrument to localize small ordered domains (tau bodies) in brain tissue with low enough divergence so that diffraction spots on the detector were well separated and that the diffraction signal was well resolved above the diffuse background from amorphous material in the beam. Another application of micro-diffraction is in the study of artificially orientated fibrous specimens, typical of amyloid systems. Fig. 3(a) ▶ shows a view of the sample that was used to contrast diffraction quality from the conventional wide-angle diffraction instrument on 18-ID (∼60 µm × 60 µm beam size at the sample) with that collected with the micro-diffraction instrument. The large box in Fig. 3(a) ▶ shows the location of the region interrogated with the conventional fiber diffraction instrument (see Fig. 3d ▶) and the smaller boxes inside show the approximate location of sample regions interrogated with the micro-diffraction instrument. As Fig. 3(b) ▶ shows, even closely spaced regions may not show any significant degree of order from which minimal or no X-ray diffraction can be recorded [Fig. 3(b), (i) and (ii) ▶] whereas nearby (of the order of 10 µm) there may be significantly better ordered sample regions [Fig. 3(b), (iii) and (iv) ▶]. It would seem in this example that the wide-angle fiber diffraction pattern (Fig. 3d ▶) is also collecting significant ‘background’ from the poorly ordered domains of the sample in the larger beam cross-section. With the microbeam (Fig. 3c ▶), the signal-to-noise ratio is better and the background minimized {[as indicated by the much better contrast of the cross-beta diffraction of the inter-filament spacing [as shown by arrows in Figs. 3(c) and 3(d) ▶]}.
Figure 2.

Micro-diffraction data from lamprey notochord. (a) Hydrated room-temperature data collection from lamprey notochord. The sample was scanned to find regions containing better ordered domains and, once located, the sample stretched to improve the alignment of the collagen fiber orientation and order in the direction of the stretch. This procedure will also yield poorer diffraction for the population of fibers in the perpendicular direction to the stretch, e.g. for the pattern third from left top [see part (b)]. In so doing it is possible to isolate higher quality diffraction then would be possible otherwise [see part (b)]. (b) Medium-wide angle X-ray diffraction pattern of collagen type II fibrils from lamprey notochord with 15–20 Å resolution (the meridional reflections series). {This figure is reused here with permission (under a creative commons license). This research was originally published in J. Biol. Chem. [Antipova & Orgel (2010). J. Biol. Chem. 285, 7087–7096]; © the American Society for Biochemistry and Molecular Biology.}
Figure 3.

X-ray diffraction of Iowa alzheimer amyloid mutant sample in normal and micro-diffraction WAX modes. (a) Light microscope view of the sample. D23N Abeta amyloid, the white mass in the sample tube, was scanned at intervals shown by light red boxes with ∼10 µm beam [(b) (i)–(iv)] with the best diffracting example (iv) shown more clearly in (c), and was also diffracted with a ∼100 µm × 100 µm-sized beam [larger red box, (d)]. The signal-to-noise improvement of the use of the smaller source size [compare (c) with (d)] is seen most noticeably for the lower-order reflections. [Reprinted with permission from Tycko et al. (2009). Biochemistry, 48, 6072–6084. Copyright 2009 American Chemical Society.]
In the standard configuration at 12 keV the accessible d-spacing range is 134–3.5 Å. We recently established that it is straightforward, with minor adjustments, to change energy to 8 keV to access a d-spacing range of ∼600–5 Å making it adequate for many small-angle experiments including muscle diffraction. One of the developments allowing this large Q-range was upgrades to the existing Mar165 detector allowing to be used in unbinned full-resolution mode (4k × 4k 39.9 µm pixels) and a thinner phosphor to give it a smaller point spread function (∼65 µm FWHM). This combination is much better for fiber diffraction applications where one also needs to distinguish weak closely spaced diffraction features in the presence of high diffuse backgrounds from amorphous regions in the sample. Smaller pixel sizes also make it easier to record strong reflections in the presence of weak ones without saturating the detector by spreading the intensity of the strong reflections over more pixels.
In parallel with the development of the micro-diffraction capabilities was that of cryo-preservation techniques for fibrous protein systems. Smaller beam sizes allow for better signal-to-noise ratios by selecting the most crystalline parts of the fibrous sample, but also require longer exposure times to record weaker reflections from some samples such as those from connective tissues. Soft tissues dry or suffer heat damage rapidly at room temperature, but the millimeter-scale size of the sample (necessary to preserve the native structures that disappear with over-dissection) make the rapid cooling required challenging. By using either flash-freezing or plunge-freezing as described above, we have been successful in preserving connective tissue samples in the X-ray beam for extended periods of time, up to an hour or more. Recently, we have used this approach to record diffraction to exceed a previous best for high-resolution data from rat-tail tendon [∼0.51 nm (Orgel et al., 2006 ▶) now improved to 0.4 nm (Fig. 4 ▶)]. The operating temperature range was approximately 82–100 K and the cryo-protectant for collagen was 15% glycerol in PBS buffer or neat mineral oil, or, in the case of liquid propane, no cryo-protection was required. Together, these developments have allowed data to be obtained with unprecedented resolution for a natural crystallite within its native tissue, showing continuous data recorded to better then 0.4 nm for the crystalline section of type I collagen.
Figure 4.

Cryo- and micro-diffraction data collected from rat-tail tendon. (a) Cryo-frozen native rat-tail tendon data extending past 4 Å. The pattern was collected with an exposure time of 5 min using the microfocus diffraction set-up on the BioCAT beamline 18-ID. (b) The arrow points to the 4 Å helix merdional, whilst meridional reflections from the 670 Å repeat are clearly seen extending from low resolution (a) to just below the water ice ring [red arrow in (c) and labels in green therein]. (c) Background-subtracted, folded and smoothed pattern to more clearly show data in (b). *1 labels 160th reflection; *2 data from −10l row line (off-equatorial reflection series) both extend past 4.2 Å.
4. Conclusions and future directions
We have shown that the micro-diffraction instrument on the BioCAT beamline 18-ID allows access to what might be a critical threshold of X-ray beam size as compared with the sizes of crystallites or ordered domains containing fibrous protein structures within biological samples. This has enabled the collection of significantly better X-ray diffraction data than previously possible, in particular the high-angle collagen data being reported here for the first time. Once one has the capability of collecting high-quality X-ray diffraction patterns from micrometer-scale regions, X-ray scanning microscopy becomes practical and this is the most common use of the technology with micro-focus beamlines elsewhere (Paris, 2008 ▶). X-ray scanning microscopy has been used to study the organization of collagen in eyes, teeth, bone, mineral of bone, skin, neurological myelin and to determine the location of amyloid or neurofibrillar tangles in nervous tissue (Landahl et al., 2011 ▶; Meek & Boote, 2009 ▶; Simmons et al., 2013 ▶; Seidel et al., 2008 ▶, 2013 ▶). In many of these studies, diffraction features such as total scattered intensity, presence or absence of specific diffraction features, and/or degree of orientation can be quantified as a function of position in the sample and used to produce an ‘image’ of diffraction or scattering patterns. Since these images originate from an X-ray ‘probe’ (the beam) on the micrometer scale, they can be compared with data from other scanning microscopies, such as X-ray fluorescence microscopy [for which our instrument was originally designed (Barrea et al., 2010 ▶)] or scanning UV/Vis spectroscopy mapping, as was used in the aforementioned study by Landahl et al. (2011 ▶) of tau bodies in brain tissue. Such studies will benefit from a high degree of automation in in-line processing the large volume of diffraction images. These capabilities are being added to the BioCAT micro-diffraction instrument and will be the subject of future reports.
Acknowledgments
Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357. This project was supported by grants (9 P41 GM103622-18) from the National Institute of General Medical Sciences of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Center for Research Resources or the National Institutes of Health.
References
- Antipova, O. & Orgel, J. P. R. O. (2010). J. Biol. Chem. 285, 7087–7096. [DOI] [PMC free article] [PubMed]
- Antipova, O. & Orgel, J. P. R. O. (2012). PLoS ONE, 7, e32241. [DOI] [PMC free article] [PubMed]
- Ayad, S., Boot-Handford, R. P., Humphries, M. J., Kadler, K. E. & Shuttleworth, C. A. (1998). The Extracellular Matrix Facts Book, Vol. 2. New York: Academic Press.
- Barrea, R. A., Davidson, M., Orgel, J., Weng, T., Kondrashkina, E., Gore, D., Heurich, R., Vukonich, R. M. & Irving, T. C. (2005). SPring-8 Users Meeting 2005, SPring-8, Japan.
- Barrea, R. A., Gore, D., Kujala, N., Karanfil, C., Kozyrenko, S., Heurich, R., Vukonich, M., Huang, R., Paunesku, T., Woloschak, G. & Irving, T. C. (2010). J. Synchrotron Rad. 17, 522–529. [DOI] [PMC free article] [PubMed]
- Barrea, R. A., Huang, R., Cornaby, S., Bilderback, D. H. & Irving, T. C. (2009). J. Synchrotron Rad. 16, 76–82. [DOI] [PMC free article] [PubMed]
- Bella, J. (2010). J. Struct. Biol. 170, 377–391. [DOI] [PubMed]
- Bella, J., Eaton, M., Brodsky, B. & Berman, H. M. (1994). Science, 266, 75–81. [DOI] [PubMed]
- Chandrasekaran, R. & Stubbs, G. (2001). International Tables for Crystallography, Vol. F, pp. 444–450. Berlin: Springer.
- Chiti, F. & Dobson, C. M. (2006). Annu. Rev. Biochem. 75, 333–366. [DOI] [PubMed]
- Cloe, A. L., Orgel, J. P. R. O., Sachleben, J. R., Tycko, R. & Meredith, S. C. (2011). Biochemistry, 50, 2026–2039. [DOI] [PMC free article] [PubMed]
- Dejus, R., Lai, B., Khounsary, A., Savoy, R., Moog, L. & Gluskin, E. (1994). Technical Bulletin ANL/APS TB-17. ANL, APS, Argonne, IL, USA.
- Eng, P. J., Newville, M., Rivers, M. L. & Sutton, S. R. (1998). Proc. SPIE, 3449, 145–156.
- Fan, P., Li, M. H., Brodsky, B. & Baum, J. (1993). Biochemistry, 32, 13299–13309. [DOI] [PubMed]
- Fischetti, R., Stepanov, S., Rosenbaum, G., Barrea, R., Black, E., Gore, D., Heurich, R., Kondrashkina, E., Kropf, A. J., Wang, S., Zhang, K., Irving, T. C. & Bunker, G. B. (2004). J. Synchrotron Rad. 11, 399–405. [DOI] [PubMed]
- Inouye, H. & Kirschner, D. A. (1998). J. Struct. Biol. 122, 247–255. [DOI] [PubMed]
- Kadler, K. E., Baldock, C., Bella, J. & Boot-Handford, R. P. (2007). J. Cell Sci. 120, 1955–1958. [DOI] [PubMed]
- Kramer, R. Z., Bella, J., Brodsky, B. & Berman, H. M. (2001). J. Mol. Biol. 311, 131–147. [DOI] [PubMed]
- Lai, B., Khounsary, A., Savoy, R., Moog, L. & Gluskin, E. (1993). Technical Bulletin ANL/APS TB-3. ANL, APS, Argonne, IL, USA.
- Landahl, E., Antipova, O., Bongaarts, A., Barrea, R., Berry, R., Binder, L., Irving, T., Orgel, J., Vana, L. & Rice, S. (2011). Nucl. Instrum. Methods Phys. Res. A, 695, 184–187. [DOI] [PMC free article] [PubMed]
- Malinchik, S. B., Inouye, H., Szumowski, K. E. & Kirschner, D. A. (1998). Biophys. J. 74, 537–545. [DOI] [PMC free article] [PubMed]
- Meek, K. M. & Boote, C. (2009). Prog. Retin. Eye Res. 28, 369–392. [DOI] [PubMed]
- Okuyama, K. (2008). Connect. Tissue Res. 49, 299–310. [DOI] [PubMed]
- Okuyama, K., Takayanagi, M., Ashida, T. & Kakudo, M. (1971). Polym. J. 9, 341–343.
- Orgel, J. P. R. O. & Irving, T. C. (2014). Encycl. Anal. Chem. In the press.
- Orgel, J. P. R. O., Irving, T. C., Miller, A. & Wess, T. J. (2006). Proc. Natl Acad. Sci. USA, 103, 9001–9005. [DOI] [PMC free article] [PubMed]
- Orgel, J. P. R. O., Persikov, A. V. & Antipova, O. (2014). PLos ONE, 9, e89519. [DOI] [PMC free article] [PubMed]
- Orgel, J. P. R. O., San Antonio, J. D. & Antipova, O. (2011). Connect. Tissue Res. 52, 2–17. [DOI] [PubMed]
- Paris, O. (2008). Biointerphases, 3, FB16. [DOI] [PubMed]
- Perumal, S., Antipova, O. & Orgel, J. P. R. O. (2008). Proc. Natl Acad. Sci. USA, 105, 2824–2829. [DOI] [PMC free article] [PubMed]
- Rainey, J. K. & Goh, M. C. (2002). Protein Sci. 11, 2748–2754. [DOI] [PMC free article] [PubMed]
- Rich, A. & Crick, F. H. (1961). J. Mol. Biol. 3, 483–506. [DOI] [PubMed]
- San Antonio, J. D., Parkin JD, Savige, J., Orgel, J. P. & Jacenko, O. (2012). Extracellular Matrix: Pathobiology and Signaling, edited by R. Tenni and N. Karamanos, pp. 575–591. Berlin: DeGruyter Press.
- Seidel, R., Gourrier, A., Burghammer, M., Riekel, C., Jeronimidis, G. & Paris, O. (2008). Micron, 39, 198–205. [DOI] [PubMed]
- Seidel, T., Hammer, N., Garnov, N., Schneider, G. & Steinke, H. (2013). Magn. Reson. Med. 69, 1594–1602. [DOI] [PubMed]
- Simmons, L. M., Montgomery, J., Beaumont, J., Davis, G. R. & Al-Jawad, M. (2013). Arch. Oral Biol. 58, 1726–1734. [DOI] [PubMed]
- Soto, C. (2003). Nat. Rev. Neurosci. 4, 49–60. [DOI] [PubMed]
- Tsuruta, H. & Irving, T. C. (2008). Curr. Opin. Struct. Biol. 18, 601–608. [DOI] [PMC free article] [PubMed]
- Tycko, R., Sciarretta, K. L., Orgel, J. P. R. O. & Meredith, S. C. (2009). Biochemistry, 48, 6072–6084. [DOI] [PMC free article] [PubMed]
- Tycko, R. & Wickner, R. B. (2013). Acc. Chem. Res. 46, 1487–1496. [DOI] [PMC free article] [PubMed]
- Vera, L. & Stura, E. A. (2014). Cryst. Growth Des. 14, 427–435.
- Vestergaard, B., Groenning, M., Roessle, M., Kastrup, J. S., van de Weert, M., Flink, J. M., Frokjaer, S., Gajhede, M. & Svergun, D. I. (2007). PLoS Biol. 5, e134. [DOI] [PMC free article] [PubMed]
- Warkentin, M., Berejnov, V., Husseini, N. S. & Thorne, R. E. (2006). J. Appl. Cryst. 39, 805–811. [DOI] [PMC free article] [PubMed]
- Weissmann, C. (2004). Nat. Rev. Microbiol. 2, 861–871. [DOI] [PubMed]