

Abstract
Although quinolones have been in clinical use for decades, the mechanism underlying drug activity and resistance has remained elusive. However, recent studies indicate that clinically relevant quinolones interact with Bacillus anthracis (Gram-positive) topoisomerase IV through a critical water–metal ion bridge and that the most common quinolone resistance mutations decrease drug activity by disrupting this bridge. As a first step toward determining whether the water–metal ion bridge is a general mechanism of quinolone–topoisomerase interaction, we characterized drug interactions with wild-type Escherichia coli (Gram-negative) topoisomerase IV and a series of ParC enzymes with mutations (S80L, S80I, S80F, and E84K) in the predicted bridge-anchoring residues. Results strongly suggest that the water–metal ion bridge is essential for quinolone activity against E. coli topoisomerase IV. Although the bridge represents a common and critical mechanism that underlies broad-spectrum quinolone function, it appears to play different roles in B. anthracis and E. coli topoisomerase IV. The water–metal ion bridge is the most important binding contact of clinically relevant quinolones with the Gram-positive enzyme. However, it primarily acts to properly align clinically relevant quinolones with E. coli topoisomerase IV. Finally, even though ciprofloxacin is unable to increase levels of DNA cleavage mediated by several of the Ser80 and Glu84 mutant E. coli enzymes, the drug still retains the ability to inhibit the overall catalytic activity of these topoisomerase IV proteins. Inhibition parallels drug binding, suggesting that the presence of the drug in the active site is sufficient to diminish DNA relaxation rates.
Ciprofloxacin and other quinolones kill bacteria by increasing levels of DNA strand breaks generated by type II topoisomerases.1−6 Nearly all bacteria encode two type II topoisomerases, gyrase and topoisomerase IV.2,6−14 Both enzymes are comprised of two protomer subunits and have an A2B2 quaternary structure.2,6−9,11,12,15 Gyrase consists of two GyrA subunits (that contain the active site tyrosine residues that mediate DNA cleavage and ligation) and two GyrB subunits (that bind ATP, which is required for overall catalytic activity). Topoisomerase IV consists of two ParC and two ParE subunits that are homologous to GyrA and GyrB, respectively.6−9,12 Gyrase and topoisomerase IV alter DNA topology by passing an intact double helix through a transient break that they generate in a separate segment of DNA.6−9,11−13,15 Both type II enzymes are essential for cell survival6−9,11−13 and are physiological targets for quinolone antibacterials.1,6,16−22
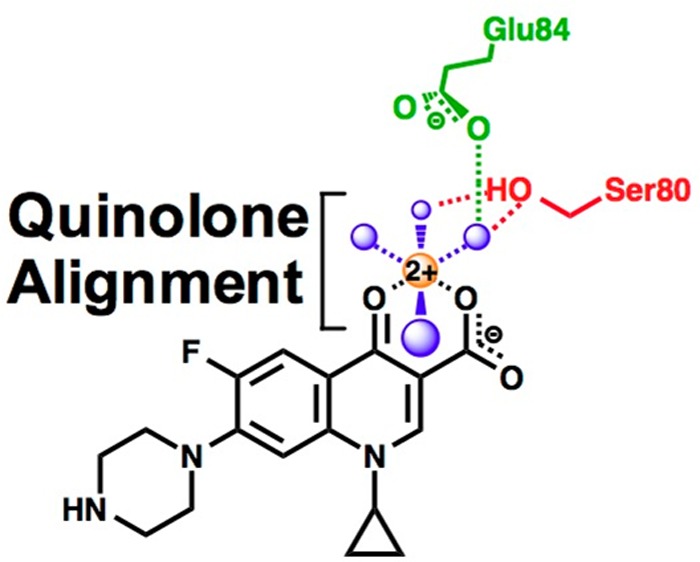
Although quinolones have been in clinical use against both Gram-positive and Gram-negative bacteria for several decades, the mechanism by which they interact with bacterial type II topoisomerases was determined only recently.23−29 Structural work with Acinetobacter baumannii topoisomerase IV suggests that the quinolone C3/C4 keto acid chelates a divalent metal ion, which interacts with the protein through water molecules that are coordinated by two highly conserved residues: a serine and an acidic amino acid located four positions downstream.26 Functional studies demonstrated that the proposed “water–metal ion bridge” exists and acts as the primary conduit by which clinically relevant quinolones interact with Bacillus anthracis (Gram-positive) topoisomerase IV (Figure 1).27−29 Partial disruption of the bridge, resulting from mutation of the serine or acidic residue, significantly decreased the potency (i.e., affinity) of clinically relevant quinolones for the enzyme but had relatively little effect on drug efficacy (i.e., maximal drug-induced cleavage).27,28 Mutation of both amino acid residues, which completely disrupted bridge function, abrogated the ability of quinolones to increase topoisomerase IV-mediated DNA cleavage.28
Figure 1.
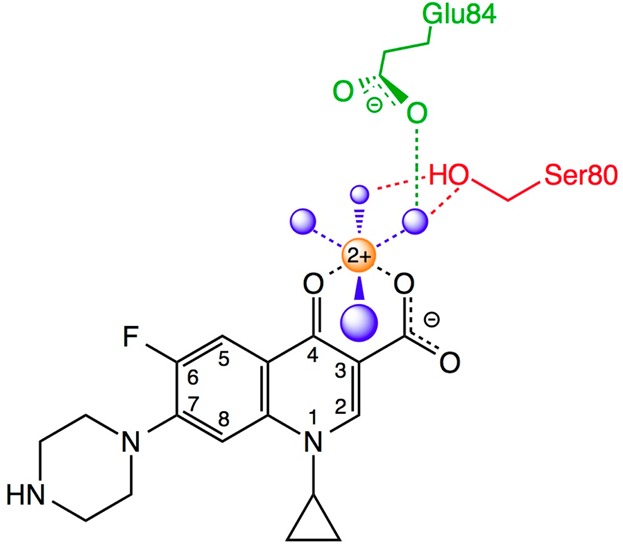
Schematic of the water–metal ion bridge that mediates quinolone–topoisomerase IV interaction. For the sake of simplicity, only interactions with the protein are shown. The residue numbering is that of Escherichia coli topoisomerase IV. Ciprofloxacin is colored black. Water molecules are colored blue. Mg2+ is colored orange. The coordinating serine and glutamic acid residues are colored red and green, respectively. Blue dashed lines indicate the octahedral coordination sphere of the divalent metal ion interacting with four water molecules and the C3/C4 keto acid of the quinolone. The red dashed lines represent hydrogen bonds between the serine side chain hydroxyl group and two of the water molecules. The green dashed line represents a hydrogen bond between the glutamic acid side chain carboxyl group and one of the water molecules. Adapted from refs (26) and (28).
Quinolone resistance has risen steadily since the 1990s and is a threat to the continued clinical use of this drug class.5,6,30 Resistance is most often associated with specific mutations in topoisomerase IV and/or gyrase.1−3,5,6,17 Overwhelmingly, the most commonly mutated amino acid residues in Gram-positive and Gram-negative quinolone-resistant strains are the conserved serine (originally described as Ser83 in Escherichia coli GyrA31,32) and acidic residue that anchor the water–metal ion bridge.1−3,5,6,17,33−41 The role of these residues in drug resistance underscores the importance of the bridge in mediating the clinical effects of quinolones.
Despite the broad-spectrum nature of quinolones, the ability (both potency and efficacy) of these drugs to enhance DNA cleavage mediated by topoisomerase IV and gyrase can differ substantially between species. Furthermore, while mutation of the serine and acidic residue is consistent across species, the substituted amino acids vary.1−3,5,6,17,33−41 For example, the most common resistance mutation is Ser → Phe in B. anthracis topoisomerase IV,35−37 but it is Ser → Ile in the E. coli enzyme.38−41
The differences in quinolone activity against bacterial type II enzymes raise the question of whether the water–metal ion bridge is utilized or functions in the same manner across species. Therefore, to address this issue, the interactions of ciprofloxacin and related drugs (quinazolinediones) with wild-type E. coli topoisomerase IV and a series of enzymes with mutations in the predicted bridge-anchoring residues (ParC Ser80 and Glu84) were analyzed. Results provide strong evidence that the water–metal ion bridge facilitates interaction between ciprofloxacin and the Gram-negative enzyme and is critical for quinolone activity. However, this interaction appears to play different roles in mediating quinolone activity in different species. Whereas the bridge facilitates the binding of clinically relevant quinolones to B. anthracis topoisomerase IV, it appears to properly align the drug in the active site of the E. coli enzyme to promote DNA cleavage.
Experimental Procedures
Enzymes and Materials
E. coli topoisomerase IV subunits (wild-type ParC, wild-type ParE, ParCS80L, ParCS80I, ParCS80F, and ParCE84K) were expressed and purified as described previously.42,43 The ParC mutants were prepared by site-directed mutagenesis using a QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) and custom-synthesized primers containing the desired mutations. The entire coding region was sequenced (Macrogen USA) to ensure the presence of the desired mutation and the absence of unwanted alterations. In all assays, topoisomerase IV was used as a 1:1 ParC:ParE mixture.
Negatively supercoiled pBR322 plasmid DNA was prepared from E. coli using a Plasmid Mega Kit (Qiagen) as described by the manufacturer. Relaxed pBR322 plasmid DNA was generated by treatment with topoisomerase I for 30 min as described previously,44 followed by phenol/chloroform/isoamyl alcohol extraction, ethanol precipitation, and resuspension in 5 mM Tris-HCl (pH 8.5) and 500 μM ethylenediaminetetraacetic acid (EDTA). Histone H1 was obtained from Boehringer Mannheim.
Ciprofloxacin was obtained from LKT Laboratories, stored at −20 °C as a 40 mM stock solution in 0.1 N NaOH, and diluted 5-fold with 10 mM Tris-HCl (pH 7.9) immediately prior to use. 3-Amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-8-methyl-2,4(1H,3H)-quinazolinedione (UIJR-1-048) was synthesized using established methods as previously reported.45 For the sake of simplicity, this compound will be referred to as 8-methyl-3′-(AM)P-dione. 3-Amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-2,4(1H,3H)-quinazolinedione (UIHS-IIa-245) and 7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-8-methyl-2,4(1H,3H)-quinazolinedione (UILI-2-97) were synthesized as previously reported.29 For the sake of simplicity, these drugs will be referred to as 8-H-3′-(AM)P-dione and 8-methyl-3′-(AM)P-non-amino-dione, respectively. The quinazolinediones were stored at 4 °C as 20 mM stock solutions in 100% DMSO. All other chemicals were analytical reagent grade.
DNA Relaxation
DNA relaxation assays were based on the protocol of Fortune and Osheroff.27,46 Reaction mixtures (20 μL) contained 10 nM wild-type or mutant E. coli topoisomerase IV and 5 nM negatively supercoiled pBR322 in relaxation buffer [40 mM HEPES (pH 7.6), 100 mM potassium glutamate, 10 mM Mg(OAc)2, 50 mM NaCl, and 1 mM ATP] and were incubated at 37 °C. Relaxation was stopped at times ranging from 0 to 30 min by the addition of 3 μL of 0.77% sodium dodecyl sulfate (SDS) and 77.5 mM EDTA. Samples were mixed with 2 μL of agarose gel loading buffer [60% sucrose, 10 mM Tris-HCl (pH 7.9), 0.5% bromophenol blue, and 0.5% xylene cyanol FF], heated at 45 °C for 5 min, and subjected to electrophoresis in 1% agarose gels in 100 mM Tris-borate (pH 8.3) and 2 mM EDTA. Gels were stained with 0.75 μg/mL ethidium bromide for 30 min. DNA bands were visualized with medium-range ultraviolet light and quantified using an Alpha Innotech digital imaging system. The percent relaxed DNA was determined by the loss of supercoiled DNA substrate.
Reactions that monitored the effects of ciprofloxacin on the relaxation activities of wild-type or mutant E. coli topoisomerase IV were incubated for 10 min (wild-type, ParCS80L, and ParCS80F), 20 min (ParCS80I), or 30 min (ParCE84K). The longer times utilized with the last two mutant enzymes reflect their slower basal rates of DNA relaxation. The amount of relaxed DNA observed in the absence of drug was set to 100% to facilitate direct comparisons between the effects of ciprofloxacin on the catalytic activities of the wild-type and mutant enzymes.
DNA Catenation
Catenation assays were based on the protocol of Fortune and Osheroff47as modified by Aldred et al.28 Reaction mixtures (20 μL) contained 20 nM wild-type or mutant E. coli topoisomerase IV and 5 nM relaxed pBR322 in relaxation buffer containing 25 mM NaCl (rather than 50 mM) supplemented with 5 μg/mL histone H1 and were incubated at 37 °C. Catenation was stopped at times ranging from 0 to 30 min by the addition of 2 μL of 250 mM EDTA (pH 8.0) followed by 2 μL of 1.25% SDS. Samples were mixed with 2 μL of agarose gel loading buffer, heated at 45 °C for 5 min, and subjected to electrophoresis in 1% agarose gels in 100 mM Tris-borate (pH 8.3) and 2 mM EDTA containing 0.5 μg/mL ethidium bromide. DNA bands were visualized and quantified as described above. The percent catenated DNA was determined by the loss of relaxed monomers.
DNA Cleavage
DNA cleavage reactions were carried out using the procedure of Fortune and Osheroff.27,46 Reaction mixtures contained 10 nM wild-type, ParCS80L, ParCS80I, or ParCS80F or 25 nM ParCE84K topoisomerase IV and 10 nM negatively supercoiled pBR322 in a total of 20 μL of cleavage buffer [40 mM Tris-HCl (pH 7.9), 2.5 mM MgCl2, 50 mM NaCl, and 2.5% (v/v) glycerol]. ParCE84K was used at an increased concentration to account for its lower basal cleavage activity, so that direct comparisons could be made with the wild-type and Ser80 mutant enzymes. In some reactions, the concentration dependence of MgCl2 was examined or the divalent metal ion was replaced with CaCl2, ZnCl2, CdCl2, MnCl2, or NiCl2. Reaction mixtures were incubated at 37 °C for 10 min, and enzyme–DNA cleavage complexes were trapped by the addition of 2 μL of 5% SDS followed by 2 μL of 250 mM EDTA (pH 8.0). Proteinase K (2 μL of a 0.8 mg/mL solution) was added, and samples were incubated at 45 °C for 45 min to digest the enzyme. Samples were mixed with 2 μL of agarose gel loading buffer, heated at 45 °C for 5 min, and subjected to electrophoresis in 1% agarose gels in 40 mM Tris-acetate (pH 8.3) and 2 mM EDTA containing 0.5 μg/mL ethidium bromide. DNA bands were visualized and quantified as described above. DNA cleavage was monitored by the conversion of supercoiled plasmid to linear molecules.
Assays that monitored the DNA cleavage activities of wild-type and mutant E. coli topoisomerase IV in the absence of drugs substituted 2.5 mM CaCl2 for 2.5 mM MgCl2 in the cleavage buffer.
For assays that monitored competition between ciprofloxacin and 8-methyl-3′-(AM)P-dione, the level of DNA cleavage generated by the corresponding concentration of ciprofloxacin in the absence of the quinazolinedione was used as a baseline and was subtracted from the cleavage level seen in the presence of both compounds. Ciprofloxacin and 8-methyl-3′-(AM)P-dione were added simultaneously to reaction mixtures.
DNA Religation
DNA religation assays were carried out using the procedure of Robinson and Osheroff.27,48 Reaction mixtures (20 μL) were identical to those described above for cleavage assays. Initial DNA cleavage/religation equilibria with wild-type or mutant ParCS80LE. coli topoisomerase IV were established at 37 °C for 10 min. Religation was initiated by rapidly shifting the temperature from 37 to 75 °C. Reactions were stopped at times ranging from 0 to 135 s by the addition of 2 μL of 5% SDS followed by 2 μL of 250 mM EDTA (pH 8.0). Samples were digested with proteinase K and processed as described above for DNA cleavage assays. Levels of DNA cleavage were set to 100% at time zero, and religation was determined by the loss of the linear reaction product over time.
Persistence of Topoisomerase IV–DNA Cleavage Complexes
The persistence of topoisomerase IV–DNA cleavage complexes established in the presence of drugs was determined using the procedure of Gentry et al.27,49 Initial reaction mixtures contained 50 nM wild-type or mutant ParCS80LE. coli topoisomerase IV, 50 nM DNA, and 20 μM ciprofloxacin or 8-methyl-3′-(AM)P-dione in a total of 20 μL of DNA cleavage buffer. Reaction mixtures were incubated at 37 °C for 10 min and then diluted 20-fold with DNA cleavage buffer warmed to 37 °C. Samples (20 μL) were removed at times ranging from 0 to 30 min, and DNA cleavage was stopped with 2 μL of 5% SDS followed by 2 μL of 250 mM EDTA (pH 8.0). Samples were digested with proteinase K and processed as described above for DNA cleavage assays. Levels of DNA cleavage were set to 100% at time zero, and the persistence of cleavage complexes was determined by the loss of the linear reaction product over time.
Results
Enzymatic Activities of Wild-Type and Mutant Quinolone-Resistant E. coli Topoisomerase IV
All of the functional evidence for the existence and role of the water–metal ion bridge in mediating quinolone activity comes from studies of B. anthracis (a Gram-positive species) topoisomerase IV.27,28 As a first step toward determining whether the water–metal ion bridge is a general mechanism of quinolone–topoisomerase interaction, we characterized the interactions of drugs with wild-type, ParCS80L, ParCS80I, ParCS80F, and ParCE84K topoisomerase IV from E. coli (a Gram-negative species). All of these mutations occur at the amino acid residues predicted (by sequence alignments) to anchor the water–metal ion bridge in this species. The S80L, S80I, and E84K mutations have been identified in clinical isolates of quinolone-resistant E. coli.38−41 The S80L and E84K mutants are resistant to quinolones in vitro.(16,50,51) However, the purified ParCS80I enzyme (which contains the most common Ser80 mutation found in clinical isolates38−41) has not yet been characterized. The S80F mutation parallels the most common quinolone resistance mutation seen in B. anthracis(35−37) and provides for direct comparisons with the previously characterized Gram-positive enzyme.27,28
Quinolones require the activity of type II topoisomerases in order to kill cells.1−6 Consequently, topoisomerase IV mutations could cause quinolone resistance either by altering drug–enzyme interactions or by diminishing enzyme activity. Therefore, wild-type, ParCS80L, ParCS80I, ParCS80F, and ParCE84KE. coli topoisomerase IV were examined for their abilities to relax, catenate, and cleave DNA in the absence of drugs (Figure 2).
Figure 2.
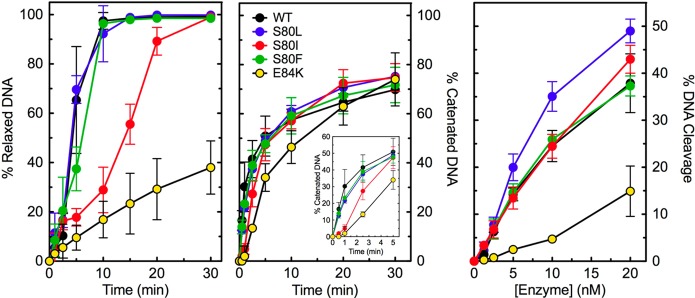
DNA relaxation, catenation, and cleavage activities of wild-type and mutant E. coli topoisomerase IV enzymes in the absence of drugs. The abilities of wild-type (WT, black), ParCS80L (S80L, blue), ParCS80I (S80I, red), ParCS80F (S80F, green), and ParCE84K (E84K, yellow) topoisomerase IV to relax (left), catenate (middle), and cleave (right) negatively supercoiled plasmid DNA are shown. The inset (middle) highlights the catenation activity of the enzymes at early time points. For relaxation and cleavage, error bars represent the standard deviation of three or more independent experiments. For catenation, error bars represent the standard error of the mean of two independent experiments.
ParCS80L and ParCS80F topoisomerase IV relaxed negatively supercoiled plasmid DNA and catenated relaxed plasmids at a rate that was nearly identical to that of the wild-type enzyme. Unexpectedly, ParCS80I relaxed negatively supercoiled plasmid DNA ∼3-fold slower than did the wild-type enzyme. This mutant also catenated relaxed plasmids slower than did the wild-type enzyme. ParCE84K relaxed and catenated DNA at a rate that was even slower than those of the wild-type enzyme and the Ser80 mutants. This mutation, as well as the equivalent mutation (E85K) in B. anthracis topoisomerase IV,28 has been shown to display decreased rates of DNA relaxation and decatenation.50 In the case of the B. anthracis topoisomerase IV E85K mutant enzyme, the decreased catalytic activity was due to a defect in the DNA strand passage event, which takes place following DNA cleavage.28
The wild-type and Ser80 mutant E. coli enzymes all displayed similar abilities to cleave DNA, suggesting that quinolone resistance conferred by these mutations results from altered drug–enzyme interactions rather than decreased enzyme activity. In contrast to results with the B. anthracis E85K mutant enzyme,28E. coli ParCE84K topoisomerase IV displayed a DNA cleavage ability that was approximately half that of the wild-type enzyme. Therefore, this topoisomerase IV mutation may cause quinolone resistance in E. coli cells by affecting the activity of the enzyme in addition to (or instead of) altering drug activity.
Effects of Ciprofloxacin and 8-Methyl-3′-(AM)P-dione on Wild-Type and Quinolone-Resistant E. coli Topoisomerase IV
To determine how the ParC Ser80 and Glu84 mutations cause quinolone resistance, the ability of ciprofloxacin to enhance DNA cleavage mediated by the wild-type and mutant E. coli enzymes was assessed (Figure 3, left). The quinolone displayed little ability to increase the level of DNA cleavage mediated by ParCS80L, ParCS80I, ParCS80F, and ParCE84K as compared to that of the wild-type enzyme. In contrast to results with the equivalent B. anthracis enzymes,27,28 the mutant E. coli topoisomerase IV enzymes displayed little to no increase in ciprofloxacin-enhanced DNA cleavage at high drug concentrations (Figure 3, right).
Figure 3.
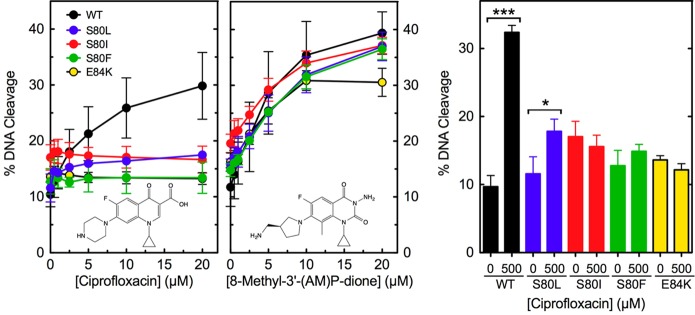
Effects of ciprofloxacin and 8-methyl-3′-(AM)P-dione on the DNA cleavage activities of wild-type and mutant E. coli topoisomerase IV enzymes. DNA cleavage mediated by wild-type (WT, black), ParCS80L (S80L, blue), ParCS80I (S80I, red), ParCS80F (S80F, green), and ParCE84K (E84K, yellow) topoisomerase IV in the presence of the quinolone (left) or quinazolinedione (middle) is shown. Drug structures are shown in the respective panels. The bar graph (right) shows the level of cleavage induced by the enzymes in the presence of 500 μM ciprofloxacin. The level of cleavage induced by each enzyme in the absence of drug is shown for comparison. Error bars represent the standard deviation of three or more independent experiments. ***p < 0.0001; *p = 0.02.
Some quinazolinediones, including 8-methyl-3′-(AM)P-dione, have been shown to maintain activity against quinolone-resistant type II enzymes and function in a manner that is independent of the water–metal ion bridge.27−29,45,52−55 Therefore, this drug was tested against wild-type and mutant ParCS80L, ParCS80I, ParCS80F, and ParCE84K topoisomerase IV. All of the enzymes maintained high sensitivity to 8-methyl-3′-(AM)P-dione (Figure 3, middle). Thus, as reported previously for B. anthracis topoisomerase IV,27,28 mutations in amino acid residues predicted to anchor the water–metal ion bridge caused resistance that was quinolone-specific.
Next, experiments were carried out to determine whether the quinolone and quinazolinedione increase the concentration of E. coli topoisomerase IV cleavage complexes by inhibiting enzyme-mediated DNA religation (Figure 4). Because ciprofloxacin was not able to enhance DNA cleavage mediated by ParCS80I, ParCS80F, or ParCE84K topoisomerase IV, only the wild-type and ParCS80L enzymes were examined. Although less dramatic than results with the Gram-positive B. anthracis(27,28) and Staphylococcus aureus(56,57) topoisomerase IV enzymes, ciprofloxacin and 8-methyl-3′-(AM)P-dione both slowed the DNA religation rate of the wild-type enzyme ∼4–5-fold. A similar effect of ciprofloxacin on the religation rate of the wild-type enzyme has been reported.58 As expected, the t1/2 for DNA religation mediated by ParCS80L in the presence of ciprofloxacin was significantly decreased, and the rate of religation was nearly indistinguishable from that observed in the absence of drug. In contrast, the quinazolinedione slowed the rate of DNA religation mediated by both wild-type and ParCS80LE. coli topoisomerase IV. Consistent with effects of drugs on DNA cleavage, these results imply that the ParCS80L resistance mutation impairs quinolone, but not quinazolinedione, function. Similar results were reported for B. anthracis enzymes containing mutations at the equivalent serine residue.27
Figure 4.
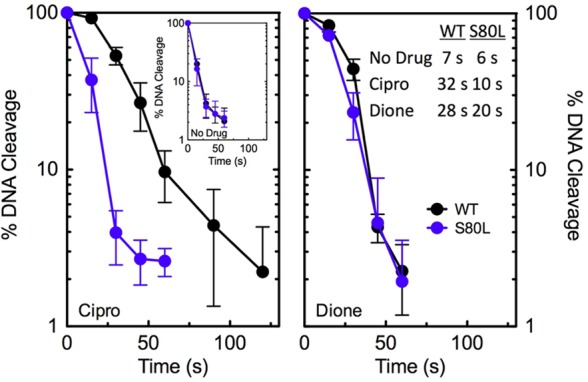
Effects of ciprofloxacin and 8-methyl-3′-(AM)P-dione on DNA religation mediated by wild-type and ParCS80LE. coli topoisomerase IV. Rates of DNA religation for wild-type (WT, black) and ParCS80L (S80L, blue) topoisomerase IV in the absence of drugs (inset, left) or in the presence of ciprofloxacin (left) or 8-methyl-3′-(AM)P-dione (right) are shown. The data table (inset, right) lists the t1/2 values of DNA religation mediated by the enzymes under each condition. In all assays, drugs were used at a concentration of 20 μM. Levels of DNA cleavage at time zero were set to 100%, and results were quantified by monitoring the loss of double-stranded DNA breaks over time. Error bars represent the standard deviation of three or more independent experiments.
To further characterize the effects of drugs on DNA cleavage mediated by E. coli topoisomerase IV, the persistence of cleavage complexes was determined (Figure 5). Once again, the only mutant enzyme examined was ParCS80L topoisomerase IV. DNA cleavage complexes formed with wild-type and mutant E. coli topoisomerase IV were significantly less stable than those formed with the equivalent B. anthracis enzymes.27,28 The t1/2 values for the decay of wild-type E. coli topoisomerase IV cleavage complexes were ∼4.4 and ∼5.3 min in the presence of ciprofloxacin and 8-methyl-3′-(AM)P-dione, respectively. The t1/2 values of wild-type B. anthracis cleavage complexes in the presence of these drugs were ∼90 and 280 min, respectively.27 The t1/2 values of cleavage complexes formed with ParCS80L topoisomerase IV in the presence of both ciprofloxacin and 8-methyl-3′-(AM)P-dione decreased severalfold as compared to that of the wild-type enzyme (Figure 5, inset). However, a much larger reduction (∼22-fold vs ∼6-fold) was seen with the quinolone. In contrast, with B. anthracis topoisomerase IV, mutations at the equivalent serine decreased the stability of quinolone-induced cleavage complexes but had little effect on the stability of those formed in the presence of the quinazolinedione.27
Figure 5.
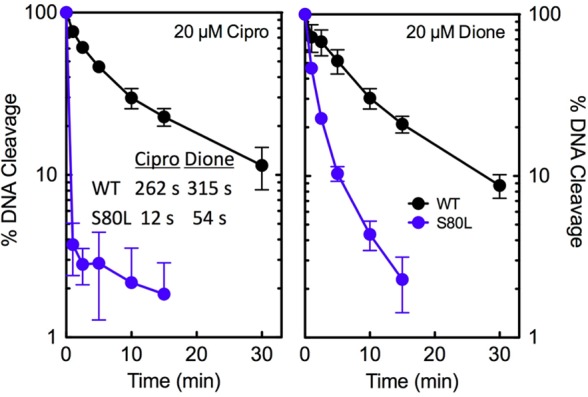
Effects of ciprofloxacin and 8-methyl-3′-(AM)P-dione on the persistence of cleavage complexes formed by wild-type and ParCS80LE. coli topoisomerase IV. The persistence of ternary enzyme–drug–DNA cleavage complexes formed with the wild-type (WT, black) and mutant (S80L, blue) enzymes in the presence of the quinolone or the quinazolinedione is shown (left and right, respectively). The data table (inset, left) lists the t1/2 values of DNA cleavage complexes formed with each drug–enzyme combination. Initial DNA cleavage–religation reactions were allowed to reach equilibrium, and the mixtures were then diluted 20-fold with DNA cleavage buffer. In all assays, drugs were used at a concentration of 20 μM. Levels of DNA cleavage at time zero were set to 100%, and results were quantified by monitoring the loss of double-stranded DNA breaks over time. Error bars represent the standard deviation of three or more independent experiments.
Correlation between Quinolone Affinity and the Inhibition of DNA Relaxation Catalyzed by Wild-Type and Mutant E. coli Enzymes
Because ciprofloxacin displayed little ability to enhance DNA cleavage mediated by ParCS80L, ParCS80I, ParCS80F, and ParCE84K topoisomerase IV even up to 500 μM drug (see Figure 3), a competition experiment was carried out to determine whether quinolone resistance was due to a lack of drug interaction. In this experiment, the ability of ciprofloxacin (0–200 μM) to compete out DNA cleavage induced by 10 μM 8-methyl-3′-(AM)P-dione was determined (Figure 6, left). With ParCS80I and ParCS80F, ciprofloxacin competed well against the quinazolinedione, decreasing the level of cleavage by 50% at ∼12 and ∼17 μM quinolone, respectively. Because these IC50 values are similar to the concentration of quinazolinedione used in the competition assay, it appears that the decreased activity of ciprofloxacin toward these two E. coli mutant enzymes does not result from a loss of drug affinity. This finding is in contrast to previous results with B. anthracis S81F topoisomerase IV. With the B. anthracis enzyme, a 7.5-fold molar excess of ciprofloxacin reduced the activity of 8-methyl-3′-(AM)P-dione by <40%, indicating a marked decrease in quinolone binding.27
Figure 6.
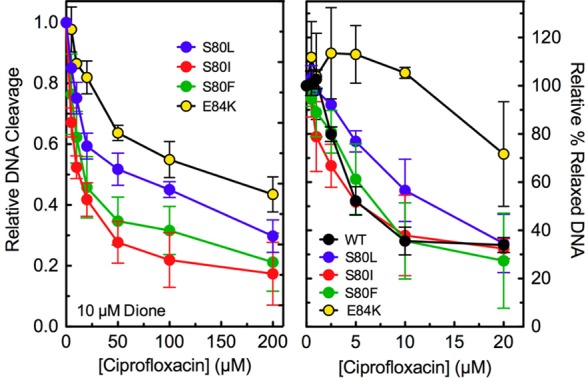
Competition between ciprofloxacin and 8-methyl-3′-(AM)P-dione and the effects of ciprofloxacin on DNA relaxation. The ability of 0–200 μM ciprofloxacin to compete with 10 μM 8-methyl-3′-(AM)P-dione for ParCS80L (S80L, blue), ParCS80I (S80I, red), ParCS80F (S80F, green), and ParCE84K (E84K, yellow) E. coli topoisomerase IV was determined using DNA cleavage assays (left). Both drugs were added to reaction mixtures simultaneously. For ParCS80L, the level of cleavage seen in the presence of ciprofloxacin alone was used as a baseline and subtracted from the total observed in the presence of both drugs. The level of DNA cleavage observed in the presence of only the quinazolinedione was set to 1.0 to facilitate direct comparisons. The effects of ciprofloxacin on the relaxation activities of wild-type (WT, black), ParCS80L (S80L, blue), ParCS80I (S80I, red), ParCS80F (S80F, green), and ParCE84K (E84K, yellow) topoisomerase IV are shown at the right. Wild-type, ParCS80L, and ParCS80F relaxation reactions were stopped at 10 min, ParCS80I reactions at 20 min, and ParCE84K reactions at 30 min to account for the different baseline DNA relaxation rates of the different enzymes. The amount of relaxed DNA observed in the absence of drug (“0” point) was set to 100% to facilitate direct comparisons. Error bars represent the standard deviation of three or more independent experiments.
In contrast to the results described above, the ability of ciprofloxacin to compete with the quinazolinedione was significantly impaired with ParCS80L and ParCE84K (Figure 6, left). IC50 values with these mutant enzymes were ∼65 and ∼143 μM quinolone, respectively. These results indicate that the S80L and E84K mutations have a significant effect on the affinity of ciprofloxacin for the E. coli topoisomerase IV–DNA complex.
Although quinolones are believed to kill bacterial cells primarily by increasing levels of DNA strand breaks, they also can impair cellular functions by inhibiting type II topoisomerases from carrying out their critical physiological functions. This catalytic inhibition may contribute to their toxicity toward bacterial cells.1−6 Therefore, the effects of ciprofloxacin on DNA relaxation mediated by ParCS80L, ParCS80I, ParCS80F, and ParCE84K topoisomerase IV were compared to those on DNA relaxation mediated by the wild-type enzyme (Figure 6, right).
Even though ciprofloxacin did not enhance DNA cleavage mediated by ParCS80I and ParCS80F topoisomerase IV (see Figure 3), the drug inhibited DNA relaxation catalyzed by the mutant enzymes with an IC50 value (∼5 μM) that was the same as that observed with wild-type E. coli topoisomerase IV. The level of inhibition was correspondingly reduced with ParCS80L and ParCE84K topoisomerase IV, which displayed lower affinities for ciprofloxacin (as determined by the competition assays described above). A similar effect of ciprofloxacin on DNA relaxation catalyzed by the mutant S80L and E84K enzymes has been reported.50 These results strongly suggest that the ability of quinolones to inhibit the overall catalytic activity of topoisomerase IV reflects drug binding in the DNA cleavage/religation active site of the enzyme. However, inhibition of DNA relaxation appears to be unrelated to the ability of quinolones to enhance enzyme-mediated DNA cleavage.
Role of the Water–Metal Ion Bridge in Facilitating Quinolone Activity against E. coli Topoisomerase IV
To determine whether quinolone interactions with E. coli topoisomerase IV are mediated by the water–metal ion bridge,26−28 three experiments were carried out to analyze the metal ion requirements of ciprofloxacin-induced and 8-methyl-3′-(AM)P-dione-induced DNA cleavage.
In the first experiment, we examined the metal ion requirement for quinolone-induced DNA cleavage with wild-type E. coli topoisomerase IV. Thus, a variety of divalent and trivalent metal ions that support enzyme function were tested for their ability to support ciprofloxacin activity. Three metal ions (Ca2+, Zn2+, and Cd2+) that supported enzyme-mediated DNA cleavage but did not support quinolone activity against the enzyme were identified (Figure 7). As a control, Ca2+, Zn2+, and Cd2+ were tested with 8-methyl-3′-(AM)P-dione, a metal ion-independent drug.26−28 In contrast to ciprofloxacin, the quinazolinedione maintained activity against the enzyme, regardless of the metal ion utilized. These findings suggest that clinically relevant quinolones such as ciprofloxacin require divalent metal ions to induce DNA cleavage mediated by E. coli topoisomerase IV.
Figure 7.
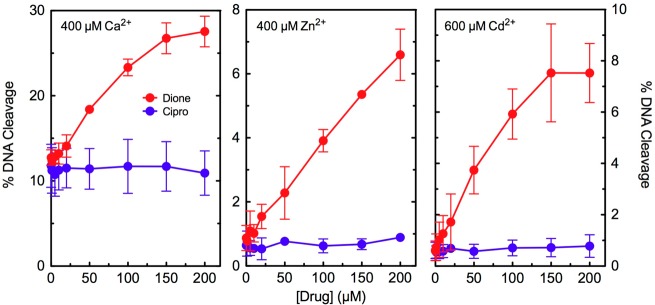
Effects of alternative metal ions on drug-induced DNA cleavage mediated by wild-type E. coli topoisomerase IV. Assays were carried out with ciprofloxacin (Cipro, blue) or 8-methyl-3′-(AM)P-dione (Dione, red). The indicated divalent metal ions were substituted for the Mg2+ used in standard assays. The concentrations of Cd2+ (600 μM, right) and Zn2+ (400 μM, middle) that were utilized gave maximal enzyme-mediated DNA cleavage activity. Ca2+ was utilized at a concentration of 400 μM to minimize the basal level of cleavage seen in the absence of drug (left). Error bars represent the standard deviation of three or more independent experiments.
In the second experiment, we determined whether a mutation in ParC Ser80 of E. coli topoisomerase IV altered metal ion utilization by ciprofloxacin. Only metal ions that could be used at millimolar concentrations were assessed to ensure that catalytic and noncatalytic sites were saturated. We found that Ni2+ could support low-level ciprofloxacin activity against the wild-type enzyme but not against the ParCS80L mutant (Figure 8). A similar result was seen with Mn2+ (not shown). As described above, 8-methyl-3′-(AM)P-dione was examined as a control. Ni2+ and Mn2+ supported the activity of the quinazolinedione against both enzymes (Figure 8 and not shown). These results indicate that a mutation at Ser80 restricts the variety of metal ions that ciprofloxacin can utilize to enhance DNA cleavage, suggesting that the residue plays a role in anchoring the water–metal ion bridge that coordinates the quinolone to E. coli topoisomerase IV.
Figure 8.
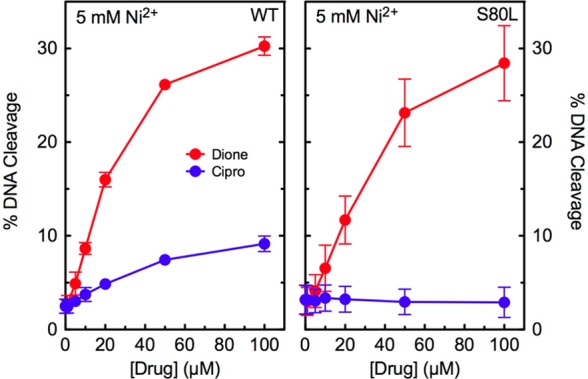
Effects of Ni2+ on drug-induced DNA cleavage mediated by wild-type and ParCS80LE. coli topoisomerase IV. Results are shown for cleavage mediated by the wild-type (WT, left) and ParCS80L (S80L, right) enzymes in the presence of ciprofloxacin (blue) or 8-methyl-3′-(AM)P-dione (red). Assays included 5 mM Ni2+, the concentration that yielded maximal enzyme activity. Error bars represent the standard deviation of three or more independent experiments.
In the third experiment, Mg2+ titrations were carried out to determine whether mutation of a bridge-anchoring residue alters the affinity of the coordinating metal ion. DNA cleavage induced by the quinolone and quinazolinedione displayed a similar requirement for Mg2+ when wild-type topoisomerase IV was used (Figure 9). In contrast, the Mg2+ requirements for DNA cleavage mediated by ParCS80L were different with the two drugs. While the metal ion utilization for 8-methyl-3′-(AM)P-dione closely resembled that seen with the wild-type enzyme, ciprofloxacin required higher levels of Mg2+ to support DNA cleavage (Figure 9). The requirement for higher Mg2+ concentrations to support quinolone- but not quinazolinedione-induced DNA cleavage by the mutant enzyme provides further evidence that interactions between quinolones (but not quinazolinediones) and E. coli topoisomerase IV are coordinated by a metal ion.
Figure 9.
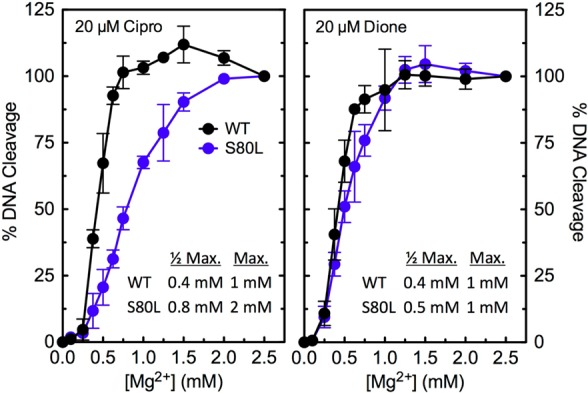
Effects of Mg2+ concentration on DNA cleavage mediated by wild-type and ParCS80L topoisomerase IV from E. coli. Results are shown for ciprofloxacin (Cipro, left) and 8-methyl-3′-(AM)P-dione (Dione, right) with the wild-type (WT, black) and ParCS80L (S80L, blue) enzymes. Drugs were used at a concentration of 20 μM. DNA cleavage for each drug–enzyme pair was normalized to 100% at 2.5 mM Mg2+ to facilitate direct comparisons. The data tables (insets) list the concentrations of Mg2+ required to achieve half-maximal and maximal DNA cleavage with each drug–enzyme pair. Error bars represent the standard deviation of three or more independent experiments.
Taken together, the results described above provide strong evidence for the existence and use of the water–metal ion bridge to mediate interactions between ciprofloxacin and E. coli topoisomerase IV. They also suggest that Ser80 plays a critical role in anchoring the bridge that coordinates the quinolone to the Gram-negative enzyme and that quinolone resistance that accompanies mutation of this residue is caused by a disruption or alteration of the bridge. Similar conclusions have been reported for B. anthracis topoisomerase IV.27,28
Contribution of the N3 and C8 Substituents to Quinazolinedione Activity against E. coli Topoisomerase IV
A C7 3′-(AM)P substituent on quinazolinediones contributes significantly to the activity of these drugs against quinolone-resistant enzymes. Unfortunately, this C7 group also promotes the activity of quinazolinediones against human topoisomerase IIα.29 Structure–activity studies were carried out to identify groups that could promote differential activity against the bacterial and human type II enzymes. C7 3′-(AM)P-quinazolinediones that lacked either the N3 amino or C8 methyl substituent displayed little activity against human topoisomerase IIα but maintained activity against wild-type and quinolone-resistant B. anthracis topoisomerase IV.29 To determine whether these relationships extend to E. coli topoisomerase IV, the activities of 8-H-3′-(AM)P-dione and 8-methyl-3′-(AM)P-non-amino-dione were compared to that of 8-methyl-3′-(AM)P-dione against the wild-type, ParCS80L, and ParCE84K enzymes (Figure 10, middle, right, and left, respectively; drug structures are shown in the corresponding panels). Both 8-H-3′-(AM)P-dione and 8-methyl-3′-(AM)P-non-amino-dione maintained activity against the wild-type and mutant E. coli enzymes. However, as seen with the B. anthracis enzymes,29 removal of the N3 or C8 substituent somewhat decreased drug activity. Thus, it may be possible to develop broad-spectrum quinolone-based drugs that can overcome defects in the water–metal ion bridge.
Figure 10.
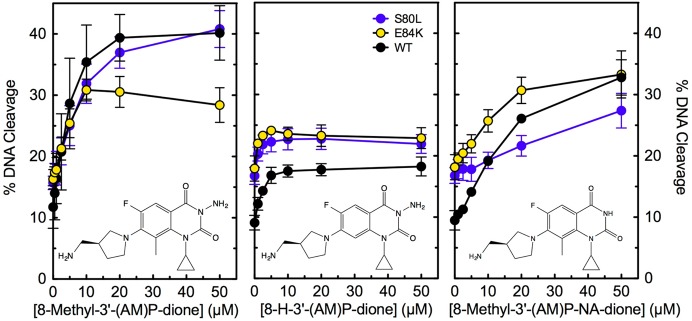
Effects of N3 and C8 substituents on DNA cleavage mediated by wild-type and quinolone-resistant E. coli topoisomerase IV. 8-H-3′-(AM)P-dione and 8-methyl-3′-(AM)P-non-amino-dione (middle and right, respectively) were tested for the ability to enhance DNA cleavage mediated by the wild-type (WT, black) and quinolone-resistant ParCS80L (S80L, blue) and ParCE84K (E84K, yellow) enzymes. The activity of 8-methyl-3′-(AM)P-dione (left) against these enzymes is shown for comparison. The structures of the compounds are shown in their respective panels. Error bars represent the standard deviation of three or more independent experiments.
Discussion
Although quinolones have been in clinical use for decades,59 the mechanism underlying drug activity and resistance remained elusive. However, a recent structural study with A. baumannii(26) topoisomerase IV and functional studies with B. anthracis topoisomerase IV27,28 provide strong evidence that clinically relevant quinolones interact through a critical water–metal ion bridge and that the most common quinolone resistance mutations decrease drug activity by disrupting this bridge. To determine whether the water–metal ion bridge also is utilized by a Gram-negative species, we examined interactions between quinolones (and related drugs) and wild-type and mutant E. coli topoisomerase IV. All of the mutations examined were in the serine and glutamic acid residues predicted to anchor the bridge to the enzyme. Results indicate that the water–metal ion bridge is essential for the actions of quinolones against E. coli topoisomerase IV. Thus, the bridge represents a common and critical mechanism that underlies quinolone function with both a Gram-positive species and a Gram-negative species.
Despite the importance of the bridge, it appears to play different roles in mediating quinolone activity against B. anthracis and E. coli topoisomerase IV. The water–metal ion bridge forms the primary binding contact of clinically relevant quinolones with the Gram-positive enzyme.27,28 Partial disruption of the bridge dramatically decreases the level of quinolone binding, but near wild-type levels of DNA cleavage can be generated at high quinolone concentrations. In contrast, partial disruption of the bridge in E. coli topoisomerase IV abrogates drug enhancement of DNA cleavage but, in some cases, has little to no effect on quinolone affinity. This finding implies that the water–metal ion bridge plays a crucial role in orienting clinically relevant quinolones in the cleavage complex, allowing them to optimally stimulate DNA scission. However, other binding contacts with the enzyme or DNA substrate can mediate drug binding in a manner that is not optimal for inducing DNA cleavage.
Even though ciprofloxacin is unable to increase levels of DNA cleavage mediated by several of the Ser80 and Glu84 mutant E. coli enzymes, the drug still retains the ability to inhibit the overall catalytic activity of these topoisomerase IV proteins. Inhibition parallels drug affinity, suggesting that the presence of the drug in the active site is sufficient to diminish the rate of DNA relaxation. These findings further indicate that the ability of quinolones to inhibit topoisomerase IV catalysis is not caused by decreased rates of DNA religation. Quinolones can kill bacteria by stimulating gyrase- and topoisomerase IV-mediated DNA cleavage or by robbing cells of the critical catalytic functions of these enzymes. The fact that clinically observed resistance mutations in Ser80 can abolish drug-induced DNA cleavage without affecting the ability of quinolones to inhibit catalytic activity implies that cleavage enhancement is the dominant, lethal effect of the drug.
Finally, if quinolone-like drugs that overcome the most common forms of resistance are to be developed, they must display activity against a broad range of gyrase and topoisomerase IV species. Two quinazolinedione derivatives that retain activity against quinolone-resistant B. anthracis topoisomerase IV but not human topoisomerase IIα have been described.29 These same drugs also retained activity against wild-type and quinolone-resistant E. coli topoisomerase IV. Thus, it may be possible to develop novel broad-spectrum drugs that are capable of quelling the rising tide of quinolone resistance.
Acknowledgments
We are grateful to Heidi A. Schwanz and Gangqin Li for the preparation of 3-amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-8-methyl-2,4(1H,3H)-quinazolinedione (UIJR-1-048, lot UILI-2-95), 3-amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-2,4(1H,3H)-quinazolinedione (UIHS-IIa-245), and 7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-8-methyl-2,4(1H,3H)-quinazolinedione (UILI-2-97). We also thank Rachel E. Ashley and MaryJean Pendleton for critical reading of the manuscript.
Glossary
Abbreviations
- 3′-(AM)P
3′-(aminomethyl)pyrrolidinyl
- 8-methyl-3′-(AM)P-dione
3-amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-8-methyl-2,4(1H,3H)-quinazolinedione
- 8-H-3′-(AM)P-dione
3-amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-2,4(1H,3H)-quinazolinedione
- 8-methyl-3′-(AM)P-non-amino-dione
7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-8-methyl-2,4(1H,3H)-quinazolinedione.
The authors declare no competing financial interest.
K.J.A. and E.J.B. were trainees under Grants T32 CA09582 and T32 GM07628 from the National Institutes of Health, respectively. V.V. was a trainee in the Vanderbilt International Summer Research Academy. This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health (to K.C.N.), by National Institutes of Health Research Grants AI87671 (to R.J.K.) and GM33944 (to N.O.), and by United States Veterans Administration Merit Review Award I01 Bx002198 (to N.O.).
Funding Statement
National Institutes of Health, United States
References
- Hooper D. C. (1999) Mode of action of fluoroquinolones. Drugs 58(Suppl. 2), 6–10. [DOI] [PubMed] [Google Scholar]
- Anderson V. E.; Osheroff N. (2001) Type II topoisomerases as targets for quinolone antibacterials: Turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des. 7, 337–353. [DOI] [PubMed] [Google Scholar]
- Hooper D. C. (2001) Mechanisms of action of antimicrobials: Focus on fluoroquinolones. Clin. Infect. Dis. 32(Suppl. 1), S9–S15. [DOI] [PubMed] [Google Scholar]
- Drlica K.; Malik M.; Kerns R. J.; Zhao X. (2008) Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 52, 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica K.; Hiasa H.; Kerns R.; Malik M.; Mustaev A.; Zhao X. (2009) Quinolones: Action and resistance updated. Curr. Top. Med. Chem. 9, 981–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Kerns R. J.; Osheroff N. (2014) Mechanism of quinolone action and resistance. Biochemistry 53, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine C.; Hiasa H.; Marians K. J. (1998) DNA gyrase and topoisomerase IV: Biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta 1400, 29–43. [DOI] [PubMed] [Google Scholar]
- Champoux J. J. (2001) DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 70, 369–413. [DOI] [PubMed] [Google Scholar]
- Gentry A. C., and Osheroff N. (2013) DNA topoisomerases: Type II. In Encyclopedia of Biological Chemistry, pp 163–168, Elsevier Inc., Amsterdam. [Google Scholar]
- Aubry A.; Fisher L. M.; Jarlier V.; Cambau E. (2006) First functional characterization of a singly expressed bacterial type II topoisomerase: The enzyme from Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 348, 158–165. [DOI] [PubMed] [Google Scholar]
- Schoeffler A. J.; Berger J. M. (2008) DNA topoisomerases: Harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 41, 41–101. [DOI] [PubMed] [Google Scholar]
- Deweese J. E.; Osheroff M. A.; Osheroff N. (2009) DNA topology and topoisomerases: Teaching a “knotty” subject. Biochem. Mol. Biol. Educ. 37, 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Deibler R. W.; Chan H. S.; Zechiedrich L. (2009) The why and how of DNA unlinking. Nucleic Acids Res. 37, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter E. M.; Lerman J. C.; Berger J. M. (2010) A naturally chimeric type IIA topoisomerase in Aquifex aeolicus highlights an evolutionary path for the emergence of functional paralogs. Proc. Natl. Acad. Sci. U.S.A. 107, 22055–22059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese J. E.; Osheroff N. (2009) The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 37, 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodursky A. B.; Zechiedrich E. L.; Cozzarelli N. R. (1995) Topoisomerase IV is a target of quinolones in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 11801–11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier B.; Zhao X.; Lu T.; Drlica K.; Hooper D. C. (2000) Selective targeting of topoisomerase IV and DNA gyrase in Staphylococcus aureus: Different patterns of quinolone-induced inhibition of DNA synthesis. Antimicrob. Agents Chemother. 44, 2160–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz R.; De la Campa A. G. (1996) ParC subunit of DNA topoisomerase IV of Streptococcus pneumoniae is a primary target of fluoroquinolones and cooperates with DNA gyrase A subunit in forming resistance phenotype. Antimicrob. Agents Chemother. 40, 2252–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.-S.; Ambler J.; Mehtar S.; Fisher L. M. (1996) Involvement of topoisomerase IV and DNA gyrase as ciprofloxacin targets in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 40, 2321–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.-S.; Fisher L. M. (1997) Targeting of DNA gyrase in Streptococcus pneumoniae by sparfloxacin: Selective targeting of gyrase or topoisomerase IV by quinolones. Antimicrob. Agents Chemother. 41, 471–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.-S.; Fisher L. M. (1998) DNA gyrase and topoisomerase IV are dual targets of clinafloxacin action in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 42, 2810–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins C. F.; Dorman C. J.; Stirling D. A.; Waddell L.; Booth I. R.; May G.; Bremer E. (1988) A physiological role for DNA supercoiling in the osmotic regulation of gene expression in S. typhimurium and E. coli. Cell 52, 569–584. [DOI] [PubMed] [Google Scholar]
- Laponogov I.; Sohi M. K.; Veselkov D. A.; Pan X. S.; Sawhney R.; Thompson A. W.; McAuley K. E.; Fisher L. M.; Sanderson M. R. (2009) Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 16, 667–669. [DOI] [PubMed] [Google Scholar]
- Laponogov I.; Pan X. S.; Veselkov D. A.; McAuley K. E.; Fisher L. M.; Sanderson M. R. (2010) Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS One 5, e11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax B. D.; Chan P. F.; Eggleston D. S.; Fosberry A.; Gentry D. R.; Gorrec F.; Giordano I.; Hann M. M.; Hennessy A.; Hibbs M.; Huang J.; Jones E.; Jones J.; Brown K. K.; Lewis C. J.; May E. W.; Saunders M. R.; Singh O.; Spitzfaden C. E.; Shen C.; Shillings A.; Theobald A. J.; Wohlkonig A.; Pearson N. D.; Gwynn M. N. (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466, 935–940. [DOI] [PubMed] [Google Scholar]
- Wohlkonig A.; Chan P. F.; Fosberry A. P.; Homes P.; Huang J.; Kranz M.; Leydon V. R.; Miles T. J.; Pearson N. D.; Perera R. L.; Shillings A. J.; Gwynn M. N.; Bax B. D. (2010) Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 17, 1152–1153. [DOI] [PubMed] [Google Scholar]
- Aldred K. J.; McPherson S. A.; Wang P.; Kerns R. J.; Graves D. E.; Turnbough C. L. Jr.; Osheroff N. (2012) Drug interactions with Bacillus anthracis topoisomerase IV: Biochemical basis for quinolone action and resistance. Biochemistry 51, 370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; McPherson S. A.; Turnbough C. L. Jr.; Kerns R. J.; Osheroff N. (2013) Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: Mechanistic basis of quinolone resistance. Nucleic Acids Res. 41, 4628–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Schwanz H. A.; Li G.; McPherson S. A.; Turnbough C. L. Jr.; Kerns R. J.; Osheroff N. (2013) Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug-enzyme interactions. ACS Chem. Biol. 8, 2660–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalhoff A. (2012) Resistance surveillance studies: A multifaceted problem-the fluoroquinolone example. Infection 40, 239–262. [DOI] [PubMed] [Google Scholar]
- Yoshida H.; Kojima T.; Yamagishi J.; Nakamura S. (1988) Quinolone-resistant mutations of the gyrA gene of Escherichia coli. Mol. Gen. Genet. 211, 1–7. [DOI] [PubMed] [Google Scholar]
- Cullen M. E.; Wyke A. W.; Kuroda R.; Fisher L. M. (1989) Cloning and characterization of a DNA gyrase A gene from Escherichia coli that confers clinical resistance to 4-quinolones. Antimicrob. Agents Chemother. 33, 886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica K.; Zhao X. (1997) DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61, 377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Deguchi T.; Yasuda M.; Kawamura T.; Kanematsu E.; Nishino Y.; Ishihara S.; Kawada Y. (1998) Alteration in the GyrA subunit of DNA gyrase and the ParC subunit of DNA topoisomerase IV in quinolone-resistant clinical isolates of Staphylococcus epidermidis. Antimicrob. Agents Chemother. 42, 3293–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price L. B.; Vogler A.; Pearson T.; Busch J. D.; Schupp J. M.; Keim P. (2003) In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob. Agents Chemother. 47, 2362–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast D. J.; Athamna A.; Duncan C. L.; de Azavedo J. C.; Low D. E.; Rahav G.; Farrell D.; Rubinstein E. (2004) Type II topoisomerase mutations in Bacillus anthracis associated with high-level fluoroquinolone resistance. J. Antimicrob. Chemother. 54, 90–94. [DOI] [PubMed] [Google Scholar]
- Grohs P.; Podglajen I.; Gutmann L. (2004) Activities of different fluoroquinolones against Bacillus anthracis mutants selected in vitro and harboring topoisomerase mutations. Antimicrob. Agents Chemother. 48, 3024–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Linnell S. K.; Becnel Boyd L.; Steffen D.; Zechiedrich L. (2009) Mechanisms accounting for fluoroquinolone resistance in Escherichia coli clinical isolates. Antimicrob. Agents Chemother. 53, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Luo Y.; Li J.; Ma Y.; Hu C.; Jin S.; Ye L.; Cui S. (2010) Characterization of clinical Escherichia coli isolates from China containing transferable quinolone resistance determinants. J. Antimicrob. Chemother. 65, 453–459. [DOI] [PubMed] [Google Scholar]
- Lautenbach E.; Metlay J. P.; Mao X.; Han X.; Fishman N. O.; Bilker W. B.; Tolomeo P.; Wheeler M.; Nachamkin I. (2010) The prevalence of fluoroquinolone resistance mechanisms in colonizing Escherichia coli isolates recovered from hospitalized patients. Clin. Infect. Dis. 51, 280–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal S.; Tandon V. (2011) Contribution of mutations in DNA gyrase and topoisomerase IV genes to ciprofloxacin resistance in Escherichia coli clinical isolates. Int. J. Antimicrob. Agents 37, 253–255. [DOI] [PubMed] [Google Scholar]
- Hardin A. H.; Sarkar S. K.; Seol Y.; Liou G. F.; Osheroff N.; Neuman K. C. (2011) Direct measurement of DNA bending by type IIA topoisomerases: Implications for non-equilibrium topology simplification. Nucleic Acids Res. 39, 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett K. D.; Schoeffler A. J.; Thomsen N. D.; Berger J. M. (2005) The structural basis for substrate specificity in DNA topoisomerase IV. J. Mol. Biol. 351, 545–561. [DOI] [PubMed] [Google Scholar]
- Fortune J. M.; Velea L.; Graves D. E.; Utsugi T.; Yamada Y.; Osheroff N. (1999) DNA topoisomerases as targets for the anticancer drug TAS-103: DNA interactions and topoisomerase catalytic inhibition. Biochemistry 38, 15580–15586. [DOI] [PubMed] [Google Scholar]
- Malik M.; Marks K. R.; Mustaev A.; Zhao X.; Chavda K.; Kerns R. J.; Drlica K. (2011) Fluoroquinolone and quinazolinedione activities against wild-type and gyrase mutant strains of Mycobacterium smegmatis. Antimicrob. Agents Chemother. 55, 2335–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune J. M.; Osheroff N. (1998) Merbarone inhibits the catalytic activity of human topoisomerase IIα by blocking DNA cleavage. J. Biol. Chem. 273, 17643–17650. [DOI] [PubMed] [Google Scholar]
- Fortune J. M.; Osheroff N. (2001) Topoisomerase II-catalyzed relaxation and catenation of plasmid DNA. Methods Mol. Biol. 95, 275–281. [DOI] [PubMed] [Google Scholar]
- Robinson M. J.; Osheroff N. (1991) Effects of antineoplastic drugs on the post-strand-passage DNA cleavage/religation equilibrium of topoisomerase II. Biochemistry 30, 1807–1813. [DOI] [PubMed] [Google Scholar]
- Gentry A. C.; Pitts S. L.; Jablonsky M. J.; Bailly C.; Graves D. E.; Osheroff N. (2011) Interactions between the etoposide derivative F14512 and human type II topoisomerases: Implications for the C4 spermine moiety in promoting enzyme-mediated DNA cleavage. Biochemistry 50, 3240–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiasa H. (2002) The Glu-84 of the ParC subunit plays critical roles in both topoisomerase IV-quinolone and topoisomerase IV-DNA interactions. Biochemistry 41, 11779–11785. [DOI] [PubMed] [Google Scholar]
- Pfeiffer E. S.; Hiasa H. (2007) Determination of the primary target of a quinolone drug and the effect of quinolone resistance-conferring mutations by measuring quinolone sensitivity based on its mode of action. Antimicrob. Agents Chemother. 51, 3410–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T. P.; Ellsworth E. L.; Sanchez J. P.; Watson B. M.; Stier M. A.; Showalter H. D.; Domagala J. M.; Shapiro M. A.; Joannides E. T.; Gracheck S. J.; Nguyen D. Q.; Bird P.; Yip J.; Sharadendu A.; Ha C.; Ramezani S.; Wu X.; Singh R. (2007) Structure-activity relationships of 3-aminoquinazolinediones, a new class of bacterial type-2 topoisomerase (DNA gyrase and topo IV) inhibitors. Bioorg. Med. Chem. Lett. 17, 1312–1320. [DOI] [PubMed] [Google Scholar]
- German N.; Malik M.; Rosen J. D.; Drlica K.; Kerns R. J. (2008) Use of gyrase resistance mutants to guide selection of 8-methoxy-quinazoline-2,4-diones. Antimicrob. Agents Chemother. 52, 3915–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X. S.; Gould K. A.; Fisher L. M. (2009) Probing the differential interactions of quinazolinedione PD 0305970 and quinolones with gyrase and topoisomerase IV. Antimicrob. Agents Chemother. 53, 3822–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppegard L. M.; Streck K. R.; Rosen J. D.; Schwanz H. A.; Drlica K.; Kerns R. J.; Hiasa H. (2010) Comparison of in vitro activities of fluoroquinolone-like 2,4- and 1,3-diones. Antimicrob. Agents Chemother. 54, 3011–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson V. E.; Zaniewski R. P.; Kaczmarek F. S.; Gootz T. D.; Osheroff N. (1999) Quinolones inhibit DNA religation mediated by Staphylococcus aureus topoisomerase IV: Changes in drug mechanism across evolutionary boundaries. J. Biol. Chem. 274, 35927–35932. [DOI] [PubMed] [Google Scholar]
- Anderson V. E.; Zaniewski R. P.; Kaczmarek F. S.; Gootz T. D.; Osheroff N. (2000) Action of quinolones against Staphylococcus aureus topoisomerase IV: Basis for DNA cleavage enhancement. Biochemistry 39, 2726–2732. [DOI] [PubMed] [Google Scholar]
- Anderson V. E.; Gootz T. D.; Osheroff N. (1998) Topoisomerase IV catalysis and the mechanism of quinolone action. J. Biol. Chem. 273, 17879–17885. [DOI] [PubMed] [Google Scholar]
- Emmerson A. M.; Jones A. M. (2003) The quinolones: Decades of development and use. J. Antimicrob. Chemother. 51(Suppl. 1), 13–20. [DOI] [PubMed] [Google Scholar]