

Abstract
Membrane protein shedding is a critical step in many normal and pathological processes. The anti-aging protein klotho (KL), mainly expressed in kidney and brain, is secreted into the serum and CSF, respectively. KL is proteolytically released, or shed, from the cell surface by ADAM10 and ADAM17, which are the α-secretases that also cleave the amyloid precursor protein and other proteins. The transmembrane KL is a coreceptor with the FGF receptor for FGF23, whereas the shed form acts as a circulating hormone. However, the precise cleavage sites in KL are unknown. KL contains two major cleavage sites: one close to the juxtamembrane region and another between the KL1 and KL2 domains. We identified the cleavage site involved in KL release by mutating potential sheddase(s) recognition sequences and examining the production of the KL extracellular fragments in transfected COS-7 cells. Deletion of amino acids T958 and L959 results in a 50–60% reduction in KL shedding, and an additional P954E mutation results in further reduction of KL shedding by 70–80%. Deletion of amino acids 954–962 resulted in a 94% reduction in KL shedding. This mutant also had moderately decreased cell surface expression, yet had overall similar subcellular localization as that of WT KL, as demonstrated by immunofluorescence. Cleavage-resistant mutants could function as a FGFR coreceptor for FGF23, but they lost activity as a soluble form of KL in proliferation and transcriptional reporter assays. Cleavage between the KL1 and KL2 domains is dependent on juxtamembrane cleavage. Our results shed light onto mechanisms underlying KL release from the cell membrane and provide a target for potential pharmacologic interventions aimed at regulating KL secretion.
The anti-aging protein α-klotho (KL) is named after the mythical Greek goddess who “spins the thread of life”.1 KL is highly expressed in the kidney and brain and to a lesser extent in reproductive organs.1 In the brain, KL is most highly expressed in the ependymal cells of the choroid plexus and cerebellar Purkinje cells1 and is detected in cerebral white matter.2 KL knockout (KL-KO) mice exhibit many changes that also frequently occur during human aging, including arteriosclerosis, osteoporosis, and cognitive decline. KL-KO mice develop normally but die prematurely, with an average lifespan of ∼61 days,1 whereas mice overexpressing KL live 30% longer than wild-type mice.3 In our microarray analyses focusing on age-associated cognitive decline in the rhesus monkey, KL expression was decreased in the aged corpus callosum,2 and this decrease was likely due to the hypermethylation of its promoter.4 We recently discovered an important and novel role of KL in the biology of oligodendrocytes, which are responsible for myelin formation. We found that KL enhances oligodendrocyte differentiation and maturation, suggesting that KL is an important factor in proper myelination and possibly in the maintenance of myelin integrity.5
KL is a type I transmembrane protein that we reported to be cleaved by a disintegrin and metalloproteinase 10 (ADAM10) and 17 and shed from the cell surface,6 a finding confirmed by others.7 Shed KL is detectable in serum, cerebral spinal fluid (CSF),8 and urine.9 KL has two domains, KL1 and KL2, which have homology to members of the glycosidase/glucuronidase family, and has been shown to act as a sialidase on a number of channels, modifying their actions.10,11 Peripheral KL functions include regulation of FGF23 signaling, resulting in the regulation of calcium and phosphate homeostasis, and suppression of the insulin/IGF1 and Wnt signaling pathways.12 In the kidney, the transmembrane full-length KL (FL-KL) and the shed extracellular domain of KL (sKL) have distinct functions: FL-KL is a coreceptor with FGFR1 for FGF23,13 which regulates serum vitamin D levels and phosphate homeostasis, and is possibly a coreceptor for other receptors, whereas sKL functions as a hormone and/or a sialidase responsible for KL’s other functions, including ion homeostasis. In other tissues, KL has been implicated in anti-inflammation, tumor suppression (including breast and pancreas),14,15 senescence, cell differentiation, and cardiovascular functions. For a comprehensive review, see refs (16 and 17).
Protein ectodomain shedding plays a crucial role in development, inflammation, and disease. Proteinases catalyzing ectodomain release, or sheddases, comprise members of various families, including matrix metalloproteinases (MMPs), ADAMs,18,19 and serine proteases.20 ADAM10 and ADAM17 are the sheddases responsible for KL ectodomain shedding6 and are the best characterized members of the ADAM family.21 Numerous protein substrates have been identified for both ADAM10 and ADAM17, including cytokine receptors, chemokines, and adhesion molecules, such as tumor necrosis factor α (TNFα), transforming growth factor α (TGF-α), Notch, and the amyloid precursor protein (APP).22−24 Because of the important roles of ADAM10 and ADAM17 in development and maintenance of normal physiology, as well as in pathological mechanisms, both ADAM10 and ADAM17 have been explored as targets for therapeutic interventions in cancer, inflammatory disease, and Alzheimer’s disease.25−27
We have identified previously two cleavage sites in the KL protein by ADAM10 and ADAM17: one close to the juxtamembrane region (α1) and a second between the KL1 and KL2 domains (α2) (Figure 1A and ref (5)). However, the exact amino acid sequence of the recognition sites by these two sheddases is unknown. In this study, we attempted to identify the KL cleavage sites by mutating potential sheddase(s) recognition sequences, and we examined the secretion of the KL extracellular 130 and 70 kDa fragments by transfection of the mutants in COS-7 cells. We were able to identify the exact α1 cleavage site close to the juxtamembrane region of KL. Moreover, we also demonstrated that the cleavage at the α2 site is dependent on α1 cleavage.
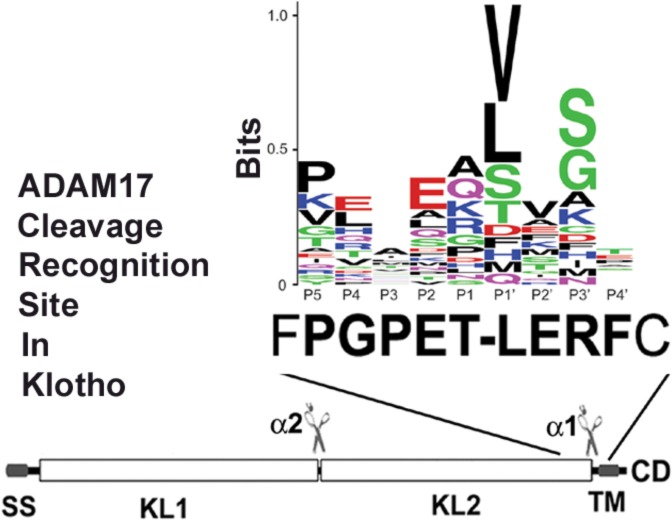
Figure 1.

ADAM10 and ADAM17 cleavage site motif prediction in KL protein. (A) Schematic diagram of the KL protein structure and its processing in COS-7 cells. KL protein contains a signal sequence (SS), two homologous domains (KL1 and KL2), a transmembrane domain (TM), and a short cytoplasmic domain (CD). The anti-KL antibody recognition region and the length of the fragments are indicated. The predicted recognition sites are in bold. Dashes (−) in the amino acid sequence indicate cleavage positions. (B) A sequence alignment of 34 known substrate proteins compiled by Caescu et al.32 was used to generate a sequence logo with the program WebLogo 3.0.38 In the logo, the height of a particular letter is proportional to the log of its frequency in the sequence alignment.
Experimental Procedures
Mutagenesis and Plasmid Construction
The mutations were introduced into KL cDNA in pcDNA3.1 vector and KL-V5 plasmid6 using the QuikChange Site-Directed mutagenesis kit (Stratagene, La Jolla, CA) with the following sense and antisense primers, respectively: KL EE mutant: 5′-TGTCCAGAAGAATTCGAAGAATGTACTGAGTGCAGTT-3′ and 5′-AACTGCACTCAGTACATTCTTCGAATTCTTCTGGACA-3′; KLΔTL mutant: 5′-GTTTCCCGGGCCCAGAAGAAAGATTTTGTCCAG-3′ and 5′-CTGGACAAAATCTTTCTTCTGGGCCCGGGAAAC-3′; KLΔTL PE mutant: 5′-GACAGCAATGGTTTCGAGGGCCCAGAAGAAAG-3′ and 5′-CTTTCTTCTGGGCCCTCGAAACCATTGCTGTC-3′; and KLΔ9 mutant: 5′-GACAGCAATGGTTTCCATATGCCAGAAACTCTGGAAAG-3′, 5′-TTCCAGAGTTTCTGGCATATGGAAACCATTGCTGTC-3′, 5′-CCAGAAACTCTGGAACATATGTGTCCAGAAGAATTC-3′, and 5′-GAATTCTTCTGGACACATATGTTCCAGAGTTTCTGG-3′. The mutagenesis of KLΔ9 was done by introducing NdeI sites to replace amino acids 954–955 and 961–962. The sequence between the two NdeI sites was removed, and the plasmid was self-ligated to produce KLΔ9 mutant with nine amino acids, 954–962, deleted. The resulting mutant contains two additional amino acids, H and M (CATATG), which were introduced to create the NdeI site. To construct KLΔ9 tagged with V5 at the C-terminus, the EcoRI–XhoI fragment from KL-V56 was ligated into the KLΔ9 plasmid. The mutated plasmids were confirmed by DNA sequencing. The mouse FGFR1c with a V5 tag in pcDNA3.1 plasmid was a gift from Dr. Kuro-o (University of Texas Southwestern Medical Center, Dallas, TX and Jichi Medical University, Tochigi, Japan).
Cell Culture, Transfection, Protein Sample Collection, and TCA Precipitation
Details of cell culture, transfections, and protein sample collection were described previously.6
Western Blotting
Details of western blotting were described previously.6 The rat anti-KL antibody KM2076 (1:2000) was described previously29 and was purchased from TransGenic Inc. (Tokyo, Japan). The antibodies to total ERK and p-ERK were from the phospho ERK pathway kit (Cell Signaling, Danvers, MA) and were used according to the manufacturer’s protocol. The mouse anti-V5 antibody (1:5000) was from Invitrogen.
Immunofluorescence
Forty-eight hours post-transfection, cells were fixed in 4% paraformaldehyde in PBS at room temperature, rinsed with PBS, and treated for 1 h with blocking solution (PBS supplemented with 1% BSA with 0.1% Triton-X 100). Cells were incubated 45 min at room temperature with the primary antibody diluted in blocking solution. Cells were stained with antibodies to rabbit anti-V5 antibody (1:500, Abcam, Cambridge, UK). Subsequently, cells were rinsed and incubated with the relevant secondary antibody (Alexa 594) (Molecular Probes) for 45 min at room temperature. Immunofluorescence images were obtained by a Nikon Eclipse 660 microscope and a SPOT-cooled CCD digital camera (Diagnostic Instruments).
Cell-Surface Biotinylation Assay
COS-7 cells in 6-well plates were transfected with plasmids expressing KL980,6 KLWT, or KLΔ9. Forty-eight hours later, cells were washed twice with PBS and incubated with a 1 mM solution of cell impermeable NHS-SS-Biotin (Pierce, Rockford, IL) in PBS for 30 min on ice. Afterward, the cells were washed twice with ice-cold PBS, and the cell lysates were collected. After collection and centrifugation, the BCA protein assay (Pierce, Rockford, IL) was used to determine the protein concentration of the samples. To precipiatate biotinylated proteins, 100 μg of protein from each sample was incubated at 4 °C with neutravidin beads (Thermo Scientific, Rockford, IL) overnight. Bound protein was eluted from the beads by incubating in 2× Laemmli sample buffer and separated by 10% SDS-PAGE.
FGF23 Signaling Assay
Details for the FGF23 signaling assay were described previously.30 Briefly, HEK293 cells were transfected with empty vector control (Ctrl) or plasmids expressing either KLWT or KLΔ9. Twenty-four hours after transfection, cells were incubated in serum-free medium for 2 h and then either 50 ng/mL of bFGF (basic fibroblast growth factor) or 10 ng/mL of FGF23 (R&D Systems, Minneapolis, MN) was added to the wells. The cells were incubated for 15 min at 37 °C and then were immediately washed in PBS and lysed in RIPA buffer containing protease and phosphatase inhibitors (Roche, Mannheim, Germany). Various incubation times and FGF23 concentrations were used as indicated in the p-ERK phosphorylation kinetics and FGF23 dose–response curve experiments. After lysis, samples were prepared for SDS-PAGE as described previously.6
Cell Viability Assay
Details for the cell viability assay were described previously.31 MO3.13 cells were plated in 96-well plates at a density of 0.65 × 106 cells/plate 1 day before the experiment. The cells were treated with serum-free conditioned medium collected from 48 to 72 h post-transfection of COS-7 cells transfected with either empty vector control, KL980, KL WT, or KL Δ9. Twenty-four hours after treatment, cells were assayed using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI).
Luciferase Transcription Factor Reporter Assay
Details for the luciferase reporter assay were described previously.31 MO3.13 cells were plated in 96-well plates at a density of 0.65 × 106 cells/plate for transfection on the following day. The cells were transfected with Sp1 and NF-κB reporter plasmids (Qiagen, Valencia, CA) and incubated with conditioned medium from COS-7 cells transfected with either empty vector control, KL980, KL WT, or KL Δ9, as described above. Twenty-four hours post-transfection, cells were assayed for luciferase activity.
Results
Determination of the Potential Shedding Sites in KL
KL contains a signal sequence (SS), two homologous domains (KL1 and KL2), a transmembrane domain (TM), and a short cytoplasmic domain (CD) (Figure 1A). We have identified two cleavage sites for ADAM10 and ADAM17:6 one close to the transmembrane (TM) region and another one between the KL1 and KL2 domains (Figure 1A). However, the exact sequences of the cleavage sites are not known. To predict the ADAM10 and ADAM17 recognition sites in KL, we examined highly conserved residues found at cleavage sites in 34 reported substrate proteins as compiled by Caescu et al.32 Sequence alignments of these cleavage sites were used to generate sequence logos representing the preferred amino acid residues for each primed and unprimed position (Figure 1B). This analysis suggested that residues at positions P5, P2, P1′, and P3′ are most predictive of cleavage by ADAM17, whereas positions P1, P1′, P2′, and P3′ are most predictive for ADAM10 (Figure 1B). Residues found with high frequency at ADAM10 and ADAM17 cleavage sites include either valine or leucine at the P1′ position. In addition, known ADAM17 substrates were enriched for Pro at the P5 position, which was also selected by both ADAM10 and ADAM17 in peptide library experiments.32 Unlike serine proteases, for which the P1 position is often critical for cleavage site recognition, ADAM10 and ADAM17 do not appear to be highly selective at this position. On the basis of these predicted recognition patterns, there are two potential ADAM10/ADAM17 cleavage sites close to the TM region (Figure 1A). In order to identify which of the two potential sites is actually cleaved by these sheddases, we mutated both sites and examined the KL secretion after transfection of mutants in COS-7 cells, cells that express both ADAM10 and ADAM176 (Figure 2). Replacement of Thr-968 and Val-969 with two glutamic acid residues (KL EE) or deletion of both amino acids (KLΔTV) did not reduce KL secretion (Figure 2A). In fact, in the case of the KLΔTV mutation, there was more secreted KL detected in the medium (Figure 2A, lane 4). For the KL EE mutation, there was more of the 70 kDa KL fragment detected in the cell lysate compared to KL WT (Figure 2A, lane 3). In contrast, deletion of Thr-958 and Leu-959 (KLΔTL) resulted in 50–60% reduction in KL shedding (Figure 2A,B,D), and an additional mutation of Pro-954 to Glu (KLΔTLPE) resulted in a further reduction in KL shedding by 70–80% (Figure 2B,D). These results suggest that KL cleavage occurs primarily at the more upstream site, possibly between Thr-958 and Leu-959. Furthermore, deletion of the nine amino acids, 954–962 (KLΔ9), encompassing the more upstream juxtamembrane site resulted in a 94% inhibition of cleavage near the TM (α1 site) and between KL1 and KL2 (α2 site). When KLΔ9 was expressed, the 70 kDa band in the cell lysate and the 130 kDa band in the medium were barely detectable (Figure 2C,D). Thus, mutations close to the TM region result in reduction of both the 130 and 70 kDa forms of sKL (Figure 2B,C), suggesting that cleavage between the KL1 and KL2 domains is dependent on the cleavage near the TM domain.
Figure 2.

Determination of the potential shedding sites and sheddases in KL in COS-7 cells. (A–C, E) Western blots from COS-7 cells transiently transfected with empty vector control (Ctrl) or with the KL plasmids as indicated. Forty-eight hours post-transfection, cells were incubated in serum-free media for 2 h, and the protein samples were collected from either the cell lysate (Cell lysate) or the medium (Medium). The medium samples in panel A were from 48 h post-transfection conditioned medium (CM), whereas in panels B and C, the serum-free medium was collected after 2 h incubation and TCA-precipitated as described in the Experimental Procedures. The estimated molecular weights of the KL fragments in the medium are indicated (130 and 70 kDa). (D) Statistical analysis of the results from panels A–C. The intensities of the 130 kDa bands were analyzed and normalized to that of the KL bands from the tissue lysate using the average intensity of the controls as 100% from 3 to 4 independent experiments. Error bar indicates standard deviation. Significance of results was determined using Student’s t-test: *, p < 0.05; **, p < 0.005. (E) Cotransfection experiments of KL WT and KL D9 with either ADAM10 or ADAM17. Anti-HA antibody detects HA-tagged ADAM10, whereas anti-V5 antibody detects V5-tagged ADAM17. (F) Statistical analysis of the results from panel E. The intensities of the 70 kDa bands in the medium were analyzed and normalized to the that of total KL bands from the tissue lysate using the average intensity of the controls as 100% from 3 independent experiments. Error bar indicates standard deviation. Significance of results was determined using Student’s t-test: *, p < 0.05.
We demonstrated previously that ADAM10 and ADAM17 are both responsible for KL shedding in COS-7 cells.6 To determine which of these shedding enzymes is responsible for the observed cleavage at the nine amino acids (954–962) site, we cotransfected KL WT or KLΔ9 with either ADAM10 or ADAM17. Cotransfection with ADAM17 increased KL WT shedding by 7–8-fold but had no significant effect on KLΔ9 shedding (Figure 2E and F). Coexpression with ADAM10 did not increase KL shedding (Figure 2E,F). These results suggested that ADAM17 is the main protease responsible for KL cleavage at amino acids 954–962 in COS-7 cells.
Subcellular Localization of the KL Mutant
To examine the effect of the deletion in KL on its subcellular localization, we performed immunofluorescence on COS-7 cells transfected with either WT V5-tagged KL (KL WT) or KLΔ9. The results showed that the KLΔ9 mutant distributed normally and similarly to that of KL WT and other typical membrane proteins (Figure 3A). Furthermore, when we cotransfected KL GFP and KLΔ9 V5-tagged into COS-7 cells and immunostained the cells with anti-V5 antibody, the results showed that KL GFP and KLΔ9 colocalized (Figure 3B), suggesting that the KLΔ9 mutation does not affect the subcellular localization, which was similar to that of KL WT.
Figure 3.

KLΔ9 mutant colocalization with KL WT and cell-surface distribution in COS-7 cells. (A) Indirect immunofluorescence showed subcellular localization pattern of KL WT and KLΔ9 mutant. COS-7 cells transfected with KL WT or KLΔ9 mutant for 48 h were fixed, and indirect immunofluorescence was performed using anti-V5 monoclonal antibody. Green: anti-V5 (Alexa 488); blue: DAPI nucleus staining. (B) Colocalization of KL GFP and KLΔ9 mutant with V5-tag. Green: GFP; red: anti-V5 (Alexa 594); blue: DAPI. (C) Western blots from COS-7 cells transiently transfected with the KL plasmids as indicated. Forty-eight hours post-transfection, the conditioned medium (Medium) was collected. The cells were surface-labeled with biotin, and the protein samples were collected from the cell lysates (Cell lysate). The biotinylated samples (Membrane) were pulled down by neutravidin beads. (D) Statistical analysis of the results from panel C. The intensities of the 130 kDa bands from the cell surface were analyzed and normalized to that of the total KL bands from the tissue lysate using the average intensity of the controls as 100% from 3 independent experiments. The arrowhead in the middle panel in C indicates nonspecific bands (arrowhead) at the 70 kDa position in the CM samples, likely from the interaction of the antibody with serum albumin. Error bar indicates standard deviation. Significance of results was determined using Student’s t-test: *, p < 0.05.
To quantify the amount of KL WT and KLΔ9 expressed on the cell surface, we conducted cell-surface biotinylation experiments. We used a truncated secretory form of KL lacking the TM domain, KL980,6 as a non-membrane-localized control. As expected, the cell-surface biotinylation assay revealed that KL980 did not localize to cell surface (Figure 3C). KLΔ9 showed about 60–70% cell-surface localization compared to that of KL WT (Figure 3C,D). Similar biotinylation experiments were also performed for the KLΔTL and KLΔTLPE mutants. These two mutants had comparable cell-surface distribution as that of KL WT (Figure 3D). These two mutants do not show accumulation on cell surface, likely because they either undergo more internalization or are more accessible to sheddases, as shown in Figure 2A. Taken together, these results suggested that even though KLΔ9 showed a normal distribution similar to that of KL WT, it has less cell-surface distribution compared to that of KL WT, likely due to less of the mutant protein being targeted to the plasma membrane. However, because KLΔ9 shedding was severely reduced (more than 90%), the modest observed decrease in plasma membrane targeting cannot explain the impaired shedding for this mutant.
Functional Assay of the KL Mutant
To determine whether the KLΔ9 mutation changes KL protein function, we performed the FGF23 signaling assay in the HEK293 cell system transfected with FGFR1 with KL acting as a coreceptor. Signaling was detected by assessing ERK phosphorylation.30,33 The results showed that KLΔ9 induces phosphorylation of ERK in the FGF23 signaling assay in HEK293 cells similar to that of KL WT using 10 ng/mL FGF23 at the 30 min time point (Figure 4A), indicating that the mutation does not modify the ability of KL to function as a FGFR1 coreceptor. To examine the FGF23 signaling kinetics on ERK phosphorylation of KL WT and KLΔ9, we compared ERK phosphorylation at various times up to 45 min in HEK293 cells transfected with either KL WT or KLΔ9. The results showed no difference in p-ERK kinetics of KL WT vs KLΔ9 (Figure 4B,C). In addition, we examined the effects of different FGF23 doses on KL WT and KLΔ9 and found that at higher FGF23 concentrations (50, 100, and 300 ng/mL) KLΔ9 reached only 60–70% ERK phosphorylation compared to that with KL WT (Figure 4D,E), likely because of the lower cell-surface distribution of KLΔ9 (Figure 3C,D). These results suggest that KLΔ9 mutant functions comparably as a FGFR1 coreceptor as KL WT on the cell surface.
Figure 4.

KLΔ9 mutant showed similar function as KL WT in downstream pERK signaling. HEK 293 cells transiently transfected with FGFR1c and either KL WT or KLΔ9 were treated with 10 ng/mL FGF23 for 30 min to activate FGFR1c signaling. (A) Representative western blot showing differences in ERK phosphorylation compared to total expression of ERK after transfection of KL WT or KLΔ9. Lanes 1 and 2 are negative controls without KL, and lanes 7 and 8 are positive controls with bFGF. The antibodies used are indicated. (B) Bar graph depicting no change in ERK phosphorylation normalized to total ERK expression (error bars are ±SEM; n = 3) when comparing KLΔ9 to KL WT. (C) Same experiments as in panel A with 0, 10, 20, 30, and 45 min time points for ERK phosphorylation kinetics analysis. (D) Statistical analysis of the results from panel C. The intensities of the pERK bands were normalized to that of the total ERK bands from 3 independent experiments. (E) FGF23 dose–response experiments. Similar experiments as those in panels A and C, with different doses of FGF23 for 10 min as indicated. (F) Statistical analysis of the results from panel E. Error bar indicates standard deviation. Significance of results was determined using Student’s t-test: *, p < 0.05.
To demonstrate that the lower levels of soluble KL shed into the medium by KLΔ9 correlates with less activity compared to that of KL WT, we collected the conditioned media (CM) of COS-7 cells transfected with either KL WT or KLΔ9 and used the CM in various experiments to assess sKL activity. Recently, we demonstrated that KL has dual functions of inhibiting proliferation and enhancing differentiation in MO3.13 cells, a human oligodendrocytic hybrid cell line.31 We treated MO3.13 cells with the CM of either KL WT or KLΔ9 and found that the CM from cells transfected with KL WT can inhibit MO3.13 cell proliferation, but much less inhibition was observed with the CM from cells transfected with KLΔ9 (Figure 5A). Similar experiments were performed on MO3.13 cells transfected with the transcription factor (TF) luciferase reporters Sp1 and NF-κB.31 We found that the CM from KL WT transfected cells can inhibit MO3.13 cells Sp1 and NF-κB TF reporter activity, but much less inhibition was observed with the CM from KLΔ9 transfected cells (Figure 5B). These results demonstrate that KLΔ9 loses the activity of sKL compared to KL WT, likely due to impaired shedding.
Figure 5.

CM of COS-7 cells transfected with KLΔ9 has less inhibition of proliferation and transcription factor reporter activity in MO3.13 cells compared to the that from CM of KL WT. (A) MO3.13 cells were incubated with CM from either empty vector, KL WT, or KL Δ9 transfected COS-7 cells for 48 h and assayed for cell viability. Asterisks (*) indicate statistical significance of p < 0.01 by Student’s t test. Error bars indicate standard deviation. Results are from 3 independent experiments. (B) Luciferase assay of MO3.13 cells transfected with luciferase reporters as indicated. Cells were incubated with CM from either empty vector, KL WT, or KL Δ9 transfected COS-7 cells for 24 h and tested using the luciferase assay. The luminescent signals were normalized to Renilla luciferase. Luciferase activity was calculated relative to that of the control (CM from empty vector transfected COS-7 cells), which was given a value of 100%. Asterisks (*) indicate statistical significance of p < 0.01 by Student’s t test. Error bars indicate standard deviation.
Discussion
ADAM10 and ADAM17 do not appear to have distinct recognition amino acid cleavage sequence requirements, in that there is no particular residue absolutely conserved at a given position in known sites of cleavage. Many reports suggest that the position of each amino acid is more important than the sequence of the substrate cleavage site. The so-called “stalk” or linker region between the TM and the first globular part of the protein determines whether the substrate can be cleaved.34−36 However, recent analysis of peptide substrate libraries showed that both ADAM10 and ADAM17 have strong sequence preferences.32,37 Using the information from peptide library screening and systematic analysis of amino acids next to the cleavage site of known substrates, we predicted and identified the ADAM10 and ADAM17 recognition sites close to the TM region of KL to be PGPET-LERF (amino acids 954–962) (Figure 1A). This site incorporates several residues frequently found at ADAM10 and/or ADAM17 cleavage sites: a Pro residue at the P5 position, a Glu residue at the P2 position, and a Leu residue at the P1′ position. Deletion of these nine amino acids resulted in a 94% reduction in cleavage. We were not able to completely inhibit shedding, likely because other proteases may also be involved in KL cleavage since it is known that KL is a substrate for BACE1 and γ-secretase in addition to ADAM10 and ADAM17.7 We cannot exclude the possibility that ADAM10 and ADAM17 cut at other sites but less efficiently. We demonstrated previously that both ADAM10 and ADAM17 can increase KL shedding;6 however, we were not able to detect enhancement of KL shedding by overexpressing ADAM10 in COS-7 cells (Figure 2E). Thus, we conclude that in COS-7 cells ADAM17, rather than ADAM10, appears to be the major sheddase.
We reported previously that membrane anchoring is required for KL shedding.6 To identify the function of the shed KL, we initially prepared a truncated form of KL that contains 980 amino acids all the way to the lysine before the TM. This truncated form of KL contains the nine amino acid recognition site; however, it was not a substrate for sheddases since it is a soluble protein found in secretory vesicles. The membrane anchor region is required for the sheddase to interact with KL and present the recognition sequence for cleavage. Interestingly, deletion of the amino acids 968T and 969V (KLΔTV) downstream of the recognition sequence increases KL shedding (Figure 2A). It is likely that the deletion results in a conformation change in KL, which favors sheddase recognition.
When assayed on peptide substrates, cleavage activity of ADAM10 and ADAM17 was almost completely lost when alanine was used to substitute the P1′ position.32 However, when we substituted L959 with alanine, the resulting mutant had a similar shedding pattern as that of KL WT (data not shown). This result suggests that even though the chemical library provides valuable prediction on sheddase selectivity, these proteases are likely to display more promiscuity in vivo. One possibility for this discrepancy is that short peptides are typically unstructured, whereas protein substrates may have secondary or tertiary structures that influence cleavage. However, point mutations in the ADAM17 substrate TNF-α have similarly large effects on cleavage in vitro. Alternatively, comparing initial velocities of WT and mutant substrate cleavage in vitro may not properly model cleavage in cells, where sheddase activity is high and the system is not at steady state.
There are two major cleavage sites in the KL protein by sheddases: one close to the juxtamembrane region and a second between the KL1 and KL2 domains (Figure 1A). In this study, we identified the recognition site in the juxtamembrane region of KL to be PGPET-LERF (aa 954–962). There are three potential recognition sites between the KL1 and KL2 domains, PKSSA-LFYQ (aa 490–498), PENQP-LEGT (aa 510–518), and PLEGT-FPCD (aa 514–522), according to our rules of prediction. We made the two amino acid deletion mutations at the P1 and P1′ positions, and the results suggested that the second predicted site (aa 510–518) is not the recognition site since the mutant was secreted similarly to that of KL WT (data no shown). The other two mutations (D495–496 and D518–519) resulted in loss of both secreted fragments (70 and 130 kDa), likely because of protein misfolding and accumulation in a subcellular compartment (data no shown). The identification of the precise recognition site between KL1 and KL2 requires further investigation. Here, we demonstrate that mutation in the juxtamembrane region results in reduction of both the 130 and 70 kDa forms of sKL (Figure 2B,C), suggesting that the cleavage between the KL1 and KL2 domains is dependent on the cleavage near the TM domain. We also found that the two cleavages happen simultaneously, as both the 70 and 130 kDa fragments appeared at the same time in the medium in a pulse–chase experiment (SI text of ref (6)). Thus, these two cuts happen simultaneously, and the cut between KL1 and KL2 is dependent on the cleavage in the juxtamembrane region.
In this study, we provide valuable information to predict the ADAM10 and ADAM17 recognition site in KL. These general rules for shedding can be applied to search for the recognition sites in other α-secretase substrate proteins. Here, we deleted the nine amino acid recognition site and achieved 94% shedding inhibition. The mutation did not result in a 100% reduction in shedding, either because there is no stringent sequence requirement for ADAM10 and ADAM17 recognition or because the remaining shedding activities may be due to β-secretase or other sheddase(s). The secreted KL (sKL) and full-length membrane form of klotho have distinct functions. sKL plays an important role in oligodendrocyte differentiation and myelination,5 with a 50% reduction in KL in a KL+/– heterozygous mouse resulting in a strong hypomyelination phenotype in the corpus callosum.5 In future experiments, we plan to define the distinct roles of the two forms of KL by generating knockin mice that express the KLΔ9 mutant, which will mostly remain on the membrane. This knockin mouse line will be lacking sKL and can be used to test the role of sKL in myelination and all other humoral functions of sKL in vivo, including neuroprotection in Alzheimer’s disease and tumor suppression in cancer.
Glossary
Abbreviations
- KL
klotho
- ADAM
a disintegrin and metalloproteinase
- CSF
cerebrospinal fluid
- PCR
polymerase chain reaction
- DMEM
Dulbecco’s modified Eagle’s medium
- PBS
phosphate buffered saline
- FBS
fetal bovine serum
- BSA
bovine serum albumin
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
This work was supported by NIH-NIA grant AG-00001 to C.R.A.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Kuro-o M.; Matsumura Y.; Aizawa H.; Kawaguchi H.; Suga T.; Utsugi T.; Ohyama Y.; Kurabayashi M.; Kaname T.; Kume E.; Iwasaki H.; Iida A.; Shiraki-Iida T.; Nishikawa S.; Nagai R.; Nabeshima Y. I. (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51. [DOI] [PubMed] [Google Scholar]
- Duce J. A.; Podvin S.; Hollander W.; Kipling D.; Rosene D. L.; Abraham C. R. (2008) Gene profile analysis implicates klotho as an important contributor to aging changes in brain white matter of the rhesus monkey. Glia 56, 106–117. [DOI] [PubMed] [Google Scholar]
- Kurosu H.; Yamamoto M.; Clark J. D.; Pastor J. V.; Nandi A.; Gurnani P.; McGuinness O. P.; Chikuda H.; Yamaguchi M.; Kawaguchi H.; Shimomura I.; Takayama Y.; Herz J.; Kahn C. R.; Rosenblatt K. P.; Kuro-o M. (2005) Suppression of aging in mice by the hormone klotho. Science 309, 1829–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King G. D.; Rosene D. L.; Abraham C. R. (2011) Promoter methylation and age-related downregulation of klotho in rhesus monkey. Age (Dordrecht, Neth.) 34, 1405–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. D.; Sloane J. A.; Li H.; Aytan N.; Giannaris E. L.; Zeldich E.; Hinman J. D.; Dedeoglu A.; Rosene D. L.; Bansal R.; Luebke J. I.; Kuro-o M.; Abraham C. R. (2013) The antiaging protein klotho enhances oligodendrocyte maturation and myelination of the CNS. J. Neurosci. 33, 1927–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. D.; Podvin S.; Gillespie E.; Leeman S. E.; Abraham C. R. (2007) Insulin stimulates the cleavage and release of the extracellular domain of klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. U.S.A. 104, 19796–19801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch L.; Sineshchekova O.; Reichenbach D.; Reiss K.; Saftig P.; Kuro-o M.; Kaether C. (2009) Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. 583, 3221–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura A.; Iwano A.; Tohyama O.; Tsuji Y.; Nozaki K.; Hashimoto N.; Fujimori T.; Nabeshima Y. (2004) Secreted klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 565, 143–147. [DOI] [PubMed] [Google Scholar]
- Akimoto T.; Yoshizawa H.; Watanabe Y.; Numata A.; Yamazaki T.; Takeshima E.; Iwazu K.; Komada T.; Otani N.; Morishita Y.; Ito C.; Shiizaki K.; Ando Y.; Muto S.; Kuro-o M.; Kusano E. (2012) Characteristics of urinary and serum soluble klotho protein in patients with different degrees of chronic kidney disease. BMC Nephrol. 13, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha S. K.; Hu M. C.; Kurosu H.; Kuro-o M.; Moe O.; Huang C. L. (2009) Regulation of renal outer medullary potassium channel and renal K+ excretion by klotho. Mol. Pharmacol. 76, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q.; Hoefs S.; van der Kemp A. W.; Topala C. N.; Bindels R. J.; Hoenderop J. G. (2005) The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310, 490–493. [DOI] [PubMed] [Google Scholar]
- Kuro-o M. (2010) Klotho. Pfluegers Arch. 459, 333–343. [DOI] [PubMed] [Google Scholar]
- Kurosu H.; Ogawa Y.; Miyoshi M.; Yamamoto M.; Nandi A.; Rosenblatt K. P.; Baum M. G.; Schiavi S.; Hu M. C.; Moe O. W.; Kuro-o M. (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 281, 6120–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramovitz L.; Rubinek T.; Ligumsky H.; Bose S.; Barshack I.; Avivi C.; Kaufman B.; Wolf I. (2011) KL1 internal repeat mediates klotho tumor suppressor activities and inhibits bFGF and IGF-I signaling in pancreatic cancer. Clin. Cancer Res. 17, 4254–4266. [DOI] [PubMed] [Google Scholar]
- Wolf I.; Levanon-Cohen S.; Bose S.; Ligumsky H.; Sredni B.; Kanety H.; Kuro O. M.; Karlan B.; Kaufman B.; Koeffler H. P.; Rubinek T. (2008) Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 27, 7094–7105. [DOI] [PubMed] [Google Scholar]
- Kuro-o M. (2012) Klotho and betaKlotho. Adv. Exp. Med. Biol. 728, 25–40. [DOI] [PubMed] [Google Scholar]
- Hu M. C.; Shiizaki K.; Kuro-o M.; Moe O. W. (2013) Fibroblast growth factor 23 and klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Ann. Rev. Physiol. 75, 503–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss M. L.; Lambert M. H. (2002) Shedding of membrane proteins by ADAM family proteases. Essays Biochem. 38, 141–153. [DOI] [PubMed] [Google Scholar]
- Seals D. F.; Courtneidge S. A. (2003) The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 17, 7–30. [DOI] [PubMed] [Google Scholar]
- Urban S.; Lee J. R.; Freeman M. (2001) Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell 107, 173–182. [DOI] [PubMed] [Google Scholar]
- Black R. A.; White J. M. (1998) ADAMs: focus on the protease domain. Curr. Opin. Cell Biol. 10, 654–659. [DOI] [PubMed] [Google Scholar]
- Blobel C. P. (1997) Metalloprotease-disintegrins: links to cell adhesion and cleavage of TNF alpha and Notch. Cell 90, 589–592. [DOI] [PubMed] [Google Scholar]
- Peschon J. J.; Slack J. L.; Reddy P.; Stocking K. L.; Sunnarborg S. W.; Lee D. C.; Russell W. E.; Castner B. J.; Johnson R. S.; Fitzner J. N.; Boyce R. W.; Nelson N.; Kozlosky C. J.; Wolfson M. F.; Rauch C. T.; Cerretti D. P.; Paxton R. J.; March C. J.; Black R. A. (1998) An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages M.; Rojo F.; Albanell J.; Baselga J.; Arribas J. (2003) TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 22, 1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenholz F. (2007) Alpha-secretase as a therapeutic target. Curr. Alzheimer Res. 4, 412–417. [DOI] [PubMed] [Google Scholar]
- Moss M. L.; Sklair-Tavron L.; Nudelman R. (2008) Drug insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 4, 300–309. [DOI] [PubMed] [Google Scholar]
- Le Gall S. M.; Bobe P.; Reiss K.; Horiuchi K.; Niu X. D.; Lundell D.; Gibb D. R.; Conrad D.; Saftig P.; Blobel C. P. (2009) ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, l-selectin, and tumor necrosis factor alpha. Mol. Biol. Cell 20, 1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y.; Arakawa E.; Kinoshita S.; Shirai A.; Furuya A.; Yamano K.; Nakamura K.; Iida A.; Anazawa H.; Koh N.; Iwano A.; Imura A.; Fujimori T.; Kuro-o M.; Hanai N.; Takeshige K.; Nabeshima Y. (2000) Establishment of the anti-klotho monoclonal antibodies and detection of klotho protein in kidneys. Biochem. Biophys. Res. Commun. 267, 597–602. [DOI] [PubMed] [Google Scholar]
- Tucker Zhou T. B.; King G. D.; Chen C.; Abraham C. R. (2013) Biochemical and functional characterization of the klotho-VS polymorphism implicated in aging and disease risk. J. Biol. Chem. 288, 36302–36311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. D.; Li H.; Liang J.; Hixson K.; Zeldich E.; Abraham C. R. (2014) The anti-aging and tumor suppressor protein klotho enhances differentiation of a human oligodendrocytic hybrid cell line. J. Mol. Neurosci. 10.1007/s12031-014-0336-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caescu C. I.; Jeschke G. R.; Turk B. E. (2009) Active-site determinants of substrate recognition by the metalloproteinases TACE and ADAM10. Biochem. J. 424, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King G. D.; Chen C.; Huang M. M.; Zeldich E.; Brazee P. L.; Schuman E. R.; Robin M.; Cuny G. D.; Glicksman M. A.; Abraham C. R. (2012) Identification of novel small molecules that elevate klotho expression. Biochem. J. 441, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlondorff J.; Blobel C. P. (1999) Metalloprotease-disintegrins: modular proteins capable of promoting cell–cell interactions and triggering signals by protein-ectodomain shedding. J. Cell Sci. 112, 3603–3617. [DOI] [PubMed] [Google Scholar]
- Wang X.; He K.; Gerhart M.; Huang Y.; Jiang J.; Paxton R. J.; Yang S.; Lu C.; Menon R. K.; Black R. A.; Baumann G.; Frank S. J. (2002) Metalloprotease-mediated GH receptor proteolysis and GHBP shedding. Determination of extracellular domain stem region cleavage site. J. Biol. Chem. 277, 50510–50519. [DOI] [PubMed] [Google Scholar]
- Hinkle C. L.; Sunnarborg S. W.; Loiselle D.; Parker C. E.; Stevenson M.; Russell W. E.; Lee D. C. (2004) Selective roles for tumor necrosis factor alpha-converting enzyme/ADAM17 in the shedding of the epidermal growth factor receptor ligand family: the juxtamembrane stalk determines cleavage efficiency. J. Biol. Chem. 279, 24179–24188. [DOI] [PubMed] [Google Scholar]
- Lambert M. H.; Blackburn R. K.; Seaton T. D.; Kassel D. B.; Kinder D. S.; Leesnitzer M. A.; Bickett D. M.; Warner J. R.; Andersen M. W.; Badiang J. G.; Cowan D. J.; Gaul M. D.; Petrov K. G.; Rabinowitz M. H.; Wiethe R. W.; Becherer J. D.; McDougald D. L.; Musso D. L.; Andrews R. C.; Moss M. L. (2005) Substrate specificity and novel selective inhibitors of TNF-alpha converting enzyme (TACE) from two-dimensional substrate mapping. Comb. Chem. High Throughput Screening 8, 327–339. [DOI] [PubMed] [Google Scholar]
- Crooks G. E.; Hon G.; Chandonia J. M.; Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]