

Abstract

The reaction catalyzed by cyanobacterial aldehyde deformylating oxygenase is of interest both because of its potential application to the production of biofuels and because of the highly unusual nature of the deformylation reaction it catalyzes. Whereas the proton in the product alkane derives ultimately from the solvent, the identity of the proton donor in the active site remains unclear. To investigate the proton transfer step, solvent isotope effect (SIE) studies were undertaken. The rate of alkane formation was found to be maximal at pH 6.8 and to be the same in D2O or H2O within experimental error, implying that proton transfer is not a kinetically significant step. However, when the ratio of protium to deuterium in the product alkane was measured as a function of the mole fraction of D2O, a D2OSIEobs of 2.19 ± 0.02 was observed. The SIE was invariant with the mole fraction of D2O, indicating the involvement of a single protic site in the reaction. We interpret this SIE as most likely arising from a reactant state equilibrium isotope effect on a proton donor with an inverse fractionation factor, for which Φ = 0.45. These observations are consistent with an iron-bound water molecule being the proton donor to the alkane in the reaction.
Aldehyde decarbonylases1 (ADs) have attracted interest in the search for sustainable, drop-in biofuels because of their ability to produce long-chain alkanes.1,2 Few biosynthetic pathways to hydrocarbons have been identified, although they are produced by a wide range of organisms, including plants, insects, and microbes, and serve a variety of important biological roles.2−5 Long-chain alkane biosynthesis utilizes fatty acids in the form of fatty-acyl-CoA esters as precursors.6 These are converted to alkanes in a two-step process. First, a fatty-acyl-CoA reductase reduces the fatty-acyl-CoA ester to a long-chain fatty aldehyde.7 Second, in a chemically difficult reaction, AD cleaves the bond between the aldehyde carbon and the α-carbon to produce the corresponding n-1 alkane and an aldehyde carbon (C1) coproduct.8
Three mechanistically distinct types of ADs have been identified. These produce different C1 coproducts and in each case perform chemistry that is highly unusual for their respective enzyme families.9 The plant and algal ADs are integral membrane metalloenzymes that belong to the fatty acid hydroxylase superfamily. These enzymes convert the aldehyde carbon to carbon monoxide.6,8,10 Insect AD is a membrane-bound cytochrome p450 enzyme that oxidizes C1 to carbon dioxide.11−14 The recently identified cyanobacterial AD is the most widely studied enzyme because, unlike the insect or plant ADs, it is a small, stable, soluble protein.15−21 In this case, C1 is released as formate in a reaction that requires oxygen and reducing equivalents; it is now generally termed cyanobacterial aldehyde deformylating oxygenase (cADO).
cADO is a member of the non-heme diiron oxygenase family of enzymes. Typical enzymes of this class, as exemplified by methane monooxygenase, employ molecular oxygen and two reducing equivalents to oxidize their substrates through hydroxylation.22 However, cADO is unique in requiring four reducing equivalents per turnover, fully reducing one oxygen atom to water and resulting in an oxygenated C1 coproduct.16 The kinetics of alkane formation by cADO are notable for their sluggishness, and despite various attempts to optimize the reaction conditions, in no cases have steady state turnover numbers faster than ∼1 min–1 been obtained.9,17,21 Over the past few years, investigations of cADO have elucidated several key steps in the mechanism; however, it remains unclear which step(s) in the mechanism is rate-limiting.9
In the mechanism shown in Figure 1, an initial two-electron reduction of the resting diferric state to the active diferrous state (species I) initiates the reaction between cADO and molecular oxygen.15,16 This forms an intermediate peroxo-bridged diiron core (species II). Next, nucleophilic addition to the aldehyde substrate results in the formation of a peroxy-hemiacetal intermediate (species III).23 Addition of an electron results in the breakdown of this intermediate, possibly through the formation of a transient formyl radical (species IV) that undergoes homolytic cleavage of the C1–C2 aldehyde bond, resulting in the formation of a primary alkyl radical and formate (species V).24 Finally, the alkyl radical is reduced to an alkane by electron–proton transfer (EPT), and the products are released from the enzyme (species VI).
Figure 1.

Proposed mechanism for deformylation of aldehydes by cADO. The color coding indicates the origin of the oxygen atoms and the protons in the products.
Experimental evidence supporting this mechanism comes from labeling studies indicating that the aldehyde proton is retained in formate whereas the alkane proton is derived from the solvent or an exchangeable site on the enzyme,18,25 and that one atom of O2 is retained in formate.15,16 Recent spectroscopic evidence supports the formation of a peroxy-hemiacetal intermediate (species III) and its breakdown upon addition of the exogenous reductant.23 Recently, our laboratory has used both cyclopropyl and oxiranyl aldehydes designed to function as radical clocks to support a radical mechanism for C–C bond scission. From these experiments, the lifetime of the alkyl radical was estimated to be in the range of 10–100 μs, implying that the EPT step occurs relatively fast (k ∼ 104–105 s–1).19,24 This suggests the electron is transferred directly from a site on the protein rather than from the external reducing system, with the diiron center being the most likely source of electrons. However, the proton donor in the reaction remains unclear.
To investigate further the nature of the protonation step, we have undertaken solvent isotope effect (SIE) studies, which we report here. SIEs are informative in identifying proton donors in enzymatic reactions if the donating group has an unusual fractionation factor and may also inform on the nature of the rate-determining step.
Materials and Methods
Materials
Recombinant cADO from Prochlorococcus marinus MIT9313 was purified from Escherichia coli, loaded with FeII by anaerobic incubation with ferrous ammonium sulfate, and subsequently desalted as previously described.18 Spin desalting columns were from Thermo Scientific. Octadecanal, heptanal, heptadecane, hexane, phenazine methosulfate, ferrous ammonium sulfate, nicotinamide adenine dinucleotide (NADH), and deuterium oxide were obtained from Acros Organics. Potassium chloride and 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) salts were from Fisher Chemicals.
Preparation of Deuterated Buffers
HEPES buffer was dissolved in 99.8% D2O and lyophilized, twice, to produce deuterated HEPES (D-HEPES). Deuterated cADO assay buffer was prepared in 99.8% D2O with 100 mM D-HEPES (pD 6.8) and 100 mM KCl. Proteated and deuterated buffers were purged with nitrogen for 2 h before being transferred to an anaerobic chamber (Coy Laboratory Products Inc., Grass Lake, MI), where they were allowed to equilibrate for at least 0.5 h before being used. Mixed D2O/H2O buffers were prepared at various mole fractions by combining deuterated and proteated buffers at the desired ratios, and differences in molar volumes between H2O and D2O were accounted for using values of 18.126 and 18.058 mL/mol, respectively.26 The mole fraction of D2O in each assay was recalculated to account for the isotopic purity of the D2O and the addition of enzyme from the stock solution in proteated buffer. The buffers were brought to pL 6.8 using the equation pL = pHobs + 0.311x + 0.0766x2, where x is the mole fraction of deuterium.27
Enzyme Assays
All assays were prepared under microaerobic conditions in an anaerobic chamber under an atmosphere of 3% hydrogen and 97% nitrogen, at <20 ppm O2. Standard assay conditions include 10 μM FeII-loaded cADO, 100 μM PMS, 1 mM NADH, and 300 μM substrate with a total assay volume of 500 μL in either 100 mM HEPES or D-HEPES buffer containing 100 mM KCl. All assays were initiated by the addition of substrate, removed from the chamber, and shaken at 37 °C.
We note that removing the tubes from the chamber allows the slow diffusion of O2 into the sealed tubes so that the concentration of oxygen is certainly significantly higher than 20 ppm (equivalent to ∼26 nM dissolved O2). However, as the enzyme turns over very slowly, the low O2 concentration does not appear to limit the overall rate of reaction and minimizes side reactions that deplete the auxiliary reducing system, the products of which appear to inhibit the enzyme.
Assays containing octadecanal were performed in triplicate in 2 mL Eppendorf tubes for varying lengths of time. Octadecanal stock solutions were made up in DMSO and dissolved at 70 °C; the final concentration of DMSO in the assay was 2%. Assays were quenched with 500 μL of ethyl acetate, vortexed, and centrifuged to extract the heptadecane product to the organic layer, which was then combined with a tridecane internal standard for gas chromatography–mass spectrometry (GC–MS) analysis.
Assays containing heptanal were performed like those described above except that 1.5 mL gastight vials were substituted for Eppendorf tubes. At the end of the reaction period, the vials were shaken vigorously for 3 min to volatilize the hexane product. A gastight syringe was used to extract 250 μL of headspace, which was analyzed by GC–MS to quantify the total ion counts of labeled and unlabeled hexane produced.
Quantification of Hydrocarbon Products by GC–MS
Quantification of hydrocarbon products was performed using a Shimadzu QP-2010 GC–MS system equipped with a DB-5 column (30 m × 0.25 mm × 0.25 μm). During heptadecane quantification, the flow rate of the helium carrier gas was 1.0 mL/min with an injection temperature of 200 °C. Eight microliters of sample was injected in splitless mode. The initial temperature was held at 70 °C for 2 min, increased to 320 °C at a rate of 20 °C/min, and then maintained at 320 °C for 2 min. The interface temperature for MS was 250 °C, and the solvent cut time was 4.5 min. Heptadecane eluted between 8.9 and 9.1 min, and the concentration was determined using a calibration curve of heptadecane standards prepared in ethyl acetate.
For hexane quantification, the flow rate of the helium carrier gas was 4.0 mL/min with an injection temperature of 200.0 °C; 250 μL aliquots of gas were manually injected in splitless mode. The initial temperature was held at 70.0 °C for 2 min, increased to 275 °C at a rate of 20.0 °C/min, and then maintained at 275 °C for 1.75 min. The interface temperature for MS was 250 °C, and the solvent cut time was 4 min. Hexane eluted between 1.73 and 1.98 min. Total ion counts for 86 and 87 amu, corresponding to the molecular ion peaks for proteated and deuterated hexane, respectively, were generated for each sample and corrected for instrument background as well as natural 13C isotope abundance. Chromatographic data were analyzed by Shimadzu GC–MS solution software.
Results and Discussion
The crystal structures of wild-type cADO and two mutant forms have been determined with aliphatic carboxylic acids bound to the diiron center.28 The carboxylic acid side chain occupies a hydrophobic channel leading from the metal center and likely closely mimics the binding of the aldehyde substrate. Close inspection of the structure of cADO with stearate bound reveals a network of hydrogen bonding residues and water molecules that leads from the exterior of the enzyme to the active site. This suggests a potential route by which the solvent-derived proton transferred to the alkane gains access to the active site. As shown in Figure 2, there are four potential proton-donating groups within 5 Å of the α-carbon of stearate (corresponding to the carbon atom that would be the site of protonation in the reaction). These are the side chains of Gln123 and Tyr135, an iron-bound water, and a water molecule hydrogen-bonded to Gln123. We considered that SIE measurements might provide insight into the identity of the proton donor and/or the kinetic significance of the proton transfer step.
Figure 2.
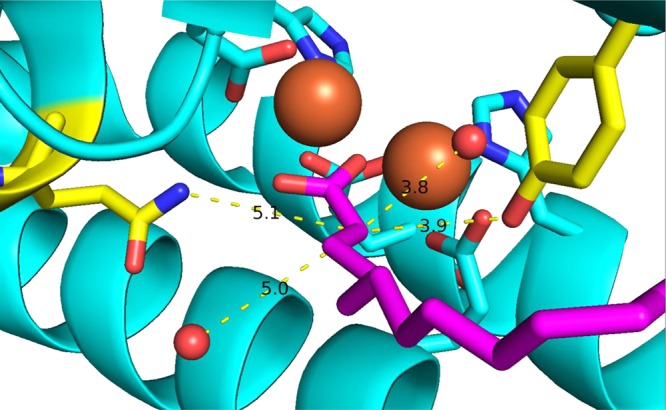
Crystal structure of cADO with stearate (purple) bound (Protein Data Bank entry 2OC5). Possible proton donors within ∼5 Å of Cα in the active site are Gln123 and Tyr135 (yellow) and the two water molecules (red spheres). The diiron core is colored orange.
Various assay conditions have been reported in mechanistic investigations of cADO.2,18,20,21,25 Although the enzyme requires oxygen, in air-saturated buffer we have found that only 1–2 equivalents of product are formed, relatively rapidly, before the reaction nearly ceases. This is because the auxiliary reducing system necessary to supply the electrons needed in the reaction also efficiently reacts with oxygen. This side reaction depletes the reducing system and produces H2O2 as a byproduct that has been shown to inhibit cADO.21 However, at low oxygen concentrations that have been termed microaerobic conditions, we have consistently found that the enzyme achieves multiple turnovers with the reaction rate remaining linear for several hours. For steady state kinetic measurements, it is, of course, necessary to take measurements over multiple turnovers, and therefore, we employed microaerobic conditions, as described in Materials and Methods, for these experiments.
Dependence of Rate on pL
SIEs can be a function of pL (L = H or D) because of kinetically significant ionizations of protic positions in the enzyme or substrate.26 Therefore, we initially investigated the rate of reaction as a function of pL in H2O and D2O, both to determine the optimal pL for SIE measurements and to examine whether any isotope-dependent shift in activity was observed. cADO was assayed in proteated and deuterated buffers at various pL values between 6.0 and 8.4 using octadecanal as the substrate. Plots of reaction rate as a function of pL are shown in Figure 3. At pL <6.0, cADO activity was too low to reliably measure. Activity increased to a maximum near pL 7.0 in either proteated or deuterated buffers, before diminishing slightly at higher pL. The activity of the enzyme appears to be slightly lower in deuterated buffer, but given the experimental errors associated with these measurements, we do not consider the differences significant. The plots provide no evidence of a kinetically significant ionization occurring during the reaction.
Figure 3.
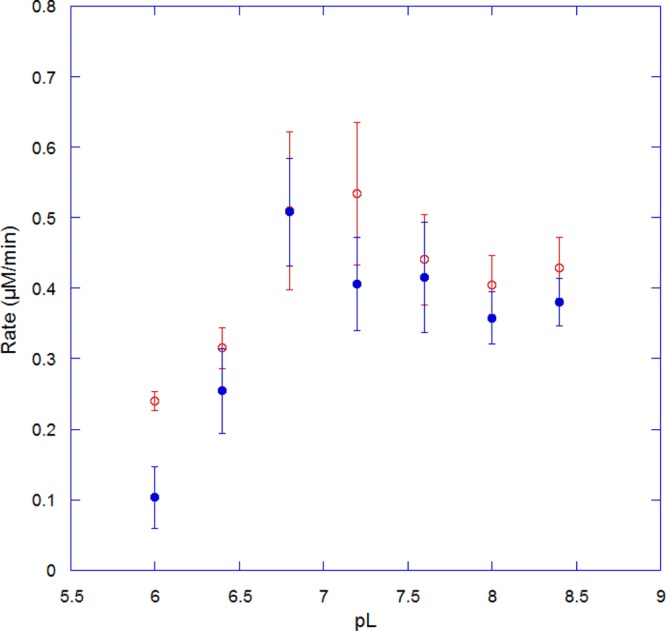
pL–rate profiles for cADO in H2O (empty red circles) and D2O (filled blue circles).
Solvent Isotope Effect on the Transfer of Deuterium to Product
On the basis of the pL–rate profile, solvent isotope effect measurements were taken at pL 6.8, where enzyme activity was found to be near maximal in both H2O and D2O. At this pL, the nonenzymatic exchange of deuterium from the solvent at the α-carbon of the aldehyde was sufficiently slow that it did not interfere with the experiment. At pL 6.8, measurements of the rates of heptadecane formation in H2O and D2O allowed the SIE on Vmax for cADO reacting with octadecanal as a substrate to be calculated as 1.0 ± 0.2. This result is consistent with the pL-dependent activity of the enzyme discussed above.
This observation indicates that proton transfer is not a kinetically significant step in the reaction. Taken together with other experimental data, the absence of a solvent isotope effect on Vmax suggests that all of the chemical steps are relatively fast compared to the steady state rate of turnover. Although spectroscopic studies of the formation of the differic-peroxide intermediate(s) appear to be complicated by multiple reactant states arising from the order of binding of aldehyde and O2, the rate constants for the formation of the activated oxygen species (II or III in Figure 1) have been measured as 0.75 and 0.2 s–1 depending on which substrate binds first.23 Subsequent reaction of species III with reduced OMePMS, with concomitant formation of formate (species V), was reported to occur in <1 s. Previous studies performed using an α-oxiranyl aldehyde as a radical clock indicated the rate at which species V was converted to VI was fast (∼104 to ∼105 s–1).19 These observations are in accord with chemical logic, in that species IV and V involve the formation of highly reactive radical species that would be expected to have very short lifetimes.
The preceding discussion points to a nonchemical step being rate-determining during turnover. We have discussed possible reasons for very slow turnover of substrates by cADO previously,9 which we briefly summarize here. These include the insoluble nature of both the substrates and products, which may limit both diffusion of the substrate into the active site and release of the product alkane from the highly hydrophobic substrate-binding channel of cADO. As noted above, the inhibition of cADO by hydrogen peroxide, which arises from the uncoupled reaction of the reducing systems employed to assay the enzyme with O2, may also slow the reaction.21 The transfer of reducing equivalents from the auxiliary reducing system, e.g., PMS or ferredoxin, to cADO during the catalytic cycle could also be rate-limiting during the reaction. In this respect, it should be noted that the physiological electron donor to cADO has yet to be identified, nor is the reason that some cyanobacteria produce alkanes understood. It is possible that the cADO may require other proteins to activate it,6 similar to the plant decarbonylase, or that the deformylating activity could be a side reaction and that the enzyme catalyzes an oxidative reaction on an as yet undiscovered substrate.
Fractionation Factor for the Transfer of Deuterium to Product
Solvent fractionation factors (Φ) have the potential to discriminate between different proton donors in enzymatic reactions.26 Therefore, we sought to generate a proton inventory for the cADO reaction that would allow Φ to be determined. If the proton donor was a free water molecule, Φ would be predicted to be near unity.29 If Tyr135 or Gln123 were the proton donor, Φ would be predicted to be greater than one based on Φ for phenol and R-NL2 (Φ = 1.13 for each).29 However, a water molecule bound to a Lewis acidic group, such as a metal ion, will behave more like a free hydroxyl group (Φ = 0.43) and will have a characteristically inverse Φ.30−33
Typically, proton inventories are calculated using the rate ratio of reactions in mixed isotopic versus pure protic solvents plotted against the mole fraction of deuterium (χ) in the mixed solvent.26 In this case, the velocity of the reaction is independent of the isotopic composition of the solvent; therefore, we determined Φ by performing an internal competition experiment in mixed isotopic solvents. We note that in this experimental design the observed fractionation factor (Φobs) is related to the observed solvent isotope effect (D2OSIEobs) on V/K by the relationship Φobs = 1/D2OSIEobs.
The ratio of protium to deuterium in the alkane product was determined as a function of χ using GC–MS. To obtain the requisite degree of precision in measuring the mass ratios of the product molecular ion peak, heptanal was used as a substrate for these experiments. Measurements with octadecanal proved to be unreliable because of the low abundance of the molecular ion peak. The ratios of unlabeled to deuterated hexane were determined at every tenth χ D2O (average of five determinations per point) and were used to plot the data shown in Figure 4. The observed solvent isotope effect (D2OSIEobs) on the reaction was calculated by fitting the data to eq 1:
| 1 |
Figure 4.
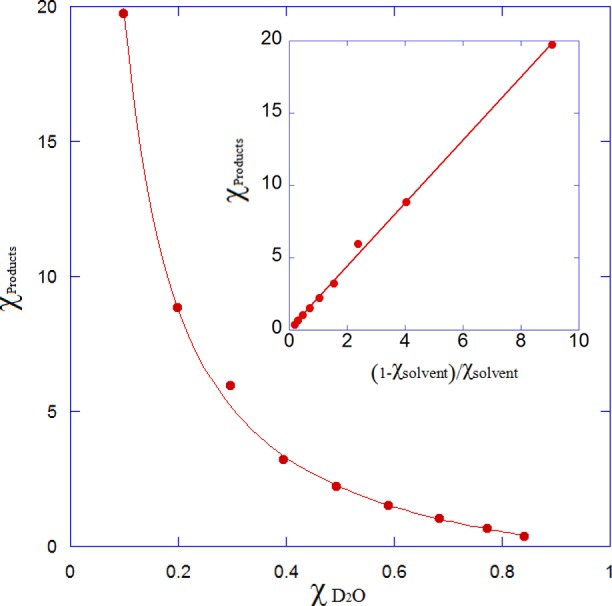
Proton inventory for cADO. The mole fraction of D2O in the solvent is plotted against the ratio of proteated to deuterated product alkane. The solid line represents the best fit to eq 1 where R2 = 0.998. Error bars are contained within the points. The inset represents a fit to the linearized form of eq 1 (R2 = 0.999).
From the data, a D2OSIEobs value of 2.19 ± 0.02 was calculated. Equation 1 assumes that D2OSIEobs does not vary as a function of χ, i.e., that only a single proton is involved in the transition state. The excellent fit of the data to eq 1 (R2 = 0.998) indicates that this is indeed the case, and fits of the data to the linearized form of eq 1 give a straight line (Figure 4, inset).
Interpretation of Φobs
Isotope effect measurements seldom allow unambiguous mechanistic interpretations to be made; here we consider two interpretations that are consistent with our data. More likely, D2OSIEobs represents a reactant state equilibrium SIE arising from a protic site that is characterized by an inverse fractionation factor, Φobs. In this interpretation, the distribution of isotope is established at the proton-donating group prior to the start of the catalytic cycle. Because the chemical steps in the reaction are irreversible, the enzyme is forced to take whichever isotope is present at the active site, and thus, the distribution of isotopes in the product alkane reflects the equilibrium distribution of isotopes at the proton-donating group prior to the reaction, i.e., Φobs. In this case, Φobs = 1/D2OSIEobs = 0.457 ± 0.004.
We are not aware of any experimentally determined fractionation factors for Fe-dependent enzymes. However, fractionation factors have been measured for two isoforms of carbonic anhydrase with CoII bound, for which the M-OL2 species has a Φ in the range of 0.81–0.9 and the M-OL– species has a Φ in the range of 0.72–0.77.26,34,35 These agree quite well with the fractionation factor for Co(H2O)62+ in solution (Φ = 0.70), and it is generally assumed that other divalent metals such as Zn and FeII have a Φ of ∼0.7.34,36 We note that Φ is calculated on a per-bond basis and is multiplicative. It is therefore diagnostic for the number of a metal-bound water molecules; for one water molecule, Φobs = Φ2 ∼ 0.49.
The low value of Φobs for cADO is consistent with an iron-bound water molecule being the proton donor in the reaction, as the experimentally determined Φobs of 0.457 is in good agreement with the predicted value of Φ2 (0.49).26 This value is similar to that determined for another non-heme iron enzyme factor inhibiting hypoxia inducible factor-1α.36 For this enzyme, fractionation factors were measured on kcat (Φobs = 0.51) and kcat/KM (Φobs = 0.40) that were associated with a kinetically significant dissociation of an FeII-bound water molecule prior to O2 binding. Thus, we conclude that the observed SIE may reasonably be interpreted as arising from a reactant state EIE due to the equilibration of H2O and D2O at the diiron core that leads to enrichment of H2O at the metal site.
An alternative explanation is that D2OSIEobs arises from a kinetic competition between two equivalent proton donors at the active site. This can give rise to isotopic discrimination even if the preceding steps in the reaction are irreversible and in favorable cases can allow intrinsic kinetic isotope effects to be determined even when proton transfer is not rate-determining.37,38 This would also be consistent with an iron-bound water molecule as the proton donor, as opposed to the active site tyrosine residue. However, in this case, the interpretation of the isotope effect would be complicated because of the expected additional discrimination against deuterium arising from the inverse fractionation factor. It is, however, unclear whether the two protons on a water molecule in the active site would behave as chemically equivalent given the strong propensity of the protons to form hydrogen bonds with polar groups at the active site. Therefore, although these findings are consistent with our experimental data, we consider this the less likely explanation, and we are not aware of a precedent in which SIEs have been demonstrated to arise by such a mechanism.
Mechanistic Implications
Interpreting the low Φobs value as arising from the donation of a proton by an iron-bound water molecule raises a question of whether the metal is in the ferric or ferrous oxidation state. Mechanistic considerations make it more likely that the proton donor is an FeII-OH2 species rather than an FeIII-OH2 species. The pKa values of FeIII-OH2 species are much lower than those of FeII-OH2 species [cf. Fe3+-(OH2)6 (pKa = 2.2) and Fe2+-(OH2)6 (pKa = 9.5)] so that at neutral pH FeIII-OH– would predominate at the active site of cADO.39 Also, donation of a proton from FeIII-OH– would result in the formation of a terminal iron oxide species or would require the concerted transfer of a second proton from another site, which is not supported by the proton inventory.
In contrast, proton transfer coupled with electron transfer from FeII-OH2 through a multiple-site electron–proton transfer (MS-EPT) reaction appears to be quite plausible (Figure 5A).40 In this case, an electron would be transferred to the alkyl radical from the metal and the proton transferred from the metal-bound water in a concerted manner. Here, oxidation of FeII to FeIII facilitates proton transfer due to the increased Lewis acidity of the metal. Previous studies using an α-oxiranyl aldehyde as a radical clock provide evidence that the intermediate alkyl radical is reduced extremely rapidly, consistent with direct electron transfer from FeII, as opposed to long-range electron transfer from an external reductant.19
Figure 5.
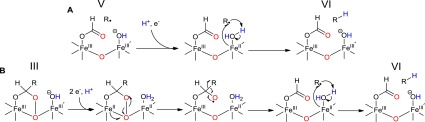
Two potential mechanisms for multiple-site electron–proton transfer (MS-EPT) from an Fe2+-OH2 species. (A) Single-electron reduction of the diferric core after deformylation resulting in a mixed valent FeIII/II species. (B) Two-electron reduction of the diferric core prior to deformylation resulting in a divalent FeII/II species that undergoes subsequent chemical steps.
The mechanism discussed above implies the formation of a mixed valent FeII/FeIII metal center during the reaction, prior to reduction of the alkyl radical. We note that the previous step in the mechanism requires an electron to facilitate C–C bond cleavage and that this electron could also be transferred from iron. It is therefore an interesting question of whether these steps occur sequentially through mixed valent metal species or whether, prior to C–C bond cleavage, complete reduction of cADO to the diferrous enzyme occurs, as shown in Figure 5B. These mechanisms may be hard to distinguish experimentally given the short lifetimes of the reactive intermediates involved, but the attractive feature of forming a diferrous intermediate is that the enzyme is primed to complete the deformylation reaction without the need to shuttle further electrons into the active site.
Lastly, we note that a rather different mechanism for cADO has been proposed on the basis of the observation of low levels of hydroxylated products when cADO is reacted with medium chain-length aldehydes such as decanal and nonanal.20 The mechanism involves the formation of a transient FeIV superoxo species that subsequently acts to hydroxylate the product alkane and does not require additional electrons to be introduced during the reaction. Whereas we regard this mechanism as less likely to be operating, at least with octadecanal and heptanal, the substrates we commonly use to assay cADO, the experiments described here do not rule out such a mechanism.
In conclusion, the solvent isotope effect measurements we have described provide information about the likely donor of the proton to the product alkane. The absence of any kinetic solvent isotope effect on the reaction, taken together with other experimental observations, points to a nonchemical step as likely being rate-determining in the reaction. With the caveat that intramolecular competition between two active site proton donors cannot be rigorously excluded, the D2OSIEobs measured in these studies most likely arises from a highly inverse reactant state fractionation factor associated with the active site group that donates the proton to the product alkane. The low Φobs value is characteristic of the pre-equilibrium binding of H2O to an iron center, pointing to an iron-bound water molecule as the likely proton donor.
Acknowledgments
We thank the reviewers of the manuscript for their suggestions about the interpretation of the solvent isotope effects reported here.
Glossary
Abbreviations
- AD
aldehyde decarbonylase
- cADO
cyanobacterial aldehyde deformylation oxygenase
- SIE
solvent isotope effect
- EPT
electron–proton transfer
- MS-EPT
multiple-site electron–proton transfer
- PMS
phenazine methosulfate
- Φ
fractionation factor.
This research was supported in part by National Science Foundation Grants CHE 1152055 and CBET 1336636, National Institutes of Health Grant GM 093088, and European Union Grant FP-7 256808.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Ghim C. M.; Kim T.; Mitchell R. J.; Lee S. K. (2010) Synthetic Biology for Biofuels: Building Designer Microbes from the Scratch. Biotechnol. Bioprocess Eng. 15, 11–21. [Google Scholar]
- Schirmer A.; Rude M. A.; Li X.; Popova E.; del Cardayre S. B. (2010) Microbial biosynthesis of alkanes. Science 329, 559–562. [DOI] [PubMed] [Google Scholar]
- Howard R. W.; Blomquist G. J. (2005) Ecological, behavioral, and biochemical aspects of insect hydrocarbons. Annu. Rev. Entomol. 50, 371–393. [DOI] [PubMed] [Google Scholar]
- Cheesebrough T. M.; Kolattukudy P. E. (1988) Microsomal Preparation from an Animal Tissue Catalyzes Release of Carbon Monoxide from a Fatty Aldehyde to Generate an Alkane. J. Biol. Chem. 263, 2738–2743. [PubMed] [Google Scholar]
- Bernard A.; Joubes J. (2013) Arabidopsis cuticular waxes: Advances in synthesis, export and regulation. Prog. Lipid Res. 52, 110–129. [DOI] [PubMed] [Google Scholar]
- Bourdenx B.; Bernard A.; Domergue F.; Pascal S.; Leger A.; Roby D.; Pervent M.; Vile D.; Haslam R. P.; Napier J. A.; Lessire R.; Joubes J. (2011) Overexpression of Arabidopsis ECERIFERUM1 promotes wax very-long-chain alkane biosynthesis and influences plant response to biotic and abiotic stresses. Plant Physiol. 156, 29–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F. M.; Das D.; Lin X. N.; Marsh E. N. G. (2013) Aldehyde-forming fatty acyl-CoA reductase from cyanobacteria: Expression, purification and characterization of the recombinant enzyme. FEBS J. 280, 4773–4781. [DOI] [PubMed] [Google Scholar]
- Cheesebrough T. M.; Kolattukudy P. E. (1984) Alkane biosynthesis by decarbonylation of aldehydes catalyzed by a particulate preparation from Pisum sativum. Proc. Natl. Acad. Sci. U.S.A. 81, 6613–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh E. N. G.; Waugh M. W. (2013) Aldehyde Decarbonylases: Enigmatic Enzymes of Hydrocarbon Biosynthesis. ACS Catal. 3, 2515–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis M.; Kolattukudy P. E. (1992) A cobalt-porphyrin enzyme converts a fatty aldehyde to a hydrocarbon and CO. Proc. Natl. Acad. Sci. U.S.A. 89, 5306–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed J. R.; Vanderwel D.; Choi S.; Pomonis J. G.; Reitz R. C.; Blomquist G. J. (1994) Unusual Mechanism of Hydrocarbon Formation in the House Fly: Cytochrome P450 Converts Aldehyde to the Sex Pheromone Component (Z)-9-Tricosene and CO2. Proc. Natl. Acad. Sci. U.S.A. 91, 10000–10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y.; Tittiger C.; Wicker-Thomas C.; Le Goff G.; Young S.; Wanjberg E.; Fricaux T.; Taquet N.; Blomquist G.; Feyereisen R. (2012) An insect-specific P450 oxidative decarbonylase for cuticular hydrocarbon biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 109, 14858–14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpuru S.; Reed J. R.; Reitz R. C.; Blomquist G. J. (1996) Mechanism of Hydrocarbon Biosynthesis from Aldehyde in Selected Insect Species: Requirement for O2 and NADPH and Carbonyl Group Released as CO2. Insect Biochem. Mol. Biol. 26, 203–208. [Google Scholar]
- Reed J. R.; Quilici D. R.; Blomquist G. J.; Reitz R. C. (1995) Proposed Mechanism for the Cytochrome P450-Catalyzed Conversion of Aldehydes to Hydrocarbons in the House Fly, Musca domestica. Biochemistry 34, 16221–16227. [DOI] [PubMed] [Google Scholar]
- Li N.; Chang W. C.; Warui D. M.; Booker S. J.; Krebs C.; Bollinger J. M. Jr. (2012) Evidence for only oxygenative cleavage of aldehydes to alk(a/e)nes and formate by cyanobacterial aldehyde decarbonylases. Biochemistry 51, 7908–7916. [DOI] [PubMed] [Google Scholar]
- Li N.; Norgaard H.; Warui D. M.; Booker S. J.; Krebs C.; Bollinger J. M. Jr. (2011) Conversion of fatty aldehydes to alka(e)nes and formate by a cyanobacterial aldehyde decarbonylase: Cryptic redox by an unusual dimetal oxygenase. J. Am. Chem. Soc. 133, 6158–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser B. E.; Das D.; Han J.; Jones P. R.; Marsh E. N. (2011) Oxygen-independent alkane formation by non-heme iron-dependent cyanobacterial aldehyde decarbonylase: Investigation of kinetics and requirement for an external electron donor. Biochemistry 50, 10743–10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D.; Eser B. E.; Han J.; Sciore A.; Marsh E. N. (2011) Oxygen-independent decarbonylation of aldehydes by cyanobacterial aldehyde decarbonylase: A new reaction of diiron enzymes. Angew. Chem. 50, 7148–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D.; Ellington B.; Paul B.; Marsh E. N. G. (2014) Mechanistic Insights from Reaction of α-Oxiranyl-Aldehydes with Cyanobacterial Aldehyde Deformylating Oxygenase. ACA Chem. Biol. 9, 570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aukema K. G.; Makris T. M.; Stoian S. A.; Richman J. E.; Munck E.; Lipscomb J. D.; Wackett L. P. (2013) Cyanobacterial aldehyde deformylase oxygenation of aldehydes yields n-1 aldehydes and alcohols in addition to alkanes. ACS Catal. 3, 2228–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre C.; Kim S. W.; Yu X. H.; Shanklin J. (2013) Fusing catalase to an alkane-producing enzyme maintains enzymatic activity by converting the inhibitory byproduct H2O2 to the cosubstrate O2. Proc. Natl. Acad. Sci. U.S.A. 110, 3191–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallar B. J.; Lipscomb J. D. (1996) Dioxygen Activation by Enzymes Containing Binuclear Non-Heme Iron Clusters. Chem. Rev. 96, 2625–2657. [DOI] [PubMed] [Google Scholar]
- Pandelia M. E.; Li N.; Norgaard H.; Warui D. M.; Rajakovich L. J.; Chang W. C.; Booker S. J.; Krebs C.; Bollinger J. M. (2013) Substrate-Triggered Addition of Dioxygen to the Diferrous Cofactor of Aldehyde-Deformylating Oxygenase to Form a Diferric-Peroxide Intermediate. J. Am. Chem. Soc. 135, 15801–15812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul B.; Das D.; Ellington B.; Marsh E. N. (2013) Probing the mechanism of cyanobacterial aldehyde decarbonylase using a cyclopropyl aldehyde. J. Am. Chem. Soc. 135, 5234–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warui D. M.; Li N.; Norgaard H.; Krebs C.; Bollinger J. M. Jr.; Booker S. J. (2011) Detection of formate, rather than carbon monoxide, as the stoichiometric coproduct in conversion of fatty aldehydes to alkanes by a cyanobacterial aldehyde decarbonylase. J. Am. Chem. Soc. 133, 3316–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn D. M., and Sutton L. D. (1991) Theoretical Basis and Mechanistic Utility of Solvent Isotope Effects. In Enzyme Mechanism from Isotope Effects, CRC Press, Boca Raton, FL. [Google Scholar]
- Vidakovic M.; Sligar S. G.; Li H. Y.; Poulos T. L. (1998) Understanding the role of the essential Asp251 in cytochrome P450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Biochemistry 37, 9211–9219. [DOI] [PubMed] [Google Scholar]
- Khara B.; Menon N.; Levy C.; Mansell D.; Das D.; Marsh E. N. G.; Leys D.; Scrutton N. S. (2013) Production of Propane and Other Short-Chain Alkanes by Structure-Based Engineering of Ligand Specificity in Aldehyde-Deformylating Oxygenase. ChemBioChem 14, 1204–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarret R. M.; Saunders M. (1985) A new method for obtaining isotopic fractionation data at multiple sites in rapidly exchanging systems. J. Am. Chem. Soc. 107, 2648–2654. [Google Scholar]
- Kresge A. J., More O’Ferrall R. A., and Powell M. F. (1987) Solvent isotope effects, fractionation factors and mechanisms of proton transfer reactions. In Isotopes in Organic Chemistry (Buncel E., and Lee C. C., Eds.) Elsevier, Amsterdam. [Google Scholar]
- Schowen K. B.; Schowen R. L. (1982) Solvent Isotope Effects on Enzyme Systems. Methods Enzymol. 87, 551–606. [PubMed] [Google Scholar]
- Galtress C. L.; Morrow P. R.; Nag S.; Smalley T. L.; Tschantz M. F.; Vaughn J. S.; Wichems D. N.; Ziglar S. K.; Fishbein J. C. (1992) Mechanism for the Solvolytic Decomposition of the Carcinogen N-Methyl-N′-nitro-N-nitrosoguanidine in Aqueous Solutions. J. Am. Chem. Soc. 114, 1406–1411. [Google Scholar]
- Gold V.; Grist S. (1972) Deuterium Solvent Isotope-Effects on Reactions Involving Aqueous Hydroxide Ion. J. Chem. Soc., Perkin Trans. 2 89–95. [Google Scholar]
- Kassebaum J. W.; Silverman D. N. (1989) Hydrogen-deuterium fractionation factors of the aqueous ligand of cobalt in Co(H2O)62+ and Co(II)-substituted carbonic-anhydrase. J. Am. Chem. Soc. 111, 2691–2696. [Google Scholar]
- Makinen M. W.; Kuo L. C.; Dymowski J. J.; Jaffer S. (1979) Catalytic Role of the metal-ion of carboxypeptidase-A in ester hydrolysis. J. Biol. Chem. 254, 356–366. [PubMed] [Google Scholar]
- Hangasky J. A.; Saban E.; Knapp M. J. (2013) Inverse Solvent Isotope Effects Arising from Substrate Triggering in the Factor Inhibiting Hypoxia Inducible Factor. Biochemistry 52, 1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M.; Song H. T.; Hakansson K.; Marsh E. N. G. (2010) Hydrogen Tunneling in Adenosylcobalamin-Dependent Glutamate Mutase: Evidence from Intrinsic Kinetic Isotope Effects Measured by Intramolecular Competition. Biochemistry 49, 3168–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M.; Kalli A.; Lee H. Y.; Hakansson K.; Marsh E. N. G. (2007) Intrinsic deuterium kinetic isotope effects in glutamate mutase measured by an intramolecular competition experiment. Angew. Chem., Int. Ed. 46, 8455–8459. [DOI] [PubMed] [Google Scholar]
- Gilson R.; Durrant M. C. (2009) Estimation of the pKa values of water ligands in transition metal complexes using density functional theory with polarized continuum model solvent corrections. J. Chem. Soc., Dalton Trans. 10223–10230. [DOI] [PubMed] [Google Scholar]
- Weinberg D. R.; Gagliardi C. J.; Hull J. F.; Murphy C. F.; Kent C. A.; Westlake B. C.; Paul A.; Ess D. H.; McCafferty D. G.; Meyer T. J. (2012) Proton-coupled electron transfer. Chem. Rev. 112, 4016–4093. [DOI] [PubMed] [Google Scholar]