

Abstract
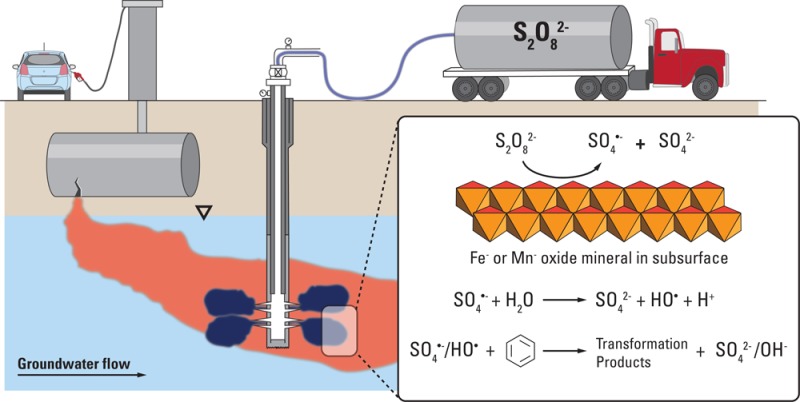
Persulfate (S2O82–) is being used increasingly for in situ chemical oxidation (ISCO) of organic contaminants in groundwater, despite an incomplete understanding of the mechanism through which it is converted into reactive species. In particular, the decomposition of persulfate by naturally occurring mineral surfaces has not been studied in detail. To gain insight into the reaction rates and mechanism of persulfate decomposition in the subsurface, and to identify possible approaches for improving its efficacy, the decomposition of persulfate was investigated in the presence of pure metal oxides, clays, and representative aquifer solids collected from field sites in the presence and absence of benzene. Under conditions typical of groundwater, Fe(III)- and Mn(IV)-oxides catalytically converted persulfate into sulfate radical (SO4•–) and hydroxyl radical (HO•) over time scales of several weeks at rates that were 2–20 times faster than those observed in metal-free systems. Amorphous ferrihydrite was the most reactive iron mineral with respect to persulfate decomposition, with reaction rates proportional to solid mass and surface area. As a result of radical chain reactions, the rate of persulfate decomposition increased by as much as 100 times when benzene concentrations exceeded 0.1 mM. Due to its relatively slow rate of decomposition in the subsurface, it can be advantageous to inject persulfate into groundwater, allowing it to migrate to zones of low hydraulic conductivity where clays, metal oxides, and contaminants will accelerate its conversion into reactive oxidants.
Introduction
Despite more than three decades of effort, contamination of groundwater with organic pollutants remains a significant threat to drinking water supplies.1 Attempts to employ ex situ treatment are often expensive and require decades to complete, while bioremediation and physical isolation of contaminants are difficult to use at many complex sites. As a result, engineers are increasingly using in situ chemical oxidation (ISCO) as an expedient approach for contaminant remediation.2−5
ISCO has been used for the treatment of chlorinated organic solvents and petroleum hydrocarbons for over three decades.6−8 Many of the early efforts to employ ISCO used permanganate (MnO4–) or hydrogen peroxide (H2O2). MnO4– reacts quickly with certain contaminants,9−11 but the formation of manganese oxides (e.g., MnO2(s)) can clog soil pores and inhibit oxidant transport in the subsurface. H2O2 reacts with minerals in the subsurface to produce hydroxyl radicals (HO•) via Fenton-like reactions.12 Because HO• reacts rapidly with H2O2 to produce O2 and H2O13 and Fenton-like reactions typically exhibit low yields at circumneutral pH values, the process is relatively inefficient.14−17 In addition, catalase enzyme activity in aquifer materials can decompose H2O2 through a nonradical pathway.13 As a result of its high reactivity, H2O2 decomposes quickly in the subsurface, which can make it difficult to remediate contaminants that are located far from the reagent injection point.
While permanganate and hydrogen peroxide are still used frequently, persulfate (S2O82–) has recently become popular as an ISCO oxidant.5,18−20 The mechanism through which S2O82– oxidizes contaminants is similar to the way in which H2O2 is converted to HO• by ultraviolet light, with thermolytic cleavage of the peroxide bond slowly producing sulfate radical (SO4•–) instead of HO•:
| 1 |
The formation of radicals from persulfate is commonly referred to as persulfate activation. However, in many cases it is difficult to measure sulfate radical concentrations and a decrease in persulfate loss is often used as a proxy for radical production. In such cases it may be more appropriate to refer to persulfate decomposition instead of activation, and this is the convention that is followed throughout this manuscript.
At 25 °C, the half-life of S2O82– is approximately 600 days, which means that groundwater must be heated if this mechanism is to be practical for remediation.21−25 Other approaches for accelerating the rate of S2O82– decomposition include UV irradiation,26−28 electrochemical systems,29 zerovalent iron,30 ferrous31 or cobalt salts,32 and base activation.33 Base activation is used most commonly in field applications.33 Each of these mechanisms has its limitations. For example, use of ultraviolet light in subsurface remediation is impractical, cobalt is a toxic metal, Fe(II) salts are rapidly oxidized to insoluble Fe(III)-oxides in the presence of persulfate, and thermal activation is energy intensive under the conditions encountered in soils and aquifers.
Compared to the mechanisms listed above, persulfate decomposition by reactions at mineral surfaces are not as well-known or understood. Results from recent studies suggest that aquifer materials and natural organic matter (NOM) can increase persulfate decomposition rates.34−36 The rate of decomposition was positively correlated with the content of amorphous iron in aquifer solids.34 Empirical studies also demonstrated that the decomposition rate was enhanced by approximately an order of magnitude by the presence of aquifer solids.21,34 Although the disappearance of persulfate during ISCO is slow compared to that of H2O2, the low reactivity of persulfate in aquifer solids can prove advantageous because it may allow the oxidant to reach source zones. It also may be useful in remediation of dilute plumes. However, there is a lack of fundamental understanding of the factors that control the rate of persulfate decomposition by aquifer materials. In addition to Fe(III) and Mn(IV) content, other factors that may affect the rate of persulfate decomposition include pH and the concentration of radical scavengers.
To provide insight into the role of naturally occurring minerals in persulfate decomposition and to develop a means of predicting the rate of persulfate loss during ISCO treatment, persulfate decomposition was studied under well-controlled conditions. Experiments were conducted using suspensions of pure minerals and aquifer solids to decompose persulfate in the presence of a pH buffer. The production of SO4•– or HO• was followed using benzene as a probe compound. Concentrations of persulfate and phenol (i.e., a benzene oxidation product) were measured under conditions expected in groundwater.
Materials and Methods
All chemicals used in this study were reagent grade or higher. All solutions were prepared using deionized water (resistivity >18.2 MΩ, Millipore system). Ferrihydrite (Fe(OH)3(s)) and pyrolusite (β-MnO2(s)) were obtained from Sigma-Aldrich. Crystallized silica (SiO2(s), i.e., pure sand, ACROS Organics, Inc.) was cleaned to remove trace amounts of metals according to the procedure described in the Supporting Information (SI) section. Goethite (α-FeOOH(s)) was synthesized by mixing 1 M Fe(NO3)3 solution with 5 M KOH and then aging the precipitates at 70 °C for 60 h.37 The synthesized goethite particles were rinsed with deionized water and dried using a Labconco Freezone 4.5 freeze-dry system (Labconco Corp., Kansas City, MO). The synthesized goethite was characterized by X-ray diffraction (XRD) to confirm its purity (SI Figure S1).
Two clay materials were obtained from the Clay Minerals Society: Australian nontronite (26.0% Fe by weight) and Wyoming montmorillonite (2.59% Fe by weight). In addition, aquifer materials were obtained from five sites in California and Arizona. Details of the physicochemical characteristics of pure minerals, clays, and aquifer materials are provided in SI Table S1. Additional information was provided previously.17 Prior to use, each material was crushed with a mortar and pestle and sieved to collect particles with sizes between 38 and 150 μm (sieves with mesh 400 and 100, respectively). The sieved particles were autoclaved at 120 °C and 20 psi for 45 min and stored under sterile conditions.
Persulfate decomposition experiments were carried out under chemical conditions typical of groundwater. Prior to initiation of each experiment, suspensions were prepared with solids concentrations between 12.5 and 125 g/L using air-saturated deionized water or synthetic groundwater (SI Table S2). In some experiments, the suspension was purged with N2 gas to achieve O2-free conditions and tests were conducted in a glovebox. In most experiments, the solution was maintained at pH 8.0 with 50 mM borate buffer prepared by adding 15 mL of a solution of 1 M of boric acid (H2BO3) and 62.5 mM of sodium tetraborate (Na2B4O7). The use of high buffer concentration was necessary to maintain a constant pH throughout the experiment. For some experiments, additional NaHCO3 was added along with the borate buffer to reach a bicarbonate concentration between 0.1 and 10 mM.
Several experiments were conducted with benzene at concentrations ranging from 0.1 to 1 mM. Benzene solutions were prepared by diluting from a 1.2 mM stock solution that was freshly prepared by dissolving 10 μL of anhydrous benzene (purity ≥99.8%, Sigma-Aldrich Inc.) in a 1 L glass volumetric flask filled with deionized water without headspace. No benzene was volatilized during this preparation step.
To start a decomposition experiment, persulfate was added to yield initial concentrations ranging from 1 to 50 mM, using aliquots of a freshly prepared 100 mM K2S2O8 solution. After mixing, the suspension was immediately transferred to multiple sealed glass tubes with no headspace and placed on a rotating mixer (Labquake Tube Rotators, Thermo Scientific Inc.). Each tube was treated as a sacrificial reactor and discarded after sampling. Most conditions were tested in triplicate, along with a homogeneous control consisting of only persulfate. For experiments with benzene, two additional controls consisting of only benzene and benzene with solids were included. All experiments were conducted at room temperature of 23 ± 2 °C.
At predetermined sampling intervals, each sealed sacrificial tube reactor was centrifuged at 3000 g for 3 min to separate the solids from the solution. The supernatant was immediately removed and filtered through a 0.22-μm nylon filter prior to analysis for persulfate, benzene and oxidation products. The solids were resuspended in acetonitrile, agitated vigorously with a vortex mixer, and then placed on a rotating mixer for 3 h to extract benzene and phenol from the solids. The acetonitrile was separated from the solids using the same centrifugation and filtration procedure and analyzed for adsorbed benzene and phenol.
Persulfate was measured using the KI colorimetric method38 with a Lambda-14 UV spectrophotometer (PerkinElmer Inc., Waltham, MA). Benzene and phenol were analyzed on a Waters Alliance 2695 HPLC (Waters Corp., Milford, MA) equipped with a diode array detector. A Waters Symmetry-C18 column was used with a mobile phase of 40% acetonitrile and 60% 10 mM formic acid at a flow rate of 1 mL/min. Dissolved oxygen concentrations were determined using a YSI Model 58 oxygen probe (YSI Inc., Yellow Springs, OH).
Results and Discussion
Decomposition of Persulfate by Minerals
As expected, the rate of persulfate loss in the absence of solids (i.e., homogeneous control) at room temperature was very slow (Figure 1A). The small loss of persulfate in the homogeneous control was consistent with the predicted thermal decomposition of S2O82– to SO4•– (reaction 1). Using the activation energy measured previously,21 a loss of only 3% of persulfate was predicted over 30 days, which was similar to the measured loss of 2% in our experiments. The heterogeneous system with silica exhibited less than 3% persulfate loss over 32 days, which was indistinguishable from the homogeneous control. The addition of iron- and manganese-containing oxides accelerated the decomposition rate significantly, with an average of 12%, 65%, and 45% of persulfate loss over 32 days for 100 g/L suspensions of goethite, ferrihydrite, and pyrolusite, respectively (Figure 1A). The decomposition rate was affected by the mass of solids in the suspension. For example, reducing the mass of pyrolusite from 100 to 50 g/L decreased persulfate loss from 45% to 10% over 32 days, while increasing its mass from 100 to 200 and 500 g/L increased the loss to 60% and 90% over the same period, respectively (Figure 1B). Increasing the mass of silica did not affect the rate of persulfate loss (SI Figure S2).
Figure 1.
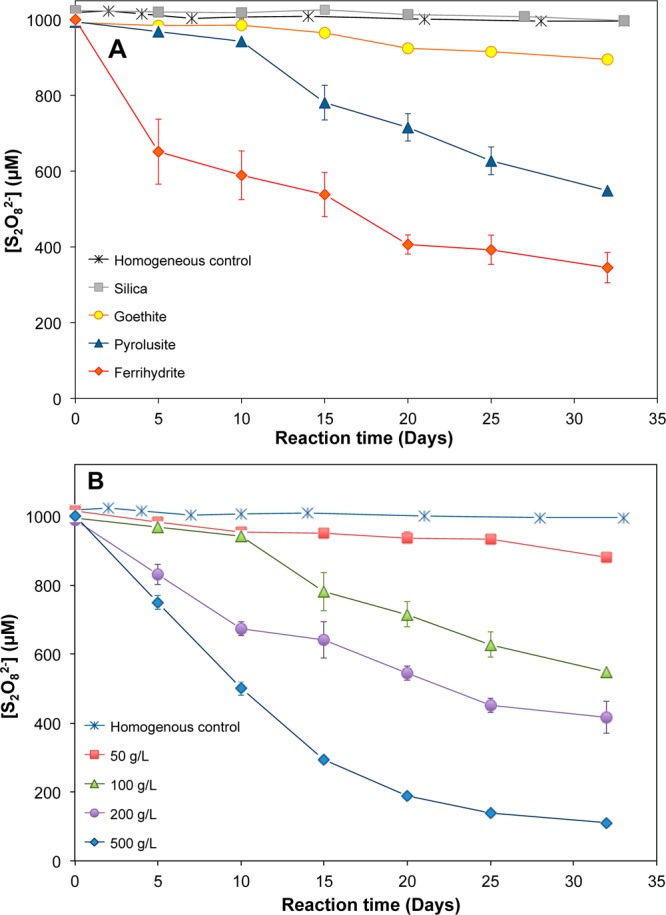
Stability and decomposition of persulfate in different systems. Initial persulfate concentration 1 mM, pH 8.0, ionic strength 50 mM. (A) Impact of different solids along with a homogeneous control, solid mass loading 100 g/L. (B) impact of pyrolusite mass loading on persulfate decomposition.
To examine the effect of suspended mineral mass on persulfate decomposition, data were converted to pseudo-first-order kinetics with the following expression:
In this expression, kobs represents the observed pseudo-first-order rate constant (s–1), kSA is the surface area normalized pseudo-first-order rate constant (L·m–2·s–1), and CS is the BET surface area of the suspended solids (m2·L–1).
The surface area normalized decomposition rate constants for different solids varied by approximately 6 orders of magnitude (Figure 2). The relative reactivity of the minerals followed the order pyrolusite > ferrihydrite > goethite > silica. Nontronite, an iron-rich clay and montmorillonite, a clay with a relatively high manganese content, were both capable of persulfate decomposition, with montmorillonite decomposing persulfate at a rate that was approximately an order of magnitude higher than that of nontronite (Figure 2). The aquifer materials exhibited decomposition rates that were similar to those observed for the iron oxides (Figure 2). These aquifer materials contained between 0.7 and 2.5% Fe and between 0.01 and 0.12% Mn. Iron- and manganese-containing minerals also decomposed persulfate at initial persulfate concentrations ranging from 5 to 50 mM with 50 g/L of each mineral (SI Figure S3).
Figure 2.
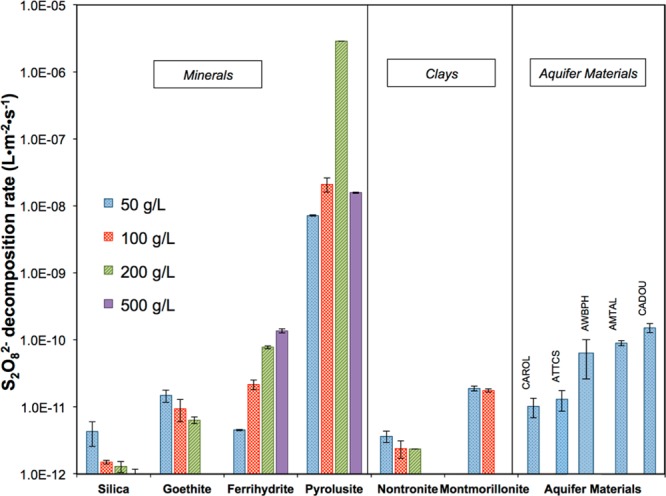
Comparison of rate constants of persulfate decomposition by different solids at varying concentrations. Rate constants are normalized by BET surface areas and corrected for thermal decomposition at room temperature. Initial persulfate 1 mM, pH 8.0. Error bar represents one standard deviation.
Reaction Mechanism for Heterogeneous Persulfate Decomposition
By analogy to the Fenton system, we propose that the rate-determining step for persulfate decomposition by an iron-containing mineral can be described by the following reaction:
| 2 |
where ≡Fe(III) represents the redox-active iron surface site. One-electron reduction of ≡Fe(III) would result in the formation of persulfate radical (S2O8•–), which could then initiate radical chain reactions. The existence of S2O8•– is supported by previous studies of the reaction between S2O82– and SO4•–, in which S2O82– was oxidized to S2O8•– via a one-electron transfer reaction.39,40
Similar to the Fenton reaction, the overall reaction rate of persulfate decomposition is likely affected by the number of reactive surface sites and electron transfer rates.41 As in the Fenton system, the surface reactivity of each type of mineral is likely affected by crystallinity and metal coordination. For example, in the Fenton system, the H2O2 decomposition rate was much higher in the presence of amorphous ferrihydrite than crystalline goethite.14 A similar trend was also observed in our experiments, with ferrihydrite exhibiting higher rate constants than goethite. Similarly, our observation of higher reactivity for manganese oxides was consistent with observations from the Fenton system.17
Previous research has demonstrated persulfate decomposition by dissolved transition metals, including Fe2+, Mn2+, Ag+, Co2+, and Ni2+, under acidic conditions (i.e., pH < 3).31,42 Acidic conditions are potentially problematic in groundwater because they can release toxic metals and alter minerals. Under circumneutral pH conditions, transition metals associated with surfaces can undergo similar reactions. After surface metals are reduced via reaction 2, ≡Fe(II) should be oxidized rapidly by persulfate to regenerate ≡Fe(III) with the production of SO4•– and sulfate:
| 3 |
Therefore, the metal catalyzed decomposition of persulfate proceeds as follows:
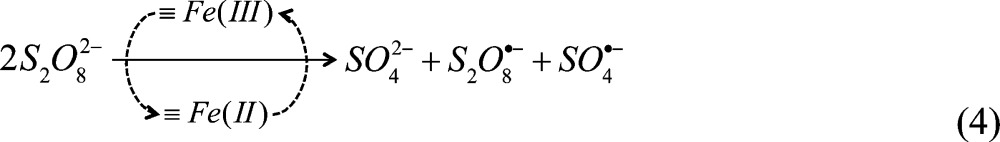 |
4 |
The fact that persulfate can be both oxidized and reduced is similar to the mechanism by which H2O2 acts as both an oxidant and reductant in the Fenton system. In addition, S2O8•– can be produced through a radical chain propagation reaction:40,43,44
| 5 |
The fate of S2O8•– is unclear. Previous studies on the fate of the radical did not examine its decomposition mechanism.45,46 One possible fate of S2O8•– is oxidation of water, which would occur through a multistep process with the following stoichiometry:
| 6 |
OH– could also serve as a source of HO• 46 and SO4•– can also oxidize water,45 and under alkaline conditions
| 7 |
| 8 |
| 9 |
In the absence of organic solutes, Reactions 6 and 9 serve as chain termination steps.
The overall reaction of persulfate decomposition in water is
| 10 |
Results from selected experiments in which sulfate production was measured along with persulfate loss showed good agreement with the overall stoichiometry of reaction 10 (SI Figure S4A). For Mn(IV)-containing materials, a similar reaction pathway can take place through a redox cycle involving Mn(IV)/Mn(III) surface species to generate sulfate radical and initiate chain reactions.
To assess the possible role of dissolved O2 in persulfate decomposition, experiments were conducted in the absence of oxygen with a pyrolusite suspension. Results showed that the presence of dissolved O2 had no effect on the rate of persulfate loss (SI Figure S4B). In addition, dissolved O2 was produced as S2O82– decomposed. The observed stoichiometric ratio of O2 produced per S2O82– lost was approximately 0.5 (SI Figure S5), which was consistent with reaction 10.
Impact of Organic Compounds on Persulfate Decomposition
In the presence of organic contaminants, the radical chain decomposition of persulfate changes. Using benzene as a representative organic contaminant, the stability of persulfate was examined (Figures 3 and 4). The rate of persulfate decomposition increased as benzene concentrations increased from 0 to 1000 μM. For example, in the presence of 50 g/L ferrihydrite, approximately 15% of persulfate was decomposed over 30 days in the absence of benzene. When 100 μM of benzene was added, persulfate decomposition accelerated significantly, with 45% loss over 30 days. At a benzene concentration of 1000 μM, approximately 75% of persulfate was decomposed over the same period (Figure 3). These results indicated that the half-life of persulfate would decrease from 170 days in the absence of benzene to 45 and 20 days in the presence of 100 and 1000 μM of benzene, respectively. A similar trend was observed for other pure minerals, with the exception of silica, which was not capable of catalyzing persulfate decomposition. For the aquifer materials, benzene appeared to have an effect, but it was harder to detect due to higher variability among replicates due to heterogeneity of the aquifer materials.
Figure 3.
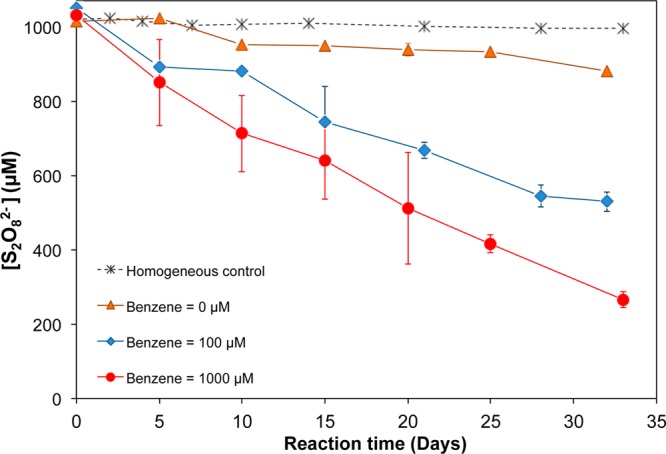
Impact of benzene concentration on the decomposition of persulfate in the presence of 50 g/L ferrihydrite, pH 8.0. No mineral was present in the homogeneous control.
Figure 4.
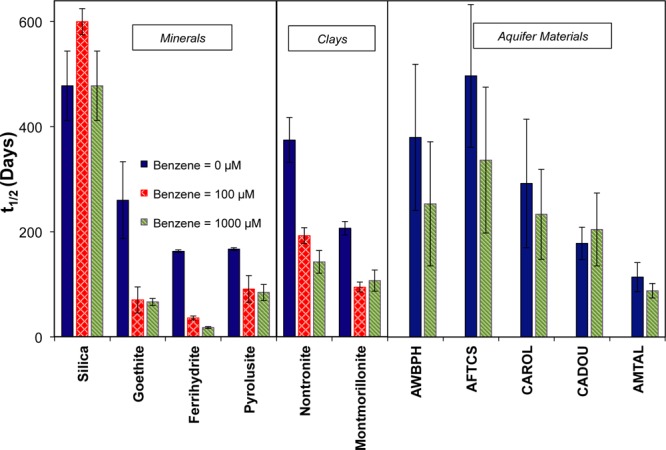
Impact of benzene concentration on the decomposition of persulfate in the presence of different solids. Solids mass loading 50 g/L, pH 8.0.
The acceleration of persulfate decomposition in the presence of benzene may be attributable to radical chain reactions initiated by oxidation of benzene. Previous research using HO• has shown that the intermediate products of benzene oxidation include organic radicals that can propagate the chain.28,48 For example, the reaction of HO• with benzene results in formation of a cationic radical that is subsequently hydrolyzed to hydroxyl-cyclohexadienyl (HCHD) radical.48 HCHD radical further reacts with oxygen to produce an organic peroxy radical that generates HO2• and phenol. Under circumneutral pH conditions, HO2• will deprotonate to superoxide (O2•–), which can undergo iron-catalyzed dismutation:
| 11 |
| 12 |
During this process, the faster rates of reduction of Fe(III) relative to oxidation of Fe(II) favor the presence of reduced Fe(II),49 thereby contributing to oxidant decomposition. H2O2 produced by superoxide dismutation reacts quickly with iron and manganese through Fenton-like reactions on mineral surfaces.13,14,17
Presumably a similar radical chain mechanism occurs during the reaction of benzene with SO4•–. A second-order rate constant of 3 × 109 M–1·s–1 has been reported for the initial step of this reaction:50
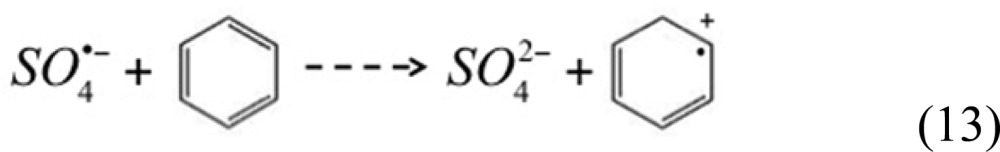 |
13 |
Because the branching ratio of reaction 13 is much bigger than those of reactions 5 and 7 under experimental conditions used in this study (i.e., k13[benzene] ≫ k5[S2O82–]; k13[benzene] ≫ k7[H2O], reaction rate constants provided in SI Table S4), essentially all SO4•– reacted with benzene instead of persulfate or water when benzene was present.
The acceleration of persulfate decomposition by benzene was also observed in experiments using synthetic groundwater that contained chloride, bromide, carbonate and natural organic matter (SI Table S2). In these experiments, the rates of persulfate decomposition by iron and manganese-containing minerals were consistent with those measured in benzene solutions prepared with borate-buffered water (SI Table S3).
Under typical groundwater conditions, additional reactions between SO4•– and chloride or bromide can take place:44,51
| 14 |
| 15 |
Cl• and Br• radicals can further react with water to produce other short-lived radicals.52−54 However, in the presence of 1 mM benzene and under typical groundwater conditions, chloride and bromide are minor sinks for sulfate radical (i.e., k13[benzene] > k14[Cl–]; k13[benzene] > k15[Br–]). Therefore, groundwater constituents only had a small impact on the scavenging of SO4•– radicals in the presence of benzene.
In addition, under groundwater conditions, SO4•– generated through reaction 3 can react with bicarbonate:47
| 16 |
CO3•– can then react with persulfate to cause further decomposition of persulfate:
| 17 |
However, for the range of concentrations studied in these experiments ([HCO3–] = 1–10 mM and [S2O82–] = 1 mM) and in the presence of 1 mM benzene, bicarbonate is not a major sink for sulfate radical (i.e., k16[HCO3–] < k13[benzene]) and reaction 17 will not be important to persulfate decomposition. This is consistent with observations from experiments in which persulfate decomposition was measured in the presence of benzene at varying concentrations of bicarbonate (SI Figure S6).
The presence of SO4•– or HO• during persulfate decomposition in heterogeneous systems was confirmed by measurements of phenol production. In heterogeneous systems with Fe(III)- and Mn(IV)-containing minerals, an initial persulfate concentration of 1 mM and an initial benzene concentration of 100 μM, phenol was formed in the presence of different solids (Figure 5). For example, approximately 25 μM of phenol was generated in the presence of 50 g/L goethite, nontronite, and montmorillonite after 30 days. Fifteen micromolar of phenol was produced in pyrolusite, and 5 μM, in ferrihydrite. Benzene loss ranged from 20 to 40 μM in these experiments.
Figure 5.
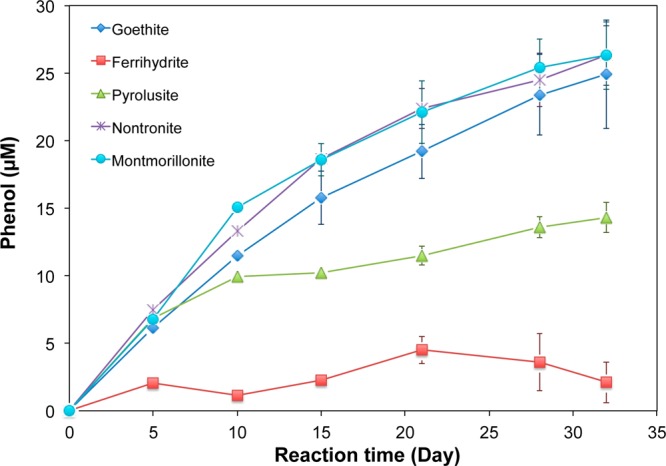
Formation of phenol as oxidation product of benzene by sulfate radical in the presence of 50 g/L different Fe- or Mn-containing oxides and aquifer materials. Initial benzene concentration 100 μM, initial persulfate concentration 1 mM, pH 8.0, ionic strength 55 mM.
Environmental Implications
The findings from this study have significant implications for the use of persulfate for ISCO applications. First, the observation that iron- and manganese-containing minerals decompose persulfate under conditions encountered in the subsurface allow for prediction of the rate at which SO4•– or HO• is produced in the absence of added activators, such as heat or base. In aquifers dominated by pure sand, persulfate is very stable with half-lives of more than 2 years. If Fe(III)- or Mn(IV)-containing oxides comprise greater than approximately 2% of aquifer solids, the rate of persulfate decomposition will increase, with half-lives decreasing to less than a year. When persulfate encounters a zone that is rich in iron- or manganese-oxides or clay, its half-life can decrease substantially.
Second, once persulfate is injected into the subsurface, its treatment efficacy may be affected by the distance that it travels in the surface. In many situations, it may be preferable for persulfate to persist in the aquifer for many months to allow it to reach contaminants distant from injection wells. Results from this study show that persulfate compares favorably to H2O2-based ISCO systems in this respect. Persulfate decomposition was much slower than hydrogen peroxide decomposition in the Fenton system, with a half-life that was more than 2 orders of magnitude longer (SI Figure S7). Hydrogen peroxide was decomposed over several hours (SI Figure S8A), whereas persulfate decomposition took several weeks (SI Figure S8B). Considering different hydraulic conductivities in the subsurface and uncertainties associated with the rate of oxidant transport in the aquifer, the high stability of persulfate in uncontaminated aquifer zones indicates more effective transport and delivery of oxidants.
Finally, the rate of persulfate decomposition accelerates when the injected oxidant reaches a contaminant plume. The half-life of persulfate in the aquifer can be more than 1 year in the absence of an organic contaminant. Once persulfate encounters a contaminant such as benzene, radical chain reactions increase the persulfate decomposition rate and its half-life can decrease to as little as a few days. Its half-life can also decrease in the presence of iron- or manganese-containing oxides or clays, which are frequently associated with low hydraulic conductivity regions where contaminants often accumulate.
Acknowledgments
This study was supported by the U.S. National Institute for Environmental Health Sciences (NIEHS) Superfund Research Program (Grant P42 ES004705) and the Superfund Research Center at University of California, Berkeley. We thank our group member Anh Pham for stimulating discussions and Dr. Urs Jans for his help in conducting initial experiments that helped us to develop a means of studying these slow reactions.
Supporting Information Available
Additional description of iron and manganese mineral preparation and eight figures on the rate of persulfate decomposition and product formation in different conditions. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
‡ Department of Chemical and Environmental Engineering, University of California at Riverside, Riverside, CA 92521, USA.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Alternatives for Managing the Nation’s Complex Contaminated Groundwater Sites; The National Academies Press, 2013. [Google Scholar]
- Siegrist R. L.; Crimi M.; Simpkin T. J.. In situ chemical oxidation: technology, description and status. In situ chemical oxidation for groundwater remediation; Springer Media, LLC: New York City, 2011; Chapter 1. [Google Scholar]
- Interstate Technology and Regulatory Council (ITRC). Technical and Regulatory Guidance for In Situ Chemical Oxidation of Contaminated Soil and Groundwater, 2nd ed.; 2005; http://www.itrcweb.org.
- Krembs F. J.; Siegrist R. L.; Crimi M. L.; Furrer R. F.; Petri B. G. ISCO for groundwater remediation: analysis of field applications and performance. Ground Water Monit. Rem. 2010, 30442–53. [Google Scholar]
- Tsitonaki A.; Petri B.; Crimi M.; Mosbaek H.; Siegrist R. L.; Bjerg P. L. In Situ Chemical Oxidation of Contaminated Soil and Groundwater Using Persulfate: A Review. Crit. Rev. in Environ. Sci. and Technol. 2010, 40155–91. [Google Scholar]
- Miller C. M.; Valentine R. L. Hydrogen peroxide decomposition and quinoline degradation in the presence of aquifer material. Water Res. 1995, 29102353–2359. [Google Scholar]
- Gates D. D.; Siegrist R. L. In-situ chemical oxidation of trichloroethylene using hydrogen-peroxide. J. Environ. Eng. 1995, 1219639–644. [Google Scholar]
- Yan Y. E.; Schwartz F. W. Oxidative degradation and kinetics of chlorinated ethylenes by potassium permanganate. J. Contam. Hydrol. 1999, 373–4343–365. [Google Scholar]
- Waldemer R. H.; Tratnyek P. G. Kinetics of contaminant degradation by permanganate. Environ. Sci. Technol. 2006, 4031055–1061. [DOI] [PubMed] [Google Scholar]
- Poulson S. R.; Naraoka H. Carbon isotope fractionation during permanganate oxidation of chlorinated ethylenes (cDCE, TCE, PCE). Environ. Sci. Technol. 2002, 36153270–3274. [DOI] [PubMed] [Google Scholar]
- Huang K. C.; Hoag G. E.; Chheda P.; Woody B. A.; Dobbs G. M. Oxidation of chlorinated ethenes by potassium permanganate: a kinetics study. J. Hazard. Mater. 2001, 871155–169. [DOI] [PubMed] [Google Scholar]
- Pignatello J. J.; Oliveros E.; MacKay A. Advanced oxidation processes for organic contaminant destruction based on the Fenton reaction and related chemistry. Crit. Rev. in Environ. Sci. Technol. 2006, 3611–84. [Google Scholar]
- Petigara B. R.; Blough N. V.; Mignerey A. C. Mechanisms of hydrogen peroxide decomposition in soils. Environ. Sci. Technol. 2002, 364639–645. [DOI] [PubMed] [Google Scholar]
- Kwan W.; Voelker B. M. Rates of hydroxyl radical generation and organic compound oxidation in mineral-catalyzed Fenton-like systems. Environ. Sci. Technol. 2003, 3763270–3274. [DOI] [PubMed] [Google Scholar]
- Xu X.; Thomson N. R. Hydrogen peroxide persistence in the presence of aquifer materials. Soil Sediment Contam. 2010, 195602–616. [Google Scholar]
- Watts R. J.; Finn D. D.; Cutler L. M.; Schmidt J. T.; Teel A. L. Enhanced stability of hydrogen peroxide in the presence of subsurface solids. J. Contam. Hydrol. 2007, 913312–326. [DOI] [PubMed] [Google Scholar]
- Pham A. L.; Doyle F. M.; Sedlak D. L. Kinetics and efficiency of H2O2 activation by iron-containing minerals and aquifer materials. Water Res. 2012, 46196454–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C.; Bruell C. J.; Marley M. C.; Sperry K. L. Thermally activated persulfate oxidation of trichloroethylene (TCE) and 1,1,1-trichloroethane (TCA) in aqueous systems and soil slurries. Soil Sediment Contam. 2003, 122207–228. [Google Scholar]
- Waldemer R. H.; Tratnyek P. G.; Johnson R. L.; Nurmi J. T. Oxidation of chlorinated ethenes by heat-activated persulfate: kinetics and products. Environ. Sci. Technol. 2007, 4131010–1015. [DOI] [PubMed] [Google Scholar]
- Costanza J.; Otaño G.; Callaghan J.; Pennell K. D. PCE oxidation by sodium persulfate in the presence of solids. Environ. Sci. Technol. 2010, 44249445–9450. [DOI] [PubMed] [Google Scholar]
- Johnson R. L.; Tratnyek P. G.; Johnson R. O. Persulfate persistence under thermal activation conditions. Environ. Sci. Technol. 2008, 42249350–9356. [DOI] [PubMed] [Google Scholar]
- Hori H.; Nagaoka Y.; Murayama M.; Kutsuna S. Efficient decomposition of perfluorocarboxylic acids and alternative fluorochemical surfactants in hot water. Environ. Sci. Technol. 2008, 42197438–7443. [DOI] [PubMed] [Google Scholar]
- Liu C. S.; Higgins C. P.; Wang F.; Shih K. Effect of temperature on oxidative transformation of perfluorooctanoic acid (PFOA) by persulfate activation in water. Sep. Purif. Technol. 2012, 91, 46–51. [Google Scholar]
- Lee Y.; Lo S.; Kuo J.; Hsieh C. Decomposition of perfluorooctanoic acid by microwave-activated persulfate: Effects of temperature, pH, and chloride ions. Front. Environ. Sci. Engr. China. 2012, 6117–25. [Google Scholar]
- Deng Y.; Ezyske C. M. Sulfate radical-advanced oxidation process (SR-AOP) for simultaneous removal of refractory organic contaminants and ammonia in landfill leachate. Water. Res. 2011, 45186189–6194. [DOI] [PubMed] [Google Scholar]
- Hori H.; Yamamoto A.; Hayakawa E.; Taniyasu S.; Yamashita N.; Kutsuna S. Efficient decomposition of environmentally persistent perfluorocarboxylic acids by use of persulfate as a photochemical oxidant. Eniron. Sci. Technol. 2005, 3972383–2388. [DOI] [PubMed] [Google Scholar]
- Lau T. K.; Chu W.; Graham N. J. D. The aqueous degradation of butylated hydroxyanisole by UV/S2O82-: study of reaction mechanisms via dimerization and mineralization. Environ. Sci. Technol. 2007, 412613–619. [DOI] [PubMed] [Google Scholar]
- Antoniou M. G.; De La Cruz A. A.; Dionysiou D. D. Intermediates and reaction pathways from the degradation of microcystin-LR with sulfate radicals. Environ. Sci. Technol. 2010, 37197238–7244. [DOI] [PubMed] [Google Scholar]
- Yuan S.; Liao P.; Alshawabkeh A. N. Electrolytic manipulation of persulfate reactivity by iron electrodes for trichloroethylene degradation in groundwater. Environ. Sci. Technol. 2014, 481656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S.; Peterson T. D.; Righter J.; Miles D. M.; Tratnyek P. G. Disinfection of ballast water with iron activated persulfate. Environ. Sci. Technol. 2013, 472011717–11725. [DOI] [PubMed] [Google Scholar]
- Liang C.; Bruell C. J.; Marley M. C.; Sperry K. L. Persulfate oxidation for in situ remediation of TCE. II. Activated by chelated ferrous ion. Chemosphere 2004, 5591225–1233. [DOI] [PubMed] [Google Scholar]
- Anipsitakis G. P.; Dionysiou D. D.; Gonzalez M. A. Cobalt-mediated activation of peroxymonosulfate and sulfate radical attack on phenolic compounds. Implications of chloride ions. Environ. Sci. Technol. 2006, 4031000–1007. [DOI] [PubMed] [Google Scholar]
- Furman O. S.; Teel A. L.; Watts R. J. Mechanism of base activation of persulfate. Environ. Sci. Technol. 2010, 44166423–6428. [DOI] [PubMed] [Google Scholar]
- Sra K. S.; Thomson N. R.; Barker J. F. Persistence of persulfate in uncontaminated aquifer materials. Environ. Sci. Technol. 2010, 4483098–3104. [DOI] [PubMed] [Google Scholar]
- Ahmad M.; Teel A. L.; Watts R. J. Persulfate activation by subsurface minerals. J. Contam. Hydrol. 2010, 115134–45. [DOI] [PubMed] [Google Scholar]
- Teel A. L.; Ahmad M.; Watts R. J. Persulfate activation by naturally occurring trace minerals. J. Haz. Mater. 2011, 196, 153–159. [DOI] [PubMed] [Google Scholar]
- Schwertmann U.; Cornell R. M.. Iron oxides in the laboratory: preparation and characterization; VCH Publisher, Inc.: New York, 1991; pp 64–71. [Google Scholar]
- Liang C.; Huang C. F.; Mohanty N.; Kurakalva R. M. A rapid spectrophotometric determination of persulfate anion in ISCO. Chemosphere 2008, 7391540–1543. [DOI] [PubMed] [Google Scholar]
- McElroy W. J.; Waygood S. J. Kinetics of the reactions of the SO4– radical with SO4–, S2O82-, H2O and Fe2+. J. Chem. Soc. Faraday Trans. 1990, 86142557–2564. [Google Scholar]
- Yu X. Y.; Bao Z. C.; Barker J. R. Free radical reactions involving Cl•, Cl2–•, and SO4•– in the 248 nm photolysis of aqueous solutions containing S2O82- and Cl–. J. Phys. Chem. A 2004, 1082295–308. [Google Scholar]
- Lin S.; Gurol M. D. Catalytic decomposition of hydrogen peroxide on iron oxide: kinetics, mechanism, and implications. Environ. Sci. Technol. 1998, 32101417–1423. [Google Scholar]
- Anipsitakis G. P.; Dionysiou D. D. Radical generation by the interaction of transition metals with common oxidants. Environ. Sci. Technol. 2004, 38133705–3712. [DOI] [PubMed] [Google Scholar]
- Schuchmann H. P.; Deeble D. J.; Olbrich G.; von Sontag C. The SO4•– -induced chain reaction of 1,3- dimethyluracil with peroxodisulphate. Int. J. Radiat. Biol. 1987, 51, 441–453. [DOI] [PubMed] [Google Scholar]
- Das T. N. Reactivity and role of SO5•– radical in aqueous medium chain oxidation of sulfite to sulfate and atmospheric sulfuric acid generation. J. Phys. Chem. A 2001, 105409142–9155. [Google Scholar]
- Herrmann H.; Reese A.; Zellner R. Time-resolved UV/VIS diode-array absorption spectroscopy of SOx– (x=3, 4, 5) radical anions in aqueous solution. J. Mol. Struct. 1995, 348, 183–186. [Google Scholar]
- Neta P.; Huie R. E.; Ross A. B. Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Ref. Data. 1988, 1731027–1284. [Google Scholar]
- Zuo Z. H.; Cai Z. L.; Katsumura Y.; Chitose N.; Muroya Y. Reinvestigation of the acid-base equilibrium of the (bi)carbonate radical and pH dependence of its reactivity with inorganic reactants. Radiat. Phys. Chem. 1999, 55115–23. [Google Scholar]
- Pan X.; Schuchmann M. N.; von Sontag C. Oxidation of benzene by the OH radical. A product and pulse radiolysis study in oxygenated aqueous solution. J. Chem. Soc. Perkin Trans. 2. 1993, 3, 289–297. [Google Scholar]
- Sedlak D. L.; Hoigne J. The role of copper and oxalate in the redox cycling of iron in atmospheric waters. Atmosph. Env. Part A. Gen. Top. 1993, 27142173–2185. [Google Scholar]
- Neta P.; Madhavan V.; Zemel H.; Fessenden R. W. Rate constants and mechanism of reaction of sulfate radical anion with aromatic compounds. J. Am. Chem. Soc. 1977, 991163–164. [Google Scholar]
- Peyton G. R. The free-radical chemistry of persulfate-based total organic carbon analyzers. Mar. Chem. 1993, 41191–103. [Google Scholar]
- Jayson G.; Parsons B.; Swallow A. J. Some simple, highly reactive, inorganic chlorine derivatives in aqueous solution. Their formation using pulses of radiation and their role in the mechanism of the Fricke dosimeter. J. Chem. Soc., Faraday Trans. 1. 1973, 69, 1597–1607. [Google Scholar]
- Grigorev A. E.; Makarov I. E.; Pikaev A. K. Formation of Cl2– in the bulk solution during the radiolysis of concentrated aqueous-solutions of chlorides. High Energy Chem. 1987, 21299–102. [Google Scholar]
- Zehavi D.; Rabani J. Oxidation of aqueous bromide ions by hydroxyl radicals. A pulse radiolytic investigation. J. Phys. Chem. 1972, 763312–319. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.