

Significance
It is increasingly apparent that bacteria in the gut are important determinants of health and disease in humans. However, we know remarkably little about how this organ transitions from a sterile/near-sterile state at birth to one that soon harbors a highly diverse biomass. We show in premature infants a patterned progression of the gut bacterial community that is only minimally influenced by mode of delivery, antibiotics, or feeds. The pace of this progression is most strongly influenced by gestational age, with the microbial population assembling slowest for infants born most prematurely. These data raise the possibility that host biology, more than exogenous factors such as antibiotics, feeds, and route of delivery, drives bacterial populations in the premature newborn infant gut.
Keywords: mixed model regression analysis, necrotizing enterocolitis, nonmetric multidimensional scaling, prematurity
Abstract
In the weeks after birth, the gut acquires a nascent microbiome, and starts its transition to bacterial population equilibrium. This early-in-life microbial population quite likely influences later-in-life host biology. However, we know little about the governance of community development: does the gut serve as a passive incubator where the first organisms randomly encountered gain entry and predominate, or is there an orderly progression of members joining the community of bacteria? We used fine interval enumeration of microbes in stools from multiple subjects to answer this question. We demonstrate via 16S rRNA gene pyrosequencing of 922 specimens from 58 subjects that the gut microbiota of premature infants residing in a tightly controlled microbial environment progresses through a choreographed succession of bacterial classes from Bacilli to Gammaproteobacteria to Clostridia, interrupted by abrupt population changes. As infants approach 33–36 wk postconceptional age (corresponding to the third to the twelfth weeks of life depending on gestational age at birth), the gut is well colonized by anaerobes. Antibiotics, vaginal vs. Caesarian birth, diet, and age of the infants when sampled influence the pace, but not the sequence, of progression. Our results suggest that in infants in a microbiologically constrained ecosphere of a neonatal intensive care unit, gut bacterial communities have an overall nonrandom assembly that is punctuated by microbial population abruptions. The possibility that the pace of this assembly depends more on host biology (chiefly gestational age at birth) than identifiable exogenous factors warrants further consideration.
The vertebrate digestive system hosts a profound transition from a state of complete or near-sterility in utero to dense bacterial colonization within weeks of birth. This event has lasting effects on the host (1), influencing health and development (2–4), infection resistance (5, 6), predisposition to inflammatory (7) and metabolic disorders (8), and immune function (9), but remarkably little is known about this process. Gut colonization has been partly characterized in term infants (10–12) who reside in open venues, and who will, therefore, experience many exposures (e.g., contact with older children, adults, and pets, varying diets, oral antibiotics) that could drive microbial population assembly (1, 11–13).
A delineation of the dynamics of the natural de novo assembly of this microbial community would form a basis for better understanding how the gut acquires its founding microbiome, and how the bacteria in the gut start their transition to population equilibrium (1, 14, 15). In view of the importance of bacterial gut colonization, we sought to determine if the initial assembly of host intestinal microbial populations follows discernible patterns, and if interventions such as antibiotics or nutrition alter this progression. A discernibly patterned progression would suggest that host biology influences bacterial community assembly more than do random encounters of individuals with microbes, whereas stochastic assembly would suggest that random encounters sculpt population structure. In this latter scenario, the gut serves as a passive culture chamber. Fine interval enumeration of gut contents from multiple subjects in as controlled an environment as possible is needed to answer this question.
Here, we demonstrate that the gut microbiota of premature infants residing in a tightly controlled environment of a neonatal intensive care unit (NICU) progresses through a choreographed succession of bacterial classes from Bacilli to Gammaproteobacteria to Clostridia interrupted by abrupt population changes. The rate of assembly is slowest for the most premature of these infants.
Results
To characterize the bacterial in-population of the human gut, we collected all stools from an informative cohort of 58 premature infants weighing ≤1,500 g at birth who resided in the St. Louis Children’s Hospital NICU (Table 1) (16). This is an environment in which staff and protocols attempt to limit exposure to microbes to the extent possible. Infants leave the NICU only for brief therapeutic or diagnostic excursions. Staff and visitors (restricted to immediate family) are drilled in assiduous hygienic techniques, and enteral nutrition is limited to breast milk or sterilized formula. Registered nurses record each major event and handle all stools, and electronic records retain extensive clinical metadata. Antibiotic use, although common, consists overwhelmingly of beta-lactams, aminoglycosides, and vancomycin infused intravenously. Intraintestinal concentrations of such antimicrobials administered via this route are low (17), although it is possible that these agents affect gut microbes by altering host physiology independent of their antibacterial effects (18, 19).
Table 1.
Details of subject groups
| Gestational ages at birth (n) | All (58) | <26 wk (15) | 26–28 wk (20) | >28 wk (23) |
| Perinatal | ||||
| Vaginal birth, n (%) | 13 (22) | 4 (27) | 6 (30) | 3 (13) |
| Birth weight g, median (IQR) | 960 (800, 1220) | 750 (690, 900) | 895 (770, 980) | 1,250 (1,110, 1,445) |
| Gestational age at birth weeks, median (IQR) | 27.1 (25.6, 29.0) | 25 (24.9, 25.3) | 26.7 (26.3, 27.1) | 29.1 (28.9, 31.6) |
| Male/Female | 24/34 | 4/11 | 10/10 | 10/13 |
| Caucasian/African American | 30/28 | 5/10 | 10/10 | 15/8 |
| APGAR 5 min, median (IQR) | 7.0 (6.0, 8.0) | 6.0 (4.0, 7.0) | 7.0 (5.5, 8.0) | 8.0 (6.0, 9.0) |
| Postnatal | ||||
| First antibiotic free day of life, median (IQR) | 5 (4, 9) | 5 (4, 9) | 8.5 (4, 9.5) | 4 (2, 5) |
| Percent of days of life on antibiotics,* median (IQR) | 14 (5.8, 25.6) | 22.2 (11.4, 30.6) | 17.3 (8.6, 29.0) | 6.4 (5.4, 14.3) |
| Day of life on which enteral feedings initiated, median (IQR) | 5.5 (4.0, 8.0) | 7.0 (6.0, 9.0) | 5 (4.0, 7.0) | 4.0 (3.0, 7.0) |
| Human milk use | ||||
| None†, n (%) | 2 (3) | 0 | 1 (5) | 1 (4) |
| <10%, n (%) | 16 (28) | 4 (27) | 8 (40) | 4 (17) |
| 10–50%, n (%) | 18 (31) | 3 (20) | 4 (20) | 11 (48) |
| >50%, n (%) | 22 (38) | 8 (53) | 7 (35) | 7 (31) |
| Outcome | ||||
| Discharged to home from NICU, n (%) | 57 (98) | 15 (100) | 19 (95) | 23 (100) |
| Day of life discharged home (IQR) | 61 (49, 73) | 78 (75, 81) | 65 (60, 71) | 47 (37, 51) |
| Days to death (n) | 66 (1) | N/A | 66 (1) | N/A |
APGAR score, a measure of physical condition at birth scored from 0 to 10, 10 being highest; N/A, not applicable.
Percent of days of life antibiotics were administered before each specimen.
Percentage of enteral volume provided by human milk.
We sequenced 922 specimens from these 58 infants, none of whom experienced serious prematurity-related intraabdominal pathology such as necrotizing enterocolitis (although seven subjects had one or more positive blood culture). On any one study day, there was a median of one enrolled subject [interquartile range (IQR) 0–3 subjects]. Sequencing generated a median (IQR) of 7,183 (5,243–9,314) reads per specimen, and 400–450 nucleotides per read. Bacilli (19.3%), Gammaproteobacteria (54.0%), and Clostridia (18.4%) represented 91.7% of all bacterial sequences (Dataset S1) and together account for >90% of the sequences generated in 85% of all specimens. Within these three classes, there are 34, 52, and 40 different genera, respectively. Only two infants produced stools where these three classes did not predominate. In these two subjects, each of whom had unremarkable clinical courses, stool bacteria were largely Bacteroidia (Fig. S1).
Within almost all specimens, one or two classes predominate (a single subject example is shown Fig. 1). There are frequent abrupt shifts in bacterial composition (Movie S1). The abrupt shifts are unpredictable in direction and magnitude: some are saltatory changes aligned with the progression from Bacilli to Gammaproteobacteria to Clostridia abundance, and others are random short-lived deviations from the directional progression pattern.
Fig. 1.

Bacterial taxa composition at class level as function of day of life of a single subject. These samples are from subject 32 who was born after 29 wk gestation. Each bar represents the bacterial proportions within single samples. From left to right, bars correspond to stools obtained on days of life 3, 4, 6, 8, 9, 10, 12, 14, 14, 16, 17, 19, 25, and 39, respectively. The predominant classes are Bacilli (dark blue), Gammaproteobacteria (red), and Clostridia (green). Eleven additional classes (light blue) were also present, mostly in stools obtained on days of life 3 and 4.
In multidimensional scaling plots related to day of life, Bacilli are found chiefly in the earliest samples (blue dots surrounding the arrowhead labeled “Bacilli” in Fig. S2A). Clostridia are in greatest abundance in stools from older infants (green dots near Clostridia arrowhead; Fig. S2A), but blue and red dots representing stools from younger children are also well represented in this region of the plot. However, infants in this cohort were born after a wide range of gestational duration (23–33 wk). Therefore, we were concerned that by using day of life as the independent variable, we obscured associations between bacterial content and day of sampling. For this reason, we reportrayed the data by categorizing specimens according to postconceptional age, which is the biologically relevant sum of gestational age at birth plus chronological age (also termed postmenstrual age; ref. 20). In this second portrayal (Fig. S2B), specimens retain their coordinates on the scaling plot reflecting bacterial content, but the dots in the vicinity of the Clostridia arrowhead are more monochromatic (i.e., green). These green symbols represent samples from infants who had attained postconceptional ages between 33 and 36 wk, even though their age since birth varied by up to 11 wk.
This observation prompted us to compare bacterial composition in alternate formats in Table 2. This portrayal demonstrates the trend inferred in Fig. 1 and Fig. S2; i.e., Bacilli are best represented in the earliest in life samples, and Clostridia are most abundant in samples obtained in the 33–36 wk postconceptional age interval. This table also demonstrates that depicting bacterial population distribution by age postconception differs from depicting these populations by day of life.
Table 2.
Distributions of sequences by class and intervals of sampling
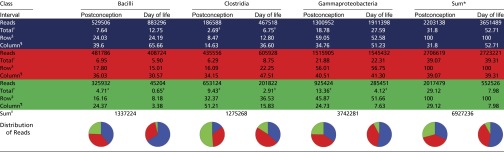 |
Colored rows correspond to age postconception [<30 (blue), 30–33 (red), and > 33 (green) wk] and day of life [<28 (blue), 28–56 (red), and >56 (green) d] groupings.
Sum of all reads across all classes for each interval. Values in these columns include the sums of the Bacilli, Clostridia, and Gammaproteobacteria reads in this table, as well as the 572,463 reads that are assigned to 38 other classes (including those designated “unclassified”) as provided in the extended dataset.
Percentage of reads for each class within each designated interval divided by the 6,927,236 reads in study.
Comparison between adjacent age postconception and day of life values differ by more than twofold.
Percentage of reads within each class within each designated interval divided by the sum of all reads for the respective intervals (Sum columns values for age postconception and day of life are denominators).
Percentage of reads within each class within each designated interval divided by the total number of reads for that class (Sum row values are denominators).
Sum of all reads, each class. Pie charts represent proportions of classes, by age postconception or day of life intervals, as designated at top of columns. Segments are earliest (blue), middle (red), and latest (green) age postconception or day of life intervals in which samples obtained.
The central tendencies of Bacilli to decrease, Clostridia to slowly increase, and Gammaproteobacteria to flourish soon after birth and remain abundant as infants age are shown with even greater clarity among infants in the three individual gestational age subcohorts (Fig. 2 and Dataset S1). The progression to Clostridial abundance is most gradual for children born most prematurely, but Clostridia are well represented by the time infants are between 33 and 36 wk postconceptional age, independent of gestational age at birth.
Fig. 2.

Percent bacterial class abundance. Proportion of each bacterial class is represented in y axes in each graph. The x axes represent samples obtained at various postconceptional ages (weeks). The columns represent class proportions, as indicated. (A–C) All infants in cohort (n = 58 subjects). (D–F) Infants in <26 wk gestational age subcohort (n = 15 subjects). (G–I) Infants in 26–28 wk gestational age subcohort (n = 20 subjects). (J–L) Infants in >28 wk gestational age subcohort (n = 23 subjects). To reduce the effect of random fluctuations in the data (noise), the lines were produced by fitting a cubic smooth spline (thick black line) to the data with 95% confidence bands (thinner lines) generated using a nonparametric bootstrap method.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways putatively encoded by the gut microbial populations do not differ significantly between the three subcohorts when comparing the first or the last stools. In contrast, 23 of the 328 KEGG pathways differed in abundance within subcohorts, when comparing the first and the last specimen’s sequences (q values ≤ 0.1) (Table S1). Several intracohort shifts in these pathways are potentially relevant to the newborn digestive system. There is decreasing capacity to degrade bisphenols in all three subcohorts, and to synthesize carotenoids in two subcohorts, and increasing capacity to synthesize lipopolysaccharide, isoquinoline alkaloids, and N glycans in the subcohort born most prematurely.
In each gestational age subcohort, day of life is significantly associated with decreasing proportions of Bacilli and increasing proportions of Clostridia (Table 3), abrupt shifts in population structure notwithstanding. The slope (beta coefficients) of the progression to a state of Clostridial abundance accelerates as gestational age increases (0.007 → 0.009 → 0.010), as suggested in Fig. 2. There were no other consistently significant changes across subcohorts: Antibiotic use was associated with increased proportions of Gammaproteobacteria, but only for infants born after gestational intervals ≥26 wk, and decreased percentages of Clostridia in stools, but only for infants born after ≤28 wk gestation. Breast feeds were associated with increasing proportions of Gammaproteobacteria but not for infants born after 28 wk gestation. However, importantly, these potential exogenous drivers of gut microbial content did not fundamentally alter the trend in population evolution, only its pace. Also, housing of subjects in single vs. open rooms distributed throughout the 75-bed, 4,580 m2 NICU did not affect microbial composition.
Table 3.
Relationship of clinical factors to predominant taxa (class level)
| Variable | GA at birth (wk) | Bacilli | Clostridia | Gammaproteobacteria |
| Day of life | <26 | −0.007 (−0.011, −0.004)↓ | 0.007 (0.004, 0.011)↑ | 0.000 (−0.004, 0.004) |
| 26–28 | −0.007 (−0.010, −0.003)↓ | 0.009 (0.005, 0.012)↑ | 0.000 (−0.005, 0.003) | |
| >28 | −0.010 (−0.014, −0.006)↓ | 0.010 (0.005, 0.014)↑ | 0.000 (−0.004, 0.005) | |
| Days on antibiotics* | <26 | 0.004 (−0.005, 0.013) | −0.012 (−0.021, −0.003)↓ | 0.009 (0.000, 0.0182) |
| 26–28 | −0.001 (−0.011, 0.010) | −0.016 (−0.026, −0.005)↓ | 0.017 (0.006, 0.027)↑ | |
| >28 | −0.012 (−0.031, 0.007) | −0.008 (−0.027, 0.012) | 0.021 (0.002, 0.040)↑ | |
| Breast milk† | <26 | −0.045 (−0.116, 0.026) | −0.031 (−0.102, 0.040) | 0.073 (0.001, 0.143)↑ |
| 26–28 | −0.080 (−0.185, 0.025) | −0.048 (−0.153, 0.0577) | 0.115 (0.010, 0.221)↑ | |
| >28 | −0.016 (−0.101, 0.070) | −0.074 (−0.159, 0.012) | 0.079 (−0.007, 0.164) | |
| C-section | <26 | −0.187 (−0.346, −0.028)↓ | −0.011 (−0.171, 0.148) | 0.201 (0.042, 0.360)↑ |
| 26–28 | 0.226 (0.027, 0.425)↑ | 0.010 (−0.189, 0.209) | −0.089 (−0.289, 0.110) | |
| >28 | 0.212 (−0.017, 0.441) | 0.021 (−0.208, 0.249) | −0.083 (−0.312, 0.146) | |
| Sampled after‡ 01/01/2011 | <26 | 0.266 (0.131, 0.401)↑ | −0.151 (−0.286, −0.016)↓ | −0.140 (−0.275, −0.005)↓ |
| 26–28 | 0.037 (−0.178, 0.252) | −0.159 (−0.373, 0.056) | −0.129 (−0.344, 0.086) | |
| >28 | 0.063 (−0.129, 0.254) | −0.049 (−0.240, 0.142) | −0.039 (−0.231, 0.152) |
A mixed model regression analysis investigated the relationship between clinical variables and taxa fractional abundance in repeated samples of subjects, by gestational age group. Statistically significant linear trends are indicated by arrows. Values are beta coefficients with 95% confidence intervals. Negative values indicate taxa decrements and positive values indicate taxa increments per unit increase in predictor variable. ↓ Significant decrease in taxa, P < 0.05; ↑ Significant increase in taxa, P < 0.05. GA, gestational age at birth.
Percent of days of life antibiotics were administered before each specimen.
Breast milk (BM) categorized by volume (0,0%; 1, <10%; 2,10–50%; 3, >50% of enteral volume).
Samples collected in first half of study or second half; positive values represent greater representation in second half.
Discussion
Our findings have three major elements. First, convergence of infant gut bacterial populations to Clostridial abundance is a nonrandom process, but not strongly correlated with mode of birth, antibiotics used, or feeds administered. Second, the overall bacterial composition is largely limited to three classes, and their distribution contrasts markedly with gut bacterial composition in older children and adults. Specifically, Bacilli, Gammaproteobacteria, and Clostridia represent 1–5%, <1%, and 45–80%, of all classes, respectively (21, 22), in later life compared with the corresponding proportions (19.3%, 54.0%, and 18.5%, respectively) in study infants. Third, maturation of the gut bacterial population structure is punctuated by abrupt shifts in microbial composition.
Our data prompt many avenues of future inquiry. First, it is important to determine where gut bacteria originate. Possibilities include an autochthonous source, especially if the fetus is colonized in utero (23) by viable progenitors of the organisms whose sequences we identified, and/or exogenous origins including nonpasteurized maternal breast milk, family or staff handlers, or fomites (24). Indeed, we recently demonstrated the simultaneous presence of specific pathogens in the stools of premature infants in proximity to each other, so patient-to-patient transmission is certainly a component (16).
It will also be important to compare data from our cohort to community infants born at term. Based on some community data, it appears that Gammaproteobacteria are found in greater proportions in the stools of these premature infants than in the stools of term infants (11, 25, 26), although one study of infants residing at home has reported a relative abundance of this taxon similar to the proportions we identified (12). Certainly, premature infants differ from term infants in kinds of exposures; whereas the NICU controls exposures to microbes to the extent possible, in the community the microbial environment is much less constrained. However, such fine interval and intense sampling of children in the community early in life, which might provide such information, has not yet been performed.
It is not known what drives this patterned progression of bacterial communities. The developing host immune system might shape the bacterial populations (27), or, alternatively, one pioneer group of bacteria (11) might metabolically prime the environment for replacement by a particular successor. Gut bacterial content is associated with term infant B-cell memory differentiation (25), and it will be informative to dissect the roles of, and interaction between, hosts and microbes, as the immune system and the gut bacterial community equilibrate in the first weeks of life.
Studies of gut bacteria in preterm infants have been limited in subject and/or sample numbers (24, 28, 29), have used cross-sectional comparisons (30), or did not stratify extensively by gestational age at birth or age postconception (30–36). Our intensive and extensive dataset suggests that limited sampling in premature neonates is problematic because of very frequent alterations in population structure. These abruptions also occur in adult and older infant gut microbial populations, although with lower frequencies (12, 37–40). Also, our data suggest that neonatal bacterial populations are more appropriately related to age postconception than to day of life.
Current data do not permit a predictive model of microbial gut colonization based on specific variables, but models proposed in future studies must account for dynamism in microbial populations, which obligates serial sampling of as many specimens as possible in future efforts to relate the premature infant intestinal biomass to various host diseases and phenotypes (41). These properties also provide an argument for very intensive and extensive sampling of control populations in this age group, and for the incorporation of computational analyses that address community changes over time (42) in dynamic microbial populations (43).
In summary, genera belonging to only three classes represent the preponderance of gut bacteria in almost all infants who spend their earliest weeks of life in a microbiologically controlled environment. Despite frequent abruptions in the relative ratios of these classes, Clostridia proportions begin to resemble those of older individuals (21, 22) by 33–36 wk postconceptional age, even if complete stability is not achieved until several years of age (1). Shifts in KEGG pathways represented by the shifting microbiota suggest ontological aspects of the maturing gut bacterial population as premature infants grow. When attempting to relate bacterial populations to outcomes or events in premature infants, trajectories of bacterial populations, rather than specific content on specific days, might be the most appropriate independent variables to enter into analysis.
Materials and Methods
This prospective observational case-cohort study was approved by the Human Research Protection Office of Washington University School of Medicine in St. Louis. Infants were enrolled after parents provided informed consent. All premature infants admitted to the St. Louis Children’s Hospital Neonatal Intensive Care Unit who weighed ≤1,500 g at birth and were expected to live beyond the first week of life were eligible. Clinical data were collected and managed using the REDCap electronic data capture tools at Washington University School of Medicine in St. Louis (44). All infant stools were collected, stored briefly (4 °C), and frozen (−80 °C) until analyzed.
The V3-5 region of the 16S rRNA gene was sequenced on the Roche-454 platform to define the composition of the bacterial community. Sample preparation, DNA isolation, sequencing, data processing followed standardized protocols developed by the Human Microbiome Project consortium (39). The minimal sequence length was 200 bp, and chimeric sequences were removed by Chimera-Slayer (45). Average quality of scores of <35 were used as the minimum to remove low-quality reads. Sequences meeting the above criteria were further classified by the Ribosomal Database Project (RDP) Naive Bayesian Classifier version 2.5 using training set 9 (46) from phylum to genus level. Phylogenetic investigation of communities by reconstruction of unobserved states was used to characterize the metabolic capacities of the microbial communities (47).
To portray specimen composition as a function of day of life and of age postconception, we performed nonmetric multidimensional scaling (Figs. S3 and S4). To attempt to associate host variables with bacterial population content, we performed a post hoc mixed-model regression analysis based on patterns discovered visually using nonmetric multidimensional analysis. The mixed model was used to adjust for repeated measures in individual subjects. All analyses used SAS (version 9.3) and R (version 3.0.1).
Detailed materials and methods are supplied in Supporting Information.
Supplementary Material
Acknowledgments
We thank families and clinical staff in the Neonatal Intensive Care Unit for their cooperation with our study; Kathie Mihindukulasuriya, Brandi Herter, Catherine Hoffmann, Brittany Kurowski, Alisa Moyer, I. Malick Ndao, Robert Cristel, and The Genome Institute production team for technical assistance; Joanne Nelson and Ally McClure for data deposition; Ariana Jasarevic and Maida Redzic for manuscript preparation; and Alexander Mellmann and Thad Stappenbeck for critical comments on our manuscript. This work was supported by the US National Institutes of Health Grants UH3AI083265, U54HG004968, and P30DK052574 (Biobank Core), along with funding from the National Institute of Allergy and Infectious Diseases, Eunice Kennedy Shriver National Institute of Child Health and Human Development, Foundation for the National Institutes of Health (made possible by support from the Gerber Foundation), and a Melvin E. Carnahan Professorship (P.I.T.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the NCBI dbGap and BioProject, www.ncbi.nlm.nih.gov (accession nos. phs000247 and PRJNA74943, respectively).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1409497111/-/DCSupplemental.
References
- 1.Yatsunenko T, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bäckhed F. Host responses to the human microbiome. Nutr Rev. 2012;70(Suppl 1):S14–S17. doi: 10.1111/j.1753-4887.2012.00496.x. [DOI] [PubMed] [Google Scholar]
- 3.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White RA, et al. Novel developmental analyses identify longitudinal patterns of early gut microbiota that affect infant growth. PLOS Comput Biol. 2013;9(5):e1003042. doi: 10.1371/journal.pcbi.1003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gill N, et al. Neutrophil elastase alters the murine gut microbiota resulting in enhanced Salmonella colonization. PLoS ONE. 2012;7(11):e49646. doi: 10.1371/journal.pone.0049646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Britton RA, Young VB. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol. 2012;20(7):313–319. doi: 10.1016/j.tim.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frank DN, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17(1):179–184. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nieuwdorp M, Gilijamse PW, Pai N, Kaplan LM. Role of the microbiome in energy regulation and metabolism. Gastroenterology. 2014;146(6):1525–1533. doi: 10.1053/j.gastro.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Olszak T, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336(6080):489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koenig JE, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jost T, Lacroix C, Braegger CP, Chassard C. New insights in gut microbiota establishment in healthy breast fed neonates. PLoS ONE. 2012;7(8):e44595. doi: 10.1371/journal.pone.0044595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5(7):e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song SJ, et al. Cohabiting family members share microbiota with one another and with their dogs. eLife. 2013;2:e00458. doi: 10.7554/eLife.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faith JJ, et al. The long-term stability of the human gut microbiota. Science. 2013;341(6141):1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Y, et al. Biogeography of the ecosystems of the healthy human body. Genome Biol. 2013;14(1):R1. doi: 10.1186/gb-2013-14-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carl MA, et al. Sepsis from the gut: The enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clin Infect Dis. 2014;58(9):1211–1218. doi: 10.1093/cid/ciu084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan A, Edlund C, Nord CE. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect Dis. 2001;1(2):101–114. doi: 10.1016/S1473-3099(01)00066-4. [DOI] [PubMed] [Google Scholar]
- 18.Al-Banna NA, et al. Acute administration of antibiotics modulates intestinal capillary perfusion and leukocyte adherence during experimental sepsis. Int J Antimicrob Agents. 2013;41(6):536–543. doi: 10.1016/j.ijantimicag.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 19.Richter J, et al. Vancomycin and to lesser extent tobramycin have vasomodulatory effects in experimental endotoxemia in the rat. Clin Hemorheol Microcirc. 2010;46(1):37–49. doi: 10.3233/CH-2010-1331. [DOI] [PubMed] [Google Scholar]
- 20.Engle WA. American Academy of Pediatrics Committee on Fetus and Newborn Age terminology during the perinatal period. Pediatrics. 2004;114(5):1362–1364. doi: 10.1542/peds.2004-1915. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA. 2009;106(7):2365–2370. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saulnier DM, et al. Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology. 2011;141(5):1782–1791. doi: 10.1053/j.gastro.2011.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aagaard K, et al. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6(237):237ra265. doi: 10.1126/scitranslmed.3008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks B, et al. Microbes in the neonatal intensive care unit resemble those found in the gut of premature infants. Microbiome. 2014;2(1):1. doi: 10.1186/2049-2618-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lundell AC, et al. Infant B cell memory differentiation and early gut bacterial colonization. J Immunol. 2012;188(9):4315–4322. doi: 10.4049/jimmunol.1103223. [DOI] [PubMed] [Google Scholar]
- 26.Azad MB, et al. Gut microbiota of healthy Canadian infants: Profiles by mode of delivery and infant diet at 4 months. Can Med Assoc J. 2013;185(5):385–394. doi: 10.1503/cmaj.121189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stockinger S, Hornef MW, Chassin C. Establishment of intestinal homeostasis during the neonatal period. Cell Mol Life Sci. 2011;68(22):3699–3712. doi: 10.1007/s00018-011-0831-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mai V, et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS ONE. 2011;6(6):e20647. doi: 10.1371/journal.pone.0020647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrow AL, et al. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome. 2013;1(1):13. doi: 10.1186/2049-2618-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaTuga MS, et al. Beyond bacteria: A study of the enteric microbial consortium in extremely low birth weight infants. PLoS ONE. 2011;6(12):e27858. doi: 10.1371/journal.pone.0027858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang JY, Shin SM, Chun J, Lee JH, Seo JK. Pyrosequencing-based molecular monitoring of the intestinal bacterial colonization in preterm infants. J Pediatr Gastroenterol Nutr. 2011;53(5):512–519. doi: 10.1097/MPG.0b013e318227e518. [DOI] [PubMed] [Google Scholar]
- 32.Morowitz MJ, et al. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc Natl Acad Sci USA. 2011;108(3):1128–1133. doi: 10.1073/pnas.1010992108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacquot A, et al. Dynamics and clinical evolution of bacterial gut microflora in extremely premature patients. J Pediatr. 2011;158(3):390–396. doi: 10.1016/j.jpeds.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 34.Claud EC, et al. Bacterial community structure and functional contributions to emergence of health or necrotizing enterocolitis in preterm infants. Microbiome. 2013;1(1):20. doi: 10.1186/2049-2618-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharon I, et al. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res. 2013;23(1):111–120. doi: 10.1101/gr.142315.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart CJ, et al. Development of the preterm gut microbiome in twins at risk of necrotising enterocolitis and sepsis. PLoS ONE. 2013;8(8):e73465. doi: 10.1371/journal.pone.0073465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960):1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zoetendal EG, Akkermans AD, De Vos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol. 1998;64(10):3854–3859. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caporaso JG, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12(5):R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murgas Torrazza R, Neu J. The developing intestinal microbiome and its relationship to health and disease in the neonate. J Perinatol. 2011;31(Suppl 1):S29–34. doi: 10.1038/jp.2010.172. [DOI] [PubMed] [Google Scholar]
- 42.Stein RR, et al. Ecological modeling from time-series inference: Insight into dynamics and stability of intestinal microbiota. PLOS Comput Biol. 2013;9(12):e1003388. doi: 10.1371/journal.pcbi.1003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marino S, Baxter NT, Huffnagle GB, Petrosino JF, Schloss PD. Mathematical modeling of primary succession of murine intestinal microbiota. Proc Natl Acad Sci USA. 2014;111(1):439–444. doi: 10.1073/pnas.1311322111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris PA, et al. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haas BJ, et al. Human Microbiome Consortium Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21(3):494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cole JR, et al. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37(Database issue):D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langille MG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.