

Significance
Sequences derived from transposable elements (TEs) are abundant in the human genome and can influence gene expression. In normal cells, most TEs are silenced by epigenetic mechanisms such as DNA methylation but, in cancer, normally dormant TEs can become active. We hypothesized that cancer-specific release of epigenetic suppression of TEs could result in gene expression perturbations, which could promote oncogenesis. Using a bioinformatics method, we identified many genes expressed in diffuse large B-cell lymphoma (DLBCL) via activation of TE promoters. Further analysis of one, FABP7, showed it was expressed in some DLBCL samples through use of a TE promoter. The TE-driven FABP7 transcript encodes a novel isoform of the protein, which is required for optimal DLBCL cell line proliferation.
Keywords: gene regulation, endogenous retroviruses, alternative promoters
Abstract
Remnants of ancient transposable elements (TEs) are abundant in mammalian genomes. These sequences harbor multiple regulatory motifs and hence are capable of influencing expression of host genes. In response to environmental changes, TEs are known to be released from epigenetic repression and to become transcriptionally active. Such activation could also lead to lineage-inappropriate activation of oncogenes, as one study described in Hodgkin lymphoma. However, little further evidence for this mechanism in other cancers has been reported. Here, we reanalyzed whole transcriptome data from a large cohort of patients with diffuse large B-cell lymphoma (DLBCL) compared with normal B-cell centroblasts to detect genes ectopically expressed through activation of TE promoters. We have identified 98 such TE-gene chimeric transcripts that were exclusively expressed in primary DLBCL cases and confirmed several in DLBCL-derived cell lines. We further characterized a TE-gene chimeric transcript involving a fatty acid-binding protein gene (LTR2-FABP7), normally expressed in brain, that was ectopically expressed in a subset of DLBCL patients through the use of an endogenous retroviral LTR promoter of the LTR2 family. The LTR2-FABP7 chimeric transcript encodes a novel chimeric isoform of the protein with characteristics distinct from native FABP7. In vitro studies reveal a dependency for DLBCL cell line proliferation and growth on LTR2-FABP7 chimeric protein expression. Taken together, these data demonstrate the significance of TEs as regulators of aberrant gene expression in cancer and suggest that LTR2-FABP7 may contribute to the pathogenesis of DLBCL in a subgroup of patients.
Cancer results from an accumulation of genetic and epigenetic abnormalities affecting both the transcriptome and proteome. Whereas next-generation sequencing has revolutionized detection of mutations in protein-coding genes (1, 2), changes in regulation of genes and noncoding RNAs are also important in conferring the malignant state (3, 4). Dysregulation of the “epigenome,” including both histone marks and DNA methylation, is a hallmark of malignancy, likely underlying many regulatory changes (5). One of the functions of epigenetic regulation is to suppress transcriptional activity of transposable elements (TEs) (6). TEs are repetitive DNA sequences present in nearly all genomes analyzed to date (7). TEs can be categorized into retrotransposons, either containing an LTR, also termed endogenous retroviruses (ERVs), or being devoid of LTRs (long/short interspersed nuclear elements; LINEs and SINEs) and DNA transposons. Whereas nearly half of the human genome is composed of TEs, only a few of these are capable of transposing and creating de novo insertional mutations in humans [0.1% of estimated spontaneous germ-line mutations (8)]. Nevertheless, although the vast majority of TEs in the human genome have lost their ability to transpose, many have retained functional regulatory sequences such as promoters and enhancers, which are able to influence the expression of nearby genes (9–11). A number of studies have demonstrated the prevalence of TEs as promoters for human genes (12–16), and the importance of TE promoters, particularly LTRs, in expression of noncoding RNAs in pluripotency/stem cells is being revealed (17–20). Most TEs are transcriptionally silent in normal somatic cells, but in cancer TEs are often released from epigenetic constraint and become transcriptionally active (21–23), potentially affecting cancer transcriptomes and contributing to the malignant state. To date, there is only one definitive report in the literature where a TE acts as an alternative promoter for a gene that plays a crucial role in human cancer. In Hodgkin lymphoma, a normally dormant THE1B ERV LTR was shown to drive expression of the proto-oncogene colony stimulating factor 1 receptor (CSF1R), which is required for growth and survival of the malignant cells (24). Although this report spurred interest, the overall prevalence and significance of TE-driven aberrant gene expression in cancer is unknown.
We hypothesize that cancer-specific release of epigenetic suppression of TEs could result in significant perturbations to the transcriptome, some of which could play a role in carcinogenesis. To address this hypothesis, we chose to examine diffuse large B-cell lymphoma (DLBCL). DLBCL is a clinically aggressive form of non-Hodgkin lymphoma that comprises about 40% of all lymphoma diagnoses in North America (25). DLBCL arises from (post-) germinal center B lymphocytes and is clinically, pathologically, and biologically heterogeneous, including over 20 subtypes and variants (26). Through the reanalysis of a large published dataset of whole transcriptome sequencing (RNA-seq) from 101 DLBCL patient samples compared with similar data from normal B-cell centroblasts (27), we were able to identify multiple genes ectopically expressed due to TE promoters (hereafter named TE-Gene chimeras). Five selected chimeric transcript predictions were confirmed in DLBCL cell lines and we conducted further analysis of one particular chimera, namely LTR2-FABP7, which we confirmed encodes a novel isoform of the FABP7 protein with an alternative first coding exon. FABP7 is normally expressed in brain, but not in blood, and is up-regulated in various solid tumor types, where it is a marker for poor prognosis (28–31). However, it has not been associated with DLBCL or any other lymphoma before this report.
Results and Discussion
Identification of TE-Gene Chimeras.
To identify TE-initiated chimeric gene transcripts in DLBCL, we mined RNA-seq data from 101 DLBCL patient samples of different subtypes (Materials and Methods) (27) and, for comparison, nine tonsil B-cell centroblast control samples (27). TE-gene chimeric candidates were initially identified by screening for paired-end chimeric reads where one end of the read maps to a TE and the other to a gene exon, using a previously published pipeline used to identify TE-promoted gene transcripts in mouse (32). In this previous mouse study (32) it was found that this method, although possibly missing some cases, was more sensitive in detecting TE-promoted chimeric transcripts than using ab initio transcript assembly via Cufflinks (33), and this was confirmed in our study (see Materials and Methods for more details). We then filtered for annotated RefSeq genes that were silent in nine normal libraries but were expressed, with three or more chimeric reads, in at least two DLBCL samples. We excluded transcripts detected as a result of transcriptional termination within TEs or due to TE-derived internal exons, which were not well differentiated from potential promoters by our initial computational pipeline (Materials and Methods). This analysis uncovered 98 TE-chimeric gene transcripts where the TE seems to be serving as the promoter (Dataset S1). The majority of these (75%) involve TE interactions with protein-coding genes, whereas the remaining involve annotated long noncoding RNAs. None of these chimeric transcripts was significantly associated with particular DLBCL subtypes.
We chose five chimeras for further study (Fig. 1 A and B and Fig. S1) involving the protein-coding genes PTPRF, EPYC, MFAP5, ALDH1L1, and FABP7. These genes have diverse functions in a range of tissues and have been implicated in various human cancers but not in DLBCL (see SI Text, section 1 for more details on each gene).
Fig. 1.

(A) Human hg18 UCSC genome browser screenshot for LTR2-FABP7 with representative paired-end reads shown. A representative DLBCL library is shown in red and a normal B-cell library in black. The transposable element track is a modified RepeatMasker track. (B) Scheme of the five TE-gene chimeric candidates. Exons are depicted as blue boxes (DNA) and blue lines (mRNA). Introns are blue dashed lines. Thinner blue boxes are untranslated sequences and ATGs are depicted in white. TEs are shown in red for LTR elements and pink for LINE copies. Black arrows depict native TSSs. Stars represent protein localization signals; NLS and FA binding (FAB) domains (D1, D2, and D3) are outlined in turquoise. (C) Heat maps of DLBCL and normal B-cell transcriptomes comparing gene expression (RPKM) and number of paired-end chimeric reads for a specific TE-gene chimera. The 101 primary DLBCL transcriptomes and 9 controls (from normal germinal center B cells) are arranged from left to right according to overall gene expression (RPKM) on the bottom half of each panel. The top of each panel shows the number of chimeric paired-end reads between the TE and the gene. The right side of each panel is the scale, which differs in each case. Spearman r correlations are depicted with P values (***P < 0.0001). Spearman’s r was calculated using the software GraphPad Prism by comparing the number of chimeric reads for each library with the respective gene RPKM value.
The TE fragments acting as potential promoters for these genes are all retrotransposons, either ERVs (LTR16A1, THE1A, MER57B, and LTR2) or an old L2 LINE fragment (Fig. 1B). Whereas LTRs intrinsically contain promoter sequences (reviewed in ref. 11), the L2 potential promoter has probably evolved after insertion, because LINE elements often lose their promoter when transposing. Moreover, human L2 copies are extremely divergent, suggesting an ancient invasion (34). With the exception of LTR2-FABP7, all chimeric transcripts potentially splice upstream of the native translational start site and therefore putatively code for a native protein (Fig. 1B). Notably, the LTR2-FABP7 transcript skips the normal ATG start codon located in the native first exon and splices directly into the second exon (Fig. 1B). Interestingly, by mining the RNA-seq data for overall gene expression, we found a significant correlation between the total expression level of these five genes and the number of TE-gene chimeric paired-end reads (Fig. 1C). Such correlations suggest that the activity of the TE promoters has a significant impact on overall gene expression levels in these DLBCL patients.
It should be noted that, because we screened only nine normal B-cell libraries compared with ∼100 DLBCL samples, we cannot rule out the possibility that the chimeric transcripts detected in DLBCL are present in a low fraction of normal samples, or indeed, in other normal or malignant cell types. However, for four of the five cases discussed above, namely FABP7, MFAP5, EPYC, and PTPRF, there are no chimeric ESTs or mRNAs in the public databases that map to the TEs involved, and perusal of available University of California Santa Cruz (UCSC) Genome Browser tracks of RNA-seq data from cultured human B-cells from 20 unrelated individuals from the Center d’Etude du Polymorphisme Humain (CEPH) collection (35) revealed no evidence of transcription from these TEs. For ALDH1L1, normally expressed in liver (SI Text, section 1), two chimeric ESTs from normal brain thalamus (DA390295 and DA409790) map to the MER57B LTR involved and the RNA-seq data from the B-cell CEPH collection does show a peak at this LTR, indicating that this chimeric transcript is not restricted to DLBCL.
Interestingly, our screening method also detected the same THE1B LTR-CSF1R chimeric transcript as reported in Hodgkin lymphoma (24) (Dataset S1), but we chose not to conduct further analysis on this case because, unlike the five cases above, the contribution of the LTR promoter is very minor (with few chimeric reads) compared with the native promoter and does not correlate with overall gene expression (Dataset S1), so the biological effect of the chimeric transcript is likely very small.
Most Prevalent TEs as Promoters for Chimeric Transcripts in DLBCL.
As mentioned above, several studies have shown that TEs can serve as promoters for human genes (12–16). Among our list of 98 TE-gene chimeras in DLBCL, we determined whether any particular TE families are overrepresented compared with their genomic abundance. In terms of overall TE class, we found that LTRs are overrepresented compared with LINEs, SINEs, or DNA elements. The ∼717,000 LTR/ERV loci in the genome comprise 16% of all ∼4.48 million TEs or TE fragments (34) (RepeatMasker Open-3.0,1996–2010; www.repeatmasker.org) but account for 45% of the chimeras (Dataset S1) (P = 1.24 × 10−10, Fisher’s exact test). This result is not surprising, given that LTRs naturally contain promoter sequences and are typically found relatively intact in the genome as solitary LTRs (36). In contrast, although L1 LINE elements can also drive chimeric transcripts (12), the vast majority of L1 loci in the genome are not full-length and lack the promoter region (34). Among the LTR cases, LTR2 elements (comprising LTR2, 2B, and 2C subgroups), which are the LTRs associated with the HERV-E family (37), are significantly overrepresented (P = 1.33 × 10−6, Fisher’s exact test). The 1,508 LTR2 elements comprise 0.21% of all LTRs in the human genome (34, 37, 38) but initiate 9.3% of the chimeric LTR transcripts (4/43), including the FABP7 case (Dataset S1). [One such locus, an LTR2B at chr6:31262047–31262525 (hg18), is involved in two different chimeric transcripts so appears twice on the list.] The other significantly overrepresented LTR family is the THE1 MaLR subfamily (39) (P = 3.06 × 10–8, Fisher’s exact test). The ∼49,000 THE1 LTR sequences comprise ∼6.85% of all genomic LTR loci yet account for 37% of the LTR-driven chimeric transcripts (16/43) (Dataset S1).
Molecular Biology Confirmation of TE-Gene Chimeras.
Using RT-PCR with transcript-specific primers, we tested for the presence of the five chosen TE-gene chimeric transcripts, as well as transcripts from the native promoter, in DLBCL and control cell lines (positive for native gene expression) (Fig. 2A). Although transcripts were detected for THE1A-MFAP5 in three DLBCL cell lines, we were unable to rule out activity from the native promoter because the THE1A transcript splices into the beginning of the first exon and it is not possible to design specific primers upstream of the chimeric splice site (Fig. 2A). L2-EPYC transcripts were observed in only one DLBCL cell line, OCl-Ly3, along with native transcripts. No LTR16A1-L2-PTPRF transcripts could be observed in the panel of nine DLBCL cell lines assessed, but we were able to amplify this chimeric transcript in a Hodgkin lymphoma-derived cell line (L591) (40) and a primary mediastinal large B-cell lymphoma-derived cell line (Med-B1) (41), thus confirming the amplification efficiency of the primers used (Fig. 2A). DLBCL cell lines harboring only chimeric transcripts for MER57B-ALDH1L1 were identified and this transcript was also detected in the lymphoblastoid cell lines IM9 (42) and LCL, in accord with the RNA-seq data from B-cell lines (35) mentioned above. Chimeric LTR2-FABP7, but no native transcript, was robustly amplified in the DLBCL cell lines SUDHL4 and DB. U251, a malignant glioma cell line previously shown to express FABP7 (43), expressed significant levels of native mRNA, and some chimeric transcripts could be amplified in this cell line, although not in every experiment, suggesting a very low level of such transcripts.
Fig. 2.
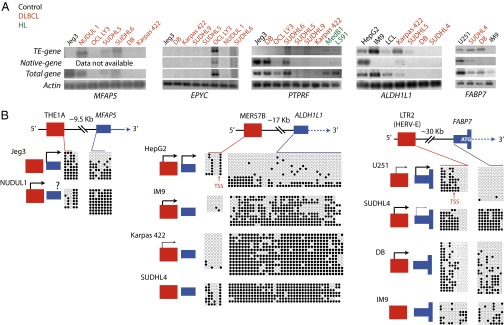
(A) RT-PCR to assess expression of native, chimera, and total MFAP5, EPYC, PTPRF, ALDH1L1, and FABP7 mRNA in DLBCL and other cell lines. Actin expression was assessed as a control. (B) Bisulfite sequencing of native and TE promoters for ALDH1L1, FABP7, and MFAP5. Gene schemes are the same as in Fig. 1B. Filled circles represent methylated CpGs, empty circles represent unmethylated CpGs, and each row represents an independent clone. Localization of CpGs relative to the DNA sequence is shown with colored lines. The activity state of native and TE promoter based on RT-PCR displayed in A is shown at the left of each bisulfite plot; the presence of an arrow depicts activity (thicker arrows represent higher activity than thinner arrows) and absence of an arrow means no transcript was detected.
The transcription start sites for MER57B-ALDH1L1 and LTR2-FABP7 were confirmed to be located within the relevant TE sequence using 5′ RACE or RT-PCR primer walking (SI Text, section 2 and Fig. S2). All available LTR-chimeric transcripts were sequenced (SI Text, section 2) and a scheme of predicted proteins is shown in Fig. 1B. In agreement with the bioinformatics analysis, most predicted chimeric proteins, with the exception of LTR-FABP7, are identical to the native form, suggesting TEs are able to ectopically activate a host gene without necessarily creating new isoforms.
Activation of TE-Gene Chimeras Is Associated with Epigenetic Derepression.
Genome-wide hypomethylation, which affects TEs, along with localized hypermethylation of gene promoters, are well-known characteristics of cancer (5). We assessed DNA methylation levels of the TE and native promoters (where sufficient CpGs were available) to determine whether methylation state correlates with transcriptional activity. In the instances studied, the native promoters of the FABP7, MFAP5, and ALDH1L1 genes were partly or heavily methylated in DLBCL cell lines (Fig. 2B and Fig. S3). Control cell lines expressing the genes from the native promoter were hypomethylated at this promoter, suggesting that the lack of native gene expression in DLBCL is related to hypermethylation of the native promoter. However, lack of appropriate transcription factors also likely plays a role because we detected no or very low levels of FABP7 transcripts from the native promoter in the DB DLBCL cell line, despite the fact that some cells have an unmethylated native promoter region. Whereas the association between methylation state and expression is clear for native promoters, the TE sequences analyzed show more variability and specific methylation patterns. For instance, the LTR2 promoter for FABP7 can be divided into two juxtaposed blocks of methylation; the block downstream of the transcription start site (TSS) is associated with promoter activity when unmethylated, whereas CpG sites upstream of the TSS are methylated even when the LTR is active. Another DLBCL cell line, SUDHL9, which was intermittently positive by RT-PCR for the LTR-FABP7 chimeric transcript using different cDNA preparations, showed highly variable LTR methylation (Fig. S3), suggesting considerable cell-to-cell heterogeneity. For the THE1A LTR driving MFAP5 expression, the three CpG sites within the LTR were hypomethylated when active and, in the case of the MER57B LTR driving ALDH1L1 expression, the LTR was generally less methylated when active. In summary, the native promoters of the gene studied are mostly methylated and silent in DLBCL cell lines and hypomethylation of certain sites within the TE promoters are associated with their transcriptional activity.
Expression of Chimeric FABP7 Protein in DLBCL Cell Lines.
We chose to conduct further analysis on the LTR2-FABP7 chimera because FABP7 has been implicated in other cancers (SI Text, section 1) and because the structure of the transcript suggests that it encodes a novel isoform. As noted above, the LTR2-FABP7 transcript skips the native first exon containing the normal translational start codon and splices directly into the second exon (Fig. 1B and SI Text, section 2). The newly formed first exon is unique and is not similar to other known sequences. Hence, the resulting chimeric protein is predicted to have a unique N-terminal sequence but would retain the native C-terminal FABP7 sequence (SI Text, section 3). To confirm expression of FABP7 protein in the SUDHL4 and DB DLBCL cell lines, as observed by RT-PCR (Fig. 2A), Western blotting was performed using a FABP7-specific antibody raised against the C-terminal end, able to recognize both the native and predicted chimeric FABP7 isoforms equally (Fig. 3A). Despite the differences in the N-terminal amino acid sequences, the predicted molecular weight of chimeric FABP7 is very similar to that of the native protein (∼15 kDa), hence the two isoforms cannot be distinguished by standard SDS/PAGE and Western blotting techniques. FABP7 expression was observed in the positive control U251 malignant glioma cell line, as reported previously (43). Importantly, robust FABP7 expression was also observed in the DLBCL cell lines positive for the LTR2-FABP7 chimera, SUDHL4 and DB, with no significant expression observed in the nonmalignant IM9 B-cell line, the DLBCL cell line SUDHL9 (which was not consistently positive by RT-PCR) or a benign, primary human lymphocyte sample (Fig. 3A).
Fig. 3.

(A) Various cell lines and a primary lymphocyte sample were lysed and subjected to SDS/PAGE and Western blotting to assess expression of FABP7. Actin expression was also assessed as a loading control. (B) SUDHL4, DB, and U251 cell lysates were subjected to isoelectric focusing and immunoblotting with anti-FABP7 antibody to confirm expression of chimeric FABP7 (**) protein in DLBCL cell lines. Native FABP7 (*) expression in U251 cells is shown as a positive control.
Despite the similarities in molecular weight, the differences in amino acid sequence between the native and chimeric protein isoforms are predicted to result in significantly different isoelectric points (pI values) (5.25–5.59 pI and 8.12–8.66 pI respectively). Hence, we performed isoelectric focusing (IEF) to confirm expression of the chimera FABP7 in DLBCL cell lines. IEF gels separate proteins based on their net charge rather than their molecular weight, so proteins migrate to their pI, the pH at which their net charge is zero (44). As shown in Fig. 3B, a single band at the expected pI for chimeric FABP7 was observed in DLBCL cell line lysates, with no detectable expression of the native FABP7 isoform. Positive control U251 cell lysates gave a band at the expected pI for native FABP7, with no detectable chimeric expression. These data confirm expression of the chimeric FABP7 in DLBCL lysates, as predicted by the transcript analysis (Fig. 2A). Moreover, because the DB and SUDHL4 DLBCL cell lines produce only the chimeric FABP7 protein they constitute good models to study the function of this novel isoform.
FABP7 Is Expressed in Primary DLBCL Patient Samples.
To confirm expression of FABP7 protein in human patient samples, a DLBCL tissue microarray from a different patient cohort was stained with anti-FABP7 antibody. FABP7 staining intensity was semiquantitatively assessed, in comparison with matched IgG controls (Fig. 4A). We detected high FABP7 expression (by immunohistochemistry) in ∼3% of cases and medium expression in another 16% of samples (Fig. 4B), indicating that the protein is expressed in a subset of DLBCL cases. Note that this experiment does not provide information on which FABP7 isoform is expressed because the antibody does not distinguish between the chimeric and native forms.
Fig. 4.

Expression of FABP7 in primary DLBCL patient samples was assessed by immunohistochemistry. Ninety-five tissue cores were stained with anti-FABP7 antibody and relative staining intensity (NovaRed) was assessed, compared with matched IgG controls. (A) Example photomicrographs showing different staining intensities are shown. Mouse brain was used a positive control, with positive staining neuronal nuclei arrowed. (B) Semiquantitative staining analysis of all samples compared with the representative staining intensity scale micrographs is shown.
Native and Chimeric FABP7 Localization upon Fatty Acid Binding.
FABP7 is a member of the FABP family of lipid chaperones, which are involved in the uptake, storage, and intracellular trafficking of fatty acids (FAs) (45). Ligand binding studies suggest that the polyunsaturated fatty acid ω-3-docosahexaenoic acid (DHA) is the preferred ligand of FABP7, but it can also bind ω-6-arachidonic acid (AA) (46). FABP7 can move between the cytosol and nucleus, where it can deliver its FA ligand. The native first exon contains one of three conserved residues of the nuclear localization signal (NLS) and the majority of residues that make up FA binding domain 1 (Fig. 1B and SI Text, section 3). As noted above, expression from the upstream LTR2 results in expression of a chimeric protein harboring an alternative exon 1 and hence missing the residues contained in the native first exon. However, the amino acid residues required for binding to DHA (46) and the FA binding domains 2 and 3 are found in the C-terminal region and hence are present in the chimeric FABP7.
We assessed the ability of the chimeric protein to respond to the natural FA ligands DHA and AA. It has previously been reported that native FABP7, endogenously expressed in U251 cells, accumulates more in the nucleus or cytoplasm, dependent on FA ligand treatment. With DHA treatment there is an increase in the proportion of FABP7 in the nucleus, compared with AA treatment, which promotes nuclear exclusion (43). We repeated this experiment using confocal microscopy and image analysis to quantify FABP7 staining intensity in nuclear and cytosolic compartments. We also observed a similarly modest, but reproducible, relocalization of native FABP7 in U251 cells, dependent on FA ligand treatment (Fig. 5 and Fig. S4A). In contrast, FA treatment of DLBCL cell lines SUDHL4 or DB (Fig. 5 and Fig. S4 B–D) did not affect the localization of chimeric FABP7, with a small proportion found in the nucleus and the majority of chimeric FABP7 in the perinuclear region of the cytoplasm, irrespective of FA treatment. It is possible that chimeric FABP7 is able to bind to and transport FA ligands to and from the nucleus but that chimeric FABP7 localization is not regulated by FA treatment; however, this cannot be confirmed in fixed cells. Chimeric FABP7 may still function as an FA ligand in these cells. Indeed, it has previously been shown that native FABP7, in which the entire NLS has been mutated, is located predominately in the cytoplasm and can still bind to FA ligands, similar to wild-type FABP7. In this case, the NLS is required for DHA-, but not AA-, mediated regulation of cell function (43). In malignant glioma cell lines native FABP7 bound to DHA is localized to the nucleus, where it is able to activate PPAR gamma downstream signaling and inhibit cell migration (43). Although our understanding of PPAR gamma signaling is not complete, activation of this pathway has been reported to have anti-cancer effects and in some cancer types can induce apoptosis and inhibit growth and invasion. In contrast, AA-bound FABP7 is retained in the cytoplasm, where it has opposing effects [e.g., promoting glioma cell migration (43)]. Because confocal analysis of chimeric FABP7 localization demonstrated that the majority of chimeric FABP7 is found outside the nucleus, regardless of FA treatment, this could mean that chimeric FABP7 is unable to promote DHA-mediated downstream signaling (e.g., PPAR-γ anti-tumorigenic signaling).
Fig. 5.
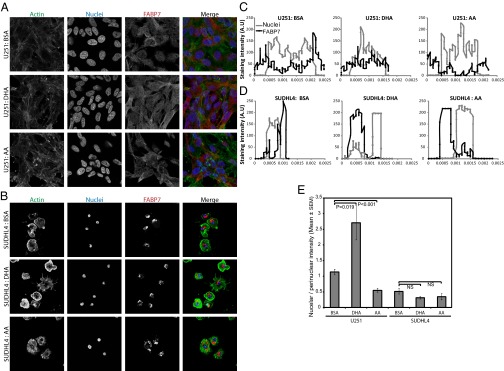
(A) U251 and (B) SUDHL4 cells were treated with BSA (vehicle), DHA, or AA, fixed, and stained with phalloidin (Actin), FABP7 antibody, and DAPI nuclear stain. FABP7 localization was assessed by confocal microscopy. IgG control staining is shown in Fig. S4A. (C and D) Representative data of FABP7 and DAPI staining intensity across the diameter of a single (C) U251 and (D) SUDHL4 cell. (E) Mean nuclear FABP7 fluorescence intensity/perinuclear FABP7 fluorescence intensity from at least 30 cells is given.
We also attempted to study the function of native versus chimeric FABP7 by expressing tagged constructs in HEK293 cells, but their overexpression resulted in transfected FABP7 (both native and chimeric) located throughout the cell in both the nucleus and cytosol, as assessed by Flag immunofluorescence, hence the physiological response to DHA and AA was lost (Fig. S5).
Depletion of FABP7 Inhibits DLBCL Cell-Line Proliferation.
Although FABP7 expression has not been linked with blood cancers before, up-regulation has been reported in various solid cancer types, including malignant glioma (28), melanoma (31), renal cell carcinoma (30), and aggressive triple negative breast cancer (47) and is associated with poor prognosis and also increased cell growth in vitro (48). To assess the requirement for chimeric FABP7 in DLBCL proliferation and cell-cycle progression in vitro, DLBCL cell lines were nucleofected to express three independent RNAi sequences targeting FABP7 and a nonsilencing scrambled control sequence. Expression of one of the shRNAs, 744, resulted in a small decrease in FABP7 expression, whereas expression of the 745 and 746 sequences resulted in robust depletion of chimeric FABP7 expression compared with scrambled control, as assessed by Western blotting (Fig. 6A). Depletion of chimeric FABP7 proportionally reduced expression of the proliferation marker proliferating cell nuclear antigen (PCNA) in these polyclonal cell lines (Fig. 6B). In addition, SUDHL4 growth, as assessed by total viable cell number, was also inhibited in chimeric FABP7-depleted SUDHL4 cells (Fig. 6C). Although there was some variation in viable cell numbers between the 745 and 746 RNAi cell lines, this variation was not statistically significant, and both RNAi cell lines displayed significant inhibition of cell growth compared with scrambled control. Incorporation of the S phase marker BrdU was also significantly reduced in SUDHL4 cells depleted of chimeric FABP7, compared with scrambled control (Fig. S6). Because exogenous overexpression of either native or chimeric FABP7 resulted in loss of differential subcellular localization (Fig. S5), which is key to its function, we could not perform a rescue experiment of the RNAi knockdown. However, to confirm these data in another cell line, the DLBCL cell line DB, which also expresses chimeric FABP7, was transiently transfected with the same RNAi sequences. Depletion of chimeric FABP7 also resulted in decreased PCNA expression (Fig. 6D) and significantly inhibited BrdU incorporation (Fig. 6E). Taken together, these data demonstrate that expression of chimeric FABP7 is required for optimal DLBCL cell growth in vitro.
Fig. 6.

SUDHL4 cells were stably transfected with RNAi vectors targeting FABP7 (744, 745, and 746) or a scrambled control. Cell lysates were assessed for (A) FABP7 and (B) PCNA expression by Western blotting. Actin expression was assessed as a loading control. (C) Stably transfected viable SUDHL4 cell growth was assessed by trypan blue counting. (t test: ***P < 0.0001, **P < 0.0003). (D) DB cells were transiently transfected with RNAi vectors targeting FABP7 (745 and 746) or a scrambled control. Cells were lysed and assessed for FABP7 and PCNA expression by Western blotting or (E) subjected to BrdU incorporation assay, assessed by FACS analysis.
Concluding Remarks.
A growing number of large-scale sequencing studies have reported detection of somatic TE insertions in some types of human cancers, and potential roles for such new insertions in malignancy are being actively investigated (49–52). Other studies have investigated potential roles for human ERV-encoded proteins in cancer (53–55) or reported general transcriptional up-regulation of ERVs or L1s in various malignancies (for reviews see refs. 56 and 57). In contrast, there has been less attention on the phenomenon of cancer-specific gene expression perturbations owing to transcriptional activation of normally dormant TEs residing in the genome. A recent study using data primarily from cell lines showed that very long noncoding RNAs (vlincRNAs) promoted by LTRs are more prevalent in cancer and embryonic stem cells compared with other tissues (18). Two groups used an experimental strategy (58) or the screening of ESTs (59) to detect chimeric transcripts driven by the antisense promoter in L1 elements that seem specific to cancer cell lines or tumors. One such transcript produces a truncated c-MET protein that may affect c-MET receptor signaling (59, 60), but a significant role in human malignancy has not been demonstrated. These latter two studies focused only on L1 elements and were not designed to screen cancer transcriptomes for all TE-initiated chimeric transcripts. The report of LTR-mediated activation of CSF1R in Hodgkin lymphoma (24), while clearly demonstrating an important role in the malignancy, is only a single case. To our knowledge our study is the first to conduct a transcriptome-wide analysis in a large primary cancer cohort specifically to detect chimeric transcripts from all types of TEs. Moreover, it is the first, to our knowledge, to suggest roles for transcriptionally activated TEs in DLBCL. We found that normally silent TEs drive ectopic expression of multiple genes in DLBCL, suggesting a significant impact on the DLBCL transcriptome. It will be interesting to conduct similar analyses in other malignancies to assess differences in TE-mediated transcriptional effects across cancer types.
We focused here on further analysis of the LTR2-FABP7 chimera that we found to be expressed in a subgroup of DLBCL patients. As discussed above, the TE promoter for this chimera, an LTR2 sequence, is overrepresented among our list of 98 cases based on the genomic abundance of this LTR family. THE1 LTRs, a member of which has been implicated in Hodgkin lymphoma through transcriptional activation of CSF1R (24), were also found to be overrepresented among our list of TE-driven chimeric transcripts, although such cases still involve a very small fraction of all related LTRs in the genome. The molecular mechanisms that underlie cancer-specific transcriptional activation of some TEs, but not others that are highly related, are poorly understood but could involve regional epigenetic perturbations or perhaps clonal selection for certain events during cancer progression.
The LTR2-FABP7 chimeric transcript produces a novel protein isoform that is required for optimal growth of DLBCL cell lines expressing this protein. Thus, it could represent a novel biomarker for a subset of DLBCL patients. In normal adults, FABP7 transcripts have been identified in brain and skeletal muscle (61); however, protein expression has only been confirmed in the brain, where it is likely a marker for neural stem cells (62). Hence, any FABP7-specific chemotherapeutic treatments that do not cross the blood–brain barrier could be highly cancer-specific, making FABP7 a potentially useful therapeutic target. FABP7 is up-regulated in several solid cancer types, including melanoma (28–31, 47), and has previously been recognized as a novel target for a melanoma treatment (48). It is worth noting that most of these studies would not have detected differential promoter use of FABP7, so it is possible that the LTR2 element might also drive expression of this gene in other cancers. A recent study of fluorescent small molecules identified a novel compound, CDr3, that can diffuse into cells to bind FABP7 intracellularly in a highly specific way and has been demonstrated as a specific marker for neural stem cells in vitro (62). Considering the limited treatment options currently available for DLBCL, and poor prognosis for patients with recurrent disease (25, 63), new therapeutics are urgently required for treatment of this disease. Targeting chimeric FABP7 could represent a new treatment avenue for DLBCL.
Materials and Methods
All reagents were purchased from Sigma unless specified. All experiments are representative of at least three independent experiments, unless specified. Statistical analysis was performed using t test, unless specified.
Patient Data.
Published RNA-seq data from 101 DLBCL patient samples of different subtypes was obtained (27). The two most common subtypes of DLBCL are germinal center B-cell (GCB) origin and activated B-cell (ABC) phenotype, with the ABC subtype recognized as having a more aggressive clinical course than the GCB subtype (64, 65). Alternative subtypes include composite lymphoma (when two or more distinctly different types of lymphoma occur in the same tissue or organ simultaneously) (66) and unclassified lymphomas, which cannot be clustered with other morphological subtypes (67). This cohort included 31 ABC, 52 GCB, 10 unclassified, and 8 samples categorized as “other or composite” (27).
Bioinformatics Analysis.
The data were mined to uncover chimeric paired-end reads and to calculate gene RPKM (reads per kilobase of transcript per million reads mapped) as previously described (32). Briefly, sequence reads were aligned to the human reference genome (hg18) using BWA v0.5.9 (68) with Smith–Waterman alignment disabled and annotated exon–exon junctions compiled from Ensembl (69), RefSEq. (70), and UCSC (71) (downloaded from http://genome.ucsc.edu on August 8, 2011). Sequence reads that could be uniquely assigned a position in the transcript resource (exon–exon junctions) were computationally repositioned to the genomic hg18 coordinates and a single merged bam file (68) generated for downstream analyses. The bam files were converted to the RPKM normalized wiggle track (WIG) files for visualization of the data in the UCSC browser. The WIG files represent the combined coverages of uniquely aligned reads as well as the multiple-aligned reads whose mates are uniquely aligned to hg18 genome. These multiple-aligned reads are retained and used to generate chimeric paired-end reads. Transcripts containing TE sequences were identified by exploiting the genomic locations of paired-end reads. Mate-pair reads separated by more than 1 SD from the mean fragment size were identified, and those mate pairs containing one read in a TE and the other in a RefSeq genic exon [downloaded from the UCSC database (72)] were enumerated. We found that discounting mate pairs mapping closer together than 1 SD from the mean fragment size reduced a specific class of false positives in which the TE is simply inserted within an exon.
To enrich for significant gene candidates we only included genes that were (i) silent in normal libraries (RPKM <1), (ii) expressed in DLBCL (RPKM ≥1), (iii) with at least three chimeric paired-end reads, and (iv) meeting these criteria in at least two DLBCL libraries. This procedure resulted in ∼600 potential TE-driven chimeric genes. This set was then visually inspected by at least two individuals using tracks created on the UCSC Genome Browser to exclude exonizations, truncations, and complex or uninterpretable cases. Cases passed this manual inspection only if three independent (nonidentical) chimeric reads splicing from a TE to an annotated RefSeq exon could be confirmed in silico with no evidence of reads/transcripts splicing into the TE from upstream. This step reduced our high-confidence candidate list to 98 genes (Dataset S1). We chose five candidate genes for further analysis based on their genome browser profile, function, and potential implication in cancer (SI Text, section 1). Heat maps correlating RPKM and chimeric paired-end reads were obtained using all available libraries and sorted by number of reads or amplitude of RPKM. Nonparametrical spearman R correlation factor statistical tests were carried out using GraphPad Prism software.
As mentioned above, we found that our method detects cases missed by ab initio assembly, particularly if the TE-derived first exon is small or the number of chimeric reads is low. We performed ab initio transcript assembly on nine DLBCL libraries chosen at random and found that Cufflinks (33) failed to identify a chimeric transcript called by our pipeline in a particular library 35% of the time (33/95 comparisons) (see last column in Dataset S1). Fig. S7 shows an example comparing the two methods. For two DLBCL libraries that were clearly called by our pipeline as having TE-initiated transcripts for ALDH1L1 (Dataset S1), only one of these was found by Tophat/Cufflinks. To further compare our method with ab initio transcript assembly, we determined how many additional libraries would have been detected using Cufflinks as harboring the chimeric transcript for our 98 cases. We found only six instances (listed in Dataset S1) where Cufflinks correctly assembled the transcript that was not detected by our pipeline in those libraries.
5′ RACE, RT-PCR, and Primer Walking.
To confirm sequence of chimeric 5′ ends, 5′RACE was performed on 1 µg RNA using a First Choice RLM-RACE kit (Ambion) per the manufacturer’s protocol. PCR to amplify 5′ ends of gene of interest was performed using primers supplied and gene-specific primers. Amplification was performed with High Fidelity Phusion polymerase (New England Biolabs) at 62 °C annealing, 25-s elongation, and 40 amplification cycles. PCR-amplified 5′ end transcripts were cloned into Promega pGEM T vector (Promega) and sequenced by Eurofins MWG Operon. For RT-PCR, RNA was extracted (at least two biological replicates per cell line) with the All Prep DNA/RNA mini kit (Qiagen) according to the manufacturer’s instructions. Five hundred nanograms of DNase-treated (Ambion Turbo DNase) RNA was used for cDNA synthesis with Vilo reverse transcriptase (Invitrogen). All primers used for cDNA amplification encompass at least one intron to check for genomic DNA contamination. For primer walking, 1 µg of cDNA was used in a 25-µL reaction, amplified using Phusion DNA polymerase (New England BioLabs) with the following cycles and primer sets: 98 °C, 30 s, (98 °C, 7 s; 62 °C, 20 s; 72 °C, 30 s) for 37 cycles. Tables S1–S4 list the primers used.
Bisulfite Analysis.
Bisulfite conversion, PCR, cloning, and sequencing were carried out as described previously (73). All of the sequences included in the analyses either displayed unique methylation patterns or unique C-to-T nonconversion errors (remaining Cs not belonging to a CpG dinucleotide) after bisulfite treatment of the genomic DNA. This avoids considering several PCR-amplified sequences from the same template molecule (provided by a single cell). All sequences had a conversion rate >95%. Sequences were analyzed with Quma free online software (74). Tables S1–S4 list the primers used.
Cell Culture.
The DLBCL-derived cell lines SUDHL4, SUDHL5, SUDHL6, and NUDUL1 were obtained from the German Collection of Microorganisms and Cell Cultures and propagated according to standard conditions (DSMZ; www.dsmz.de). The cell line MedB-1 was a kind gift from S. Brüderlein and P. Möller, University of Ulm, Germany and propagated as published (41). SUDHL9 were obtained from M. Dyer, University of Leicester, Leicester, UK. The Karpas 422 cell line was obtained from A. Karpas, Cambridge Enterprise Limited, University of Cambridge, Cambridge, UK (75). The EBV-transformed lymphoblastoid cell line (LCL) was kindly provided by U. Steidl, Einstein College of Medicine, New York. Cells were grown in RPMI media/10% FBS/1% penicillin/streptomycin (PS). OCl-Ly3 (a gift from A. Wang, BC Cancer Agency, Vancouver) and L591 cells were cultured in Iscove’s modified Dulbecco's medium/20% FBS/1% PS/1% nonessential amino acids. The HepG2 and JEG3 cell lines were grown in EMEM/10% FBS/1% PS. U251 cells (a gift from Roseline Godbout, University of Alberta, Edmonton, Canada) were cultured in DMEM/10% FBS/1% PS. Cells were maintained at 37 °C, 5% CO2.
SDS/PAGE, Western Blotting, and Isoelectric Focusing.
For standard Western blotting, cells were lysed with RIPA buffer with protease inhibitors. Forty micrograms of protein/sample were separated using 4–12% Bis-Tris gels and CAPS transfer buffer (Invitrogen). Western blotting was carried out as described (76). For isoelectric focusing, cells were lysed in RIPA buffer and 500 µg protein processed using ReadyPrep 2D cleanup kit (Bio-Rad). Samples were resuspended in rehydration buffer, and 40 µg per lane loaded onto Criterion IEF pH 3–10 gel (Bio-Rad) and run according to the manufacturer’s instructions. Proteins were transferred in 0.7% acetic acid onto PVDF membrane. Membrane was blocked in 5% milk-TBST, incubated with FABP7 antibody (AbCam) and anti-rabbit-HRP (Sigma), and visualized with X-ray film.
Antibodies.
Lamin A (Ab8980) and FABP7 (Ab104952) were from AbCam; GAPDH and β-actin were from Sigma; PCNA (610664) was from BD Transduction laboratories; rabbit IgG was from Millipore; and anti-rabbit HRP and anti-mouse-HRP were from Sigma.
Immunohistochemistry.
Human DLBCL tissue microarray (Pantomics) was subjected to citrate antigen retrieval, permeabilized with 0.2% Triton-X-100, blocked with normal rabbit serum, and then incubated overnight with rabbit-FABP7 antibody. Normal rabbit IgG was used as a control. Sections were then incubated with anti-rabbit HRP, and immunoreactivity was visualized using NovaRED reagent (SK-4800; Vector Laboratories). Sections were mounted using glycergel (C0563; Dako Canada, Inc.) and micrographs taken at 40× zoom. Semiquantitative analysis of FABP7 staining intensity was conducted, compared with an image scale of IgG control intensity as 0, low (1), or medium (2) staining intensity levels displaying increasing staining and mouse brain positive control having maximum intensity of 3.
Immunofluorescence.
U251, SUDHL4, or DB cells were plated in serum-free media and allowed to adhere to poly-l-lysine–coated glass coverslips for 24 h. Cells were treated as appropriate, fixed with 4% (wt/vol) paraformaldehyde, permeabilized with 0.2% triton-X-100 in PBS, blocked in 2% (wt/vol) BSA-PBS and stained with phalloidin-Texas Red (Sigma), FABP7 antibodies or IgG control, anti-Rabbit-FITC and Dapi. Images were captured at the same Z section (0.5 μm) using a confocal microscope (Zeiss) and processed using ImageJ. Briefly, a single line vector was drawn from one side of the cell to the other (as defined by actin staining), through the nucleus (as defined by DAPI staining). FABP7 and nuclear staining intensity across this line was quantified. Representative plots of single cells are shown. To display average data from over 30 cells, mean FABP7 staining intensity was quantified in nuclear and perinuclear areas (as defined by actin and DAPI staining) and expressed as mean nuclear FABP7 fluorescence intensity/ perinuclear FABP7 fluorescence intensity, as described previously (77).
BrdU Incorporation Assay.
Cultured cells were pulsed with 10 μM BrdU for 1 h (DB cells) or 30 min (SUDHL4 cells) in vitro before harvest. Staining was performed according to the manufacturer’s instructions (APC-BrdU kit; BD Biosciences). Acquisition was performed using a FACSCalibur (BD Biosciences) and data analyzed using FlowJo software (TreeStar).
pLKO.1 Plasmid Generation and Nucleofection.
shRNA sequences were generated by The RNAi Consortium (TRC) targeting FABP7 (744, TRCN0000059744; 745, TRCN0000059745; 746, TRCN0000059746) into the pLKO.1puro or pLKO.1GFP lentivector as previously described (78). The scrambled shRNA control (shScramble) in pLKO.1puro or pLKO.1GFP vector was a gift from A. Weng. All constructs were verified by sequencing. SUDHL4 or DB cells were nucleofected using Amaxa Cell line kit L (Lonza), program C-005 or O-017, respectively. Stably transduced cells were selected with puromycin, as applicable.
FA Treatment.
Stock solutions of DHA and AA were prepared in FA-free BSA–PBS vehicle as previously described (43, 79). Cells were treated with a final concentration of 60 µM FA, or vehicle alone, in serum-free media for 24 h.
Supplementary Material
Acknowledgments
We thank Dr. U. Naumann, Dr. Y. Zhang, Dr. R. Morin, and J. Kuo for help with bioinformatics analysis; Dr. R. Godbout for helpful discussions about FABP7 and for the U251 cells; M. Rosin and C. Kang for the tonsillitis lymphocyte isolates; and Dr. A. Weng for scrambled and pLKO.1 plasmids. This work was supported by grants from the Leukemia and Lymphoma Society of Canada (to D.L.M. and C.S.) and the Canadian Institutes of Health Research (CIHR) (to D.L.M.) with core support provided by the British Columbia Cancer Agency. This work was also partially supported by the Canadian Epigenetics, Environment and Health Research Consortium funded by CIHR and Genome BC. R.R. is supported by a postdoctoral fellowship from CIHR, M.M.K. is supported by a postdoctoral fellowship from the Michael Smith Foundation for Health Research (MSFHR), A.B. is supported by a studentship award from the Natural Sciences and Engineering Council of Canada, and C.S. is supported by a scholarship award from MSFHR.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1405507111/-/DCSupplemental.
References
- 1.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11(10):685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 2.Schweiger MR, Kerick M, Timmermann B, Isau M. The power of NGS technologies to delineate the genome organization in cancer: From mutations to structural variations and epigenetic alterations. Cancer Metastasis Rev. 2011;30(2):199–210. doi: 10.1007/s10555-011-9278-z. [DOI] [PubMed] [Google Scholar]
- 3.Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J Pathol. 2011;223(2):102–115. doi: 10.1002/path.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guil S, Esteller M. Cis-acting noncoding RNAs: Friends and foes. Nat Struct Mol Biol. 2012;19(11):1068–1075. doi: 10.1038/nsmb.2428. [DOI] [PubMed] [Google Scholar]
- 5.Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13(7):497–510. doi: 10.1038/nrc3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levin HL, Moran JV. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet. 2011;12(9):615–627. doi: 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biémont C, Vieira C. Genetics: Junk DNA as an evolutionary force. Nature. 2006;443(7111):521–524. doi: 10.1038/443521a. [DOI] [PubMed] [Google Scholar]
- 8.Burns KH, Boeke JD. Human transposon tectonics. Cell. 2012;149(4):740–752. doi: 10.1016/j.cell.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bourque G, et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008;18(11):1752–1762. doi: 10.1101/gr.080663.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunarso G, et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet. 2010;42(7):631–634. doi: 10.1038/ng.600. [DOI] [PubMed] [Google Scholar]
- 11.Rebollo R, Romanish MT, Mager DL. Transposable elements: An abundant and natural source of regulatory sequences for host genes. Annu Rev Genet. 2012;46:21–42. doi: 10.1146/annurev-genet-110711-155621. [DOI] [PubMed] [Google Scholar]
- 12.Nigumann P, Redik K, Mätlik K, Speek M. Many human genes are transcribed from the antisense promoter of L1 retrotransposon. Genomics. 2002;79(5):628–634. doi: 10.1006/geno.2002.6758. [DOI] [PubMed] [Google Scholar]
- 13.van de Lagemaat LN, Landry JR, Mager DL, Medstrand P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 2003;19(10):530–536. doi: 10.1016/j.tig.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Conley AB, Piriyapongsa J, Jordan IK. Retroviral promoters in the human genome. Bioinformatics. 2008;24(14):1563–1567. doi: 10.1093/bioinformatics/btn243. [DOI] [PubMed] [Google Scholar]
- 15.Cohen CJ, Lock WM, Mager DL. Endogenous retroviral LTRs as promoters for human genes: a critical assessment. Gene. 2009;448(2):105–114. doi: 10.1016/j.gene.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 16.Faulkner GJ, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41(5):563–571. doi: 10.1038/ng.368. [DOI] [PubMed] [Google Scholar]
- 17.Kelley D, Rinn J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012;13(11):R107. doi: 10.1186/gb-2012-13-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.St Laurent G, et al. VlincRNAs controlled by retroviral elements are a hallmark of pluripotency and cancer. Genome Biol. 2013;14(7):R73. doi: 10.1186/gb-2013-14-7-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu X, et al. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat Struct Mol Biol. 2014;21(4):423–425. doi: 10.1038/nsmb.2799. [DOI] [PubMed] [Google Scholar]
- 20.Fort A, et al. FANTOM Consortium Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat Genet. 2014;46(6):558–566. doi: 10.1038/ng.2965. [DOI] [PubMed] [Google Scholar]
- 21.Szpakowski S, et al. Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene. 2009;448(2):151–167. doi: 10.1016/j.gene.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ross JP, Rand KN, Molloy PL. Hypomethylation of repeated DNA sequences in cancer. Epigenomics. 2010;2(2):245–269. doi: 10.2217/epi.10.2. [DOI] [PubMed] [Google Scholar]
- 23.Belancio VP, Roy-Engel AM, Deininger PL. All y’all need to know ’bout retroelements in cancer. Semin Cancer Biol. 2010;20(4):200–210. doi: 10.1016/j.semcancer.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamprecht B, et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med. 2010;16(5):571–579, 1p, 579. doi: 10.1038/nm.2129. [DOI] [PubMed] [Google Scholar]
- 25.Steinhardt JJ, Gartenhaus RB. Promising personalized therapeutic options for diffuse large B-cell Lymphoma Subtypes with oncogene addictions. Clin Cancer Res. 2012;18(17):4538–4548. doi: 10.1158/1078-0432.CCR-12-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein HWR, Chan WC. Diffuse large B-cell lymphoma, not otherwise specified. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: Intl Agency Res Cancer; 2008. pp. 233–237. [Google Scholar]
- 27.Morin RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang Y, et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci USA. 2005;102(16):5814–5819. doi: 10.1073/pnas.0402870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, et al. The proteins FABP7 and OATP2 are associated with the basal phenotype and patient outcome in human breast cancer. Breast Cancer Res Treat. 2010;121(1):41–51. doi: 10.1007/s10549-009-0450-x. [DOI] [PubMed] [Google Scholar]
- 30.Seliger B, et al. Identification of fatty acid binding proteins as markers associated with the initiation and/or progression of renal cell carcinoma. Proteomics. 2005;5(10):2631–2640. doi: 10.1002/pmic.200401264. [DOI] [PubMed] [Google Scholar]
- 31.Slipicevic A, et al. The fatty acid binding protein 7 (FABP7) is involved in proliferation and invasion of melanoma cells. BMC Cancer. 2008;8:276–289. doi: 10.1186/1471-2407-8-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karimi MM, et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8(6):676–687. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lander ES, et al. International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 35.Toung JM, Morley M, Li M, Cheung VG. RNA-sequence analysis of human B-cells. Genome Res. 2011;21(6):991–998. doi: 10.1101/gr.116335.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belshaw R, et al. Rate of recombinational deletion among human endogenous retroviruses. J Virol. 2007;81(17):9437–9442. doi: 10.1128/JVI.02216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bannert N, Kurth R. The evolutionary dynamics of human endogenous retroviral families. Annu Rev Genomics Hum Genet. 2006;7:149–173. doi: 10.1146/annurev.genom.7.080505.115700. [DOI] [PubMed] [Google Scholar]
- 38.Jurka J, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1-4):462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 39.Smit AF. Identification of a new, abundant superfamily of mammalian LTR-transposons. Nucleic Acids Res. 1993;21(8):1863–1872. doi: 10.1093/nar/21.8.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diehl V, et al. Characteristics of Hodgkin’s disease-derived cell lines. Cancer Treat Rep. 1982;66(4):615–632. [PubMed] [Google Scholar]
- 41.Möller P, et al. MedB-1, a human tumor cell line derived from a primary mediastinal large B-cell lymphoma. Int J Cancer. 2001;92(3):348–353. doi: 10.1002/ijc.1211. [DOI] [PubMed] [Google Scholar]
- 42.Pellat-Deceunynk C, et al. Human myeloma cell lines as a tool for studying the biology of multiple myeloma: A reappraisal 18 years after. Blood. 1995;86(10):4001–4002. [PubMed] [Google Scholar]
- 43.Mita R, Beaulieu MJ, Field C, Godbout R. Brain fatty acid-binding protein and omega-3/omega-6 fatty acids: mechanistic insight into malignant glioma cell migration. J Biol Chem. 2010;285(47):37005–37015. doi: 10.1074/jbc.M110.170076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friedman DB, Hoving S, Westermeier R. Isoelectric focusing and two-dimensional gel electrophoresis. Methods Enzymol. 2009;463:515–540. doi: 10.1016/S0076-6879(09)63030-5. [DOI] [PubMed] [Google Scholar]
- 45.Feng L, Hatten ME, Heintz N. Brain lipid-binding protein (BLBP): A novel signaling system in the developing mammalian CNS. Neuron. 1994;12(4):895–908. doi: 10.1016/0896-6273(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 46.Balendiran GK, et al. Crystal structure and thermodynamic analysis of human brain fatty acid-binding protein. J Biol Chem. 2000;275(35):27045–27054. doi: 10.1074/jbc.M003001200. [DOI] [PubMed] [Google Scholar]
- 47.Liu RZ, et al. A fatty acid-binding protein 7/RXRβ pathway enhances survival and proliferation in triple-negative breast cancer. J Pathol. 2012;228(3):310–321. doi: 10.1002/path.4001. [DOI] [PubMed] [Google Scholar]
- 48.Goto Y, et al. A new melanoma antigen fatty acid-binding protein 7, involved in proliferation and invasion, is a potential target for immunotherapy and molecular target therapy. Cancer Res. 2006;66(8):4443–4449. doi: 10.1158/0008-5472.CAN-05-2505. [DOI] [PubMed] [Google Scholar]
- 49.Iskow RC, et al. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141(7):1253–1261. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solyom S, et al. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 2012;22(12):2328–2338. doi: 10.1101/gr.145235.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee E, et al. Cancer Genome Atlas Research Network Landscape of somatic retrotransposition in human cancers. Science. 2012;337(6097):967–971. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shukla R, et al. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell. 2013;153(1):101–111. doi: 10.1016/j.cell.2013.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hohn O, Hanke K, Bannert N. HERV-K(HML-2), the best preserved family of HERVs: Endogenisation, expression and implications in health and disease. Front Oncol. 2013;3:246. doi: 10.3389/fonc.2013.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen T, et al. The viral oncogene Np9 acts as a critical molecular switch for co-activating β-catenin, ERK, Akt and Notch1 and promoting the growth of human leukemia stem/progenitor cells. Leukemia. 2013;27(7):1469–1478. doi: 10.1038/leu.2013.8. [DOI] [PubMed] [Google Scholar]
- 55.Maliniemi P, et al. Expression of human endogenous retrovirus-w including syncytin-1 in cutaneous T-cell lymphoma. PLoS ONE. 2013;8(10):e76281. doi: 10.1371/journal.pone.0076281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Romanish MT, Cohen CJ, Mager DL. Potential mechanisms of endogenous retroviral-mediated genomic instability in human cancer. Semin Cancer Biol. 2010;20(4):246–253. doi: 10.1016/j.semcancer.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 57.Piskareva O, et al. The human L1 element: A potential biomarker in cancer prognosis, current status and future directions. Curr Mol Med. 2011;11(4):286–303. doi: 10.2174/156652411795677954. [DOI] [PubMed] [Google Scholar]
- 58.Cruickshanks HA, Tufarelli C. Isolation of cancer-specific chimeric transcripts induced by hypomethylation of the LINE-1 antisense promoter. Genomics. 2009;94(6):397–406. doi: 10.1016/j.ygeno.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 59.Wolff EM, et al. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010;6(4):e1000917. doi: 10.1371/journal.pgen.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weber B, Kimhi S, Howard G, Eden A, Lyko F. Demethylation of a LINE-1 antisense promoter in the cMet locus impairs Met signalling through induction of illegitimate transcription. Oncogene. 2010;29(43):5775–5784. doi: 10.1038/onc.2010.227. [DOI] [PubMed] [Google Scholar]
- 61.Shimizu F, Watanabe TK, Shinomiya H, Nakamura Y, Fujiwara T. Isolation and expression of a cDNA for human brain fatty acid-binding protein (B-FABP) Biochim Biophys Acta. 1997;1354(1):24–28. doi: 10.1016/s0167-4781(97)00115-2. [DOI] [PubMed] [Google Scholar]
- 62.Yun SW, et al. Neural stem cell specific fluorescent chemical probe binding to FABP7. Proc Natl Acad Sci USA. 2012;109(26):10214–10217. doi: 10.1073/pnas.1200817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maxwell SA, Mousavi-Fard S. Non-Hodgkin’s B-cell lymphoma: Advances in molecular strategies targeting drug resistance. Exp Biol Med (Maywood) 2013;238(9):971–990. doi: 10.1177/1535370213498985. [DOI] [PubMed] [Google Scholar]
- 64.Wright G, et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100(17):9991–9996. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choi WW, et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin Cancer Res. 2009;15(17):5494–5502. doi: 10.1158/1078-0432.CCR-09-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim H, Hendrickson R, Dorfman RF. Composite lymphoma. Cancer. 1977;40(3):959–976. doi: 10.1002/1097-0142(197709)40:3<959::aid-cncr2820400302>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 67.Gualco G, Natkunam Y, Bacchi CE. The spectrum of B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin lymphoma: A description of 10 cases. Mod Pathol. 2012;25(5):661–674. doi: 10.1038/modpathol.2011.200. [DOI] [PubMed] [Google Scholar]
- 68.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18(11):1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Flicek P, et al. Ensembl 2014. Nucleic Acids Res. 2014;42(Database issue):D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pruitt KD, et al. RefSeq: An update on mammalian reference sequences. Nucleic Acids Res. 2014;42(Database issue):D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rosenbloom KR, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41(Database issue):D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rhead B, et al. The UCSC Genome Browser database: Update 2010. Nucleic Acids Res. 2010;38(Database issue):D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rebollo R, et al. Epigenetic interplay between mouse endogenous retroviruses and host genes. Genome Biol. 2012;13(10):R89. doi: 10.1186/gb-2012-13-10-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumaki Y, Oda M, Okano M. QUMA: Quantification tool for methylation analysis. Nucleic Acids Res. 2008;36(Web Server issue):W170–175. doi: 10.1093/nar/gkn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dyer MJ, Fischer P, Nacheva E, Labastide W, Karpas A. A new human B-cell non-Hodgkin’s lymphoma cell line (Karpas 422) exhibiting both t(14;18) and t(4;11) chromosomal translocations. Blood. 1990;75(3):709–714. [PubMed] [Google Scholar]
- 76.Lock FE, Ryan KR, Poulter NS, Parsons M, Hotchin NA. Differential regulation of adhesion complex turnover by ROCK1 and ROCK2. PLoS ONE. 2012;7(2):e31423. doi: 10.1371/journal.pone.0031423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Drake KR, Kang M, Kenworthy AK. Nucleocytoplasmic distribution and dynamics of the autophagosome marker EGFP-LC3. PLoS ONE. 2010;5(3):e9806. doi: 10.1371/journal.pone.0009806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giambra V, et al. NOTCH1 promotes T cell leukemia-initiating activity by RUNX-mediated regulation of PKC-θ and reactive oxygen species. Nat Med. 2012;18(11):1693–1698. doi: 10.1038/nm.2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chambrier C, et al. Eicosapentaenoic acid induces mRNA expression of peroxisome proliferator-activated receptor gamma. Obes Res. 2002;10(6):518–525. doi: 10.1038/oby.2002.70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.