

Significance
Ca2+ homeostasis is indispensable for the well being of all living organisms. Ca2+ homeostasis is disrupted by α-synuclein (α-syn), whose misfolding plays a major role in neurodegenerative diseases termed synucleinopathies, such as Parkinson disease. We report that α-syn can induce sustained and highly elevated levels of cytoplasmic Ca2+, thereby activating a calcineurin (CN) cascade that results in toxicity. CN is a highly conserved Ca2+–calmodulin (CaM)-dependent phosphatase critical for sensing Ca2+ concentrations and transducing that information into cellular responses. Limiting, but not eliminating, the availability of CaM, CN and/or CN substrates directly with genetic or pharmacological tools shifts the α-syn–induced CN cascade to a protective mode. This has mechanistic implications for CN's activity and provides a therapeutic venue for the treatment of synucleinopathies.
Keywords: NFAT, TORC2, neuroinflammation, Crz1, Slm2
Abstract
Calcineurin (CN) is a highly conserved Ca2+–calmodulin (CaM)-dependent phosphatase that senses Ca2+ concentrations and transduces that information into cellular responses. Ca2+ homeostasis is disrupted by α-synuclein (α-syn), a small lipid binding protein whose misfolding and accumulation is a pathological hallmark of several neurodegenerative diseases. We report that α-syn, from yeast to neurons, leads to sustained highly elevated levels of cytoplasmic Ca2+, thereby activating a CaM-CN cascade that engages substrates that result in toxicity. Surprisingly, complete inhibition of CN also results in toxicity. Limiting the availability of CaM shifts CN's spectrum of substrates toward protective pathways. Modulating CN or CN's substrates with highly selective genetic and pharmacological tools (FK506) does the same. FK506 crosses the blood brain barrier, is well tolerated in humans, and is active in neurons and glia. Thus, a tunable response to CN, which has been conserved for a billion years, can be targeted to rebalance the phosphatase’s activities from toxic toward beneficial substrates. These findings have immediate therapeutic implications for synucleinopathies.
Cells must tightly regulate Ca2+ homeostasis to avoid pathological perturbations and cell death (1). For example, a profound disruption of Ca2+ homeostasis is seen in Parkinson disease (PD), the second most common neurodegenerative disorder. Mutations or aberrant expression of α-synuclein (α-syn), a major protein involved in the pathogenesis of PD, can induce Ca2+ overload and cell death (2–5). Additional clinical and experimental observations highlight the importance of Ca2+ homeostasis in the pathogenesis of PD. Midbrain dopaminergic (DA) neurons that overexpress Ca2+-binding proteins, which buffer intracellular Ca2+, are characteristically spared from degeneration (6). Patients with hypertension who are treated with the L-type Ca2+ channel blocker, isradipine, have a lower incidence of PD (7). Moreover, isradipine protects DA neurons incubated with α-syn fibrils and is protective in animal models of toxin-induced PD (8–10).
From yeast to mammals, calcineurin is largely responsible for transducing the signals generated by changes in Ca2+ levels (11). Calcineurin (CN) is a calmodulin (CaM)-dependent serine/threonine phosphatase composed of a catalytic subunit (calcineurin A, CNA) and an activating regulatory subunit (calcineurin B, CNB). As intracellular Ca2+ levels rise, Ca2+ binds to CNB and CaM, another key calcium signaling protein. Together, Ca2+-bound CNB and CaM bind CNA, inducing a conformational change that fully activates the phosphatase (11). Signaling through CN plays critical roles in processes ranging from stress response survival in yeast (12) to mammalian development (13).
Despite the compelling link between Ca2+ homeostasis and PD, we know little about the signaling pathways driven by sustained Ca2+ elevations and how they might lead to cell death (4, 5). Yeast provide a powerful model system for such investigations, given their genetic tractability and the remarkable conservation of Ca2+-signaling pathways from yeast to humans (14, 15). Moreover, the expression of human α-syn in yeast leads to cellular pathologies directly relevant to neurons and PD, including nitrosative stress (16, 17), defects in vesicle trafficking (18–20), and faulty mitochondrial function (21, 22).
Results
Intracellular Ca2+ Is Highly Dependent on α-Syn Dosage.
The toxicity of α-syn is extremely dosage sensitive (20, 23). We first asked if the deregulation of Ca2+ in yeast shows the same extreme sensitivity to α-syn dosage as does toxicity. We monitored relative cytosolic Ca2+ levels in strains expressing different levels of α-syn using the genetically encoded Ca2+ sensor, aequorin. Four yeast strains were transformed with a plasmid expressing aequorin (Fig. 1 and Fig. S1A): control cells (expressing yellow fluorescent protein, YFP), NoTox (expressing α-syn at low, nontoxic levels), IntTox (expressing α-syn at intermediate, moderately toxic levels), and HiTox (expressing α-syn at a higher, severely toxic level).
Fig. 1.

BSCaMIQ, a sensor and a sink for calmodulin, rescues toxicity induced by α-syn by decreasing the total levels of free Ca2+–CaM. (A) Strains of control (no α-syn), NoTox (low copy number of α-syn), IntTox (intermediate copy number of α-syn), and HiTox (high copy number of α-syn) were transformed with aequorin, a genetically encoded Ca2+ indicator. Cytosolic Ca2+ was measured by aequorin luminescence over time after α-syn induction. Cytosolic Ca2+ levels are expressed as fold induction relative to control. (B) Cmd1pfree levels assayed by FRET 0, 4, and 8 h after α-syn induction in the presence of BSCaMIQ in control (blue), HiTox strain (red), and HiTox strain transfected with cmd1 (red dashed line). Cmd1pfree = Kd [(Rmax − FRET/CFP)/FRET/CFP − Rmin] (see Supporting Information). (C) Yeast strains were spotted onto plates containing uninducing media [synthetic defined (SD) −Ura; GPD-BSCaMIQ selective; Lower] and replica platted in threefold serial dilutions onto α-syn–inducing plates containing selective media and (SGal −Ura) (Upper). YFP is used as control plasmid. (D) Yeast strains were spotted onto plates containing uninducing media (SD −Ura, Leu; BSCaMIQ and cmd1 selective; Lower) and replica platted in threefold serial dilutions onto α-syn–inducing plates containing selective media and SGal −Ura, Leu (Upper). YFP and empty vector (vec) were used as control plasmids, cmd1 = yeast calmodulin and cmd1X = D94A, E105V; unable to bind calcium at the third EF hand. (E) BSCaMIQ (green), neuronal (red MAP2 positive), and nuclear (blue, Hoechst) stainings from representative pictures of rat primary neuronal cultures coinfected with either control lentivirus LacZ, LacZ and α-SynA53T, LacZ and α-SynA53T and BSCaMIQ, or LacZ and BSCaMIQ. (F) Percentages of neurons (MAP2 positive) relative to control (LacZ infected) in the conditions described in B. *P < 0.05, one-way ANOVA, Dunnett’s multiple comparison test.
In response to α-syn induction, the NoTox strain exhibited a reproducible twofold elevation in cytosolic Ca2+. However, this was transient and Ca2+ levels rapidly returned to normal. The IntTox strain exhibited a twofold elevation in cytosolic Ca2+ that was stable for 24 h. The HiTox strain, however, reached an ∼4-fold elevation in cytosolic Ca2+ at 4 h and cytosolic Ca2+ continued to climb to an almost 65-fold increase by 8 h (Fig. 1A). The rise in Ca2+ derived from intracellular stores because it was sensitive to low concentrations of the intracellular calcium chelator 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis-acetoxymethyl ester, but not to EGTA in the extracellular medium (Fig. S1B). Importantly, the extreme elevation in intracellular Ca2+ preceded the major onset of cell death in the HiTox strain (Fig. S1C). Thus, the release of Ca2+ from intracellular stores exhibits strong dependence on the dosage of α-syn and sustained high levels of Ca2+ are subsequently associated with extensive cell death.
The Rise in Intracellular Ca2+ Is Accompanied by a Decrease in Free Calmodulin.
The delay between Ca2+ elevation and cell death in the HiTox strain suggested that high Ca2+ levels might initiate a cascade of events that are ultimately toxic. Calmodulin (CaM in mammalian cells and Cmd1p in yeast) is a ubiquitous node through which Ca2+ activates numerous downstream targets. To determine if CaM plays a direct role in α-syn toxicity, we first expressed a genetically encoded sensor for free CaM (Fig. S2 A and B). This genetically encoded sensor for free CaM (BSCaMIQ) contains a CaM-binding site flanked by CFP and YFP (24, 25). At low Ca2+ levels, CaM is largely unbound and is therefore able to bind BSCaMIQ. As cytosolic Ca2+ rises, CaM engages its targets and binding to BSCaMIQ decreases, resulting in a real-time change in fluorescence resonance energy transfer (FRET) between CFP and YFP. Given that CaM expression is not affected by α-syn in yeast (22), changes in FRET signals from BSCaMIQ provide a quantitative measure of changes in free CaM in our strains.
In the control strain, where cytosolic Ca2+ remained low (Fig. 1A), free Cmd1p levels remained high and constant (Fig. 1B). In the HiTox strain, which experienced a sharp rise in cytosolic Ca2+ (Fig. 1A), free Cmd1p levels plummeted (Fig. 1B). To confirm that the change in FRET with BSCaMIQ was indeed due to a reduction in free Cmd1p, we overexpressed cmd1. This restored normal FRET levels in HiTox cells (Fig. 1B).
Sequestering Free Calmodulin Ameliorates α-Syn Toxicity in Yeast.
The decrease in free CaM caused by α-syn suggests CaM is engaging downstream targets. If this contributes to cell death, the binding of CaM by BSCaMIQ should compete with those targets and reduce toxicity. That is, BSCaMIQ should act not only as a sensor but as a sink for CaM (25) (Fig. S2C). To test this, control and HiTox strains expressing BSCaMIQ (or a YFP control) were assayed for growth (Fig. 1C). Indeed, expression of BSCaMIQ conferred strong protection to the HiTox strain.
Protection by BSCaMIQ was not due to a trivial effect on α-syn expression (Fig. S2B). Nor was it due to a general response to proteotoxic stress because cells expressing proteins associated with other neurodegenerative diseases, polyglutamine expanded huntingtin fragment (Htt) and TDP43, at similarly toxic levels were not rescued by BSCaMIQ (Fig. S2 D and E).
Expression of BSCaMIQ did not significantly affect Ca2+ levels (Fig. S2F), suggesting that its protection against cell death was primarily due to the sequestration of Ca2+–CaM. Indeed, the overexpression of wild-type cmd1 restored toxicity, whereas overexpression of a cmd1 mutant with impaired Ca2+ binding (26) did not (Fig. 1D and Fig. S2G).
Finally, we tested a completely different CaM sink, the IQ domain from myosin cardiac light chain (CYIQ). This alternative CaM binder also strongly rescued cells from α-syn toxicity (Fig. S2H). These findings establish that signaling through Ca2+–CaM-dependent pathways plays an important role in α-syn toxicity in yeast.
Sequestering Free Calmodulin Ameliorates α-Syn Toxicity in Neurons.
Next, we asked if Ca2+–CaM was relevant to neuronal α-syn toxicity. We used primary cells derived from rat embryonic cerebral cortices as a model because they are relatively homogenous (more than 80% neurons) and more readily obtained than dopaminergic neurons. Importantly, cortical neurons are strongly affected in patients with PD (27).
Rat embryonic cortical neurons were transduced with a lentivirus that expresses α-syn A53T, a mutation causing an autosomal-dominant form of PD (28) (about 50% infected; Fig. S3A). To test the effect of α-syn on Ca2+ homeostasis in these neurons, we used the Ca2+-sensitive dye Fluo-4. Infected neurons were visualized with either mKate (control) or an mKate-α-syn fusion regulated by the synapsin promoter. Cells expressing mKate-α-syn had increased basal Ca2+ and responded less strongly to KCl-induced depolarization than cells expressing mKate alone (Fig. S3 B and C). Thus, as previously shown in other primary neuronal models (2–4), overexpression of α-syn perturbs Ca2+ homeostasis.
To determine if CaM plays a role in the toxicity of α-syn in neurons, we coinfected A53T-expressing neurons with another lentivirus encoding BSCaMIQ or a control protein, LacZ. Differences in viability were assessed by cellular ATP content as well as by counting the number of cells positive for microtubule-associated protein 2 (MAP2), a neuron-specific marker. BSCaMIQ provided a highly significant, dosage-dependent rescue of α-syn toxicity (Fig. 1 E and F and Fig. S3D).
Calcineurin Activation by Calmodulin Has both Protective and Toxic Roles.
A central player in transducing Ca2+–CaM’s signals is the highly conserved phosphatase CN (14). To investigate CN's role in α-syn toxicity, we returned to yeast. CN function can be eliminated by deleting the regulatory subunit cnb1 alone or by the combined deletion of the catalytic subunits cna1 and cna2. As expected (29), neither manipulation affected the growth of the control strain (Fig. 2A and Fig. S4A). In the HiTox cells, deleting CN eliminated BSCaMIQ protection (Fig. S4B). Because BSCaMIQ acts by titrating CaM, and CaM has many targets, this result suggests that CaM’s main effects in response to α-syn toxicity are mediated through the calcineurin pathway.
Fig. 2.

Calcineurin activation is central to α-syn toxicity in yeast. (A) Control and HiTox strains lacking calcineurin (cnb1Δ) were spotted onto plates containing uninducing media (SD −His,Trp–α-syn selective, Lower) and replica plated in threefold serial dilutions on α-syn–inducing plates containing selective media SGal (Upper). (B) Control and HiTox yeast strains were spotted onto plates containing uninducing media (SD −Ura, Leu; cna1- and cnb1 selective; Lower) and replica plated in threefold serial dilutions onto α-syn–inducing plates containing selective media and SGal (Upper). (C) Same assay as in B except control and HiTox strains was plated onto plates containing uninducing media (SD −Leu; rcn1-, or rcn2 selective, respectively). (D) Growth (described as percentage over control) of HiTox cells grown for 48 h over a range of FK506 concentrations. See also Fig. S4D.
Surprisingly, however, both deletion and overexpression of CN increased toxicity in the HiTox strain (Fig. 2 A and B). One explanation for these apparently contradictory results is that an intermediate level of CN activation is protective against α-syn, whereas either too much or too little is detrimental.
As a genetic test of this hypothesis, we used Rcn proteins as negative regulators of CN activity. Rcn1p has a higher affinity for CN than Rcn2p (30). Whereas neither protein affected the control strain, overexpressing rcn2 (but not rcn1) rescued α-syn toxicity (Fig. 2C).
As a pharmacological approach, we used FK506. Importantly, FK506 provides a means to continuously vary CN inhibition over a very broad range. FK506 inhibits CN by precisely the same mechanism in yeast and in mammals (29, 31) although higher concentrations of this drug are required in yeast than in mammalian cells (Materials and Methods). Consistent with our hypothesis, in the HiTox strain intermediate concentrations of FK506 protected against α-syn toxicity, whereas higher concentrations eliminated this protection (Fig. 2D).
Importantly, rescue by intermediate concentrations of FK506 in the HiTox strain was not due to a trivial effect on α-syn expression (Fig. S4C). Furthermore, the lack of rescue at high concentrations was not due to off-target toxicity, because FK506 had no effect on control cells even at the highest concentrations used (Fig. S4D). Moreover, FK506 effects were specific to the HiTox strain where CN activity is high: the compound had no effect on the less toxic IntTox strain, which has much lower levels of cytosolic Ca2+ (Fig. 1A and Fig. S4E). We further confirmed FK506 effects on CN activity using BSCaMIQ, which sequesters CaM and reduces CN activation. The effects of FK506 should be greatly reduced in HiTox cells carrying BSCaMIQ, and indeed they were (Fig. S4F).
Thus, both genetic and pharmacologic experiments in yeast confirm that an intermediate level of CN inhibition balances the protective versus toxic effects of Ca2+–CaM/CN in response to α-syn.
Decreasing Calcineurin Activity Protects Against α-Syn Toxicity in Neuronal Models.
Next, we asked if modulating CN activities could balance toxic versus protective responses to α-syn in primary rat cortical neurons. At all concentrations, FK506 had only a minor effect on the viability of neurons expressing the control protein LacZ (Fig. 3A). Intermediate concentrations of FK506 rescued neurons from the toxic effect of α-syn expression. (Note, in keeping with the fact that neurons have many more mechanisms for buffering calcium than yeast cells do, the concentration range for rescue was much broader in neurons than in yeast, spanning a 100-fold range.) Higher concentrations of FK506 were not protective and, in fact, increased toxicity (Fig. 3A).
Fig. 3.

Partial calcineurin activity is necessary to protect against α-syn toxicity ex vivo and in vivo. (A, Upper) MAP2 staining from representative pictures of rat primary neuronal cultures coinfected with a lentivirus carrying LacZ as control and/or α-synA53T treated with various doses of FK506 for 14 d. (Lower) Rat cortical neurons infected with α-synA53T and/or LacZ as control treated with vehicle and/or increasing concentrations of FK506 for 14 d and assayed for viability for ATP content. *P < 0.05, one-way ANOVA, Dunnett’s multiple comparison test. Each data point was normalized against control LacZ at the same FK506 dose. (B, Upper) Dopaminergic neurons from mice infected with α-synA53T and/or mKate as control treated with vehicle and/or distinct concentrations of FK506. Cell survival was measured by counting TH positive neurons after 14 d of α-synA53T infection and drug treatment. *P < 0.05, one-way ANOVA, Dunnett’s multiple comparison test. (Lower) Representative pictures of infection efficiency of mKate α-syn in TH positive neurons. (C) Representative images of C. elegans dopaminergic neurons in worms expressing GFP alone (Left, all six anterior neurons are intact) and in α-syn–expressing worms treated with RNAi control (only four anterior DA neurons remain) or with RNAi for calcineurin (CN) (Right). Arrowheads show intact dopaminergic neuron cell bodies and arrows represent areas where dopaminergic neurons are absent. (D) The population of worms with WT (wild type) numbers of neurons remaining after treatment with RNAi for calcineurin (CN) or empty vector RNAi (control) in α-syn–expressing worms. For each experiment, three independent experiments were performed, with 30 worms per trial. *P < 0.05 (Student t test). See also Fig. S5C. (E) Same assay as in D except with worms overexpressing rcn-1. Bars 1 and 2 represent independent transgenic lines. *P < 0.005, **P < 0.0005 (Student t test). See also Fig. S5B.
We next asked if FK506 was protective in dopaminergic neurons, the cell type most classically implicated in Parkinson disease. We used primary cultures containing 30–40% dopaminergic neurons assessed by tyrosine hydroxylase (TH) positivity. To allow unambiguous identification, we infected neurons with an adeno-associated virus encoding human α-synA53T tagged with mKate or with mKate alone (Fig. S5A). Fourteen days after infection, ∼30% of the dopaminergic neurons expressing α-syn had died. Intermediate levels of FK506 rescued cells from α-syn toxicity, whereas the protective effects of the drug disappeared at higher doses (Fig. 3B).
To begin moving into multicellular organisms, we took advantage of a previously established nematode model of α-syn toxicity (32). In these worms, GFP and α-syn are both under the transcriptional control of the dopaminergic neuron-specific dat-1 promoter. Wild-type (WT) worms invariably have six dopaminergic neurons. As previously reported (18), expression of α-syn caused an age- and dose-dependent degeneration of dopaminergic neurons (Fig. 3C). We down-regulated CN activity in this model using two methods: direct knockdown of CN with RNAi and overexpression of its negative regulator, rcn-1.
To allow RNAi targeting of CN in DA neurons, we expressed the dsRNA transporter SID-1 in these cells using the dat-1 promoter (33). Knockdown of the catalytic subunit of CN, tax-6, had no effect on the dopaminergic neurons of control worms (Fig. S5C). However, in α-syn–expressing worms, reducing the expression of CN by knockdown protected against toxicity (Fig. 3 C and D).
Nematodes have only one known negative regulator of CN, rcn-1 (34). Several independent transgenic lines were established with different levels of rcn-1 expression. In two transgenic lines, we found that moderate rcn-1 expression was nontoxic (Fig. S5B) and also protected dopaminergic neurons against α-syn toxicity (Fig. 3E). Thus, establishing a biological mechanism conserved from single cells (yeast and primary mammalian neurons) to whole worms, chemical and genetic approaches, demonstrate that moderating CN activity is protective against α-syn toxicity.
Protective and Toxic Downstream Substrates of Calcineurin.
Our finding that an intermediate level of CN activity is protective against α-syn suggests that some CN downstream targets provide protection, whereas others enhance toxicity. To investigate, we returned to yeast, focusing on three of CN's best characterized substrates: Hph1p, Slm2p, and Crz1p. Remarkably, the processes regulated by all of these proteins are disturbed by α-syn in yeast and in neurons. Hph1p facilitates the posttranslational translocation of proteins, especially those involved in vacuolar ion homeostasis and vesicular trafficking (35). Slm2p acts via the target of rapamycin complex 2 (TORC2) to support cytoskeletal organization and lipid homeostasis (20, 36–38). Crz1p is a transcription factor activated by calcineurin in response to various stresses (39). Importantly, overexpression of these genes under nonstress conditions had no effect on control cells (Fig. S6A).
Overexpressing hph1 or slm2 rescued α-syn toxicity in the HiTox strain (Fig. 4A). Overexpressing crz1, however, slightly enhanced toxicity. To better characterize this enhancement of toxicity, we overexpressed crz1 in the HiTox strain in the presence of BSCaMIQ. Overexpression of crz1 completely abrogated the protective effects of BSCaMIQ, confirming the role of crz1 in enhancing α-syn toxicity and validating the effect of BSCaMIQ on the CN pathway (Fig. S6B). Thus, Hph1p and Slm2p drive protective pathways in response to α-syn, whereas Crz1p drives detrimental ones.
Fig. 4.
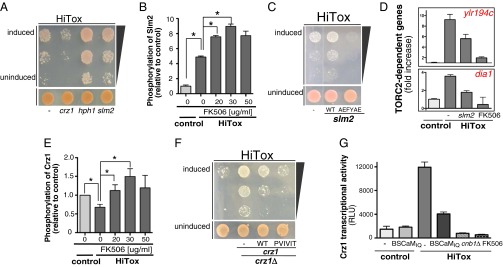
Calcineurin can activate protective and toxic substrates in yeast. (A) The HiTox yeast strain was spotted onto plates containing uninducing media (SD −Leu; vector, hph1, slm2, or crz1 selective; Lower) and replica plated in threefold serial dilutions onto α-syn–inducing plates containing selective media and SGal −Leu (Upper). (B) Peak intensity of phosphopeptide ENVD(phospho)SPR from Slm2p relative to control using SRM-mass spectrometry after phosphopeptide enrichment as described (66). Error bars reflect biological and technical variability. *P < 0.05 (Student t test). (C) Similar spotting assays as described in A but onto plates containing uninducing media [SD −Leu; slm2, slm2-AEFYAE (unable to bind calcineurin) selective; Lower] and replica plated in threefold serial dilutions onto α-syn–inducing plates containing selective media and SGal −Leu (Upper). (D) Real-time PCR for the TORC2-dependent genes ylr194c and dia1 in control and HiTox yeast strains in the presence of slm2 or a low dose of FK506 (25 μg/mL). (E) Peak intensity of phosphopeptide MDSANS(phospho)SEKISK from Crz1p relative to control using SRM-mass spectrometry after phosphopeptide enrichment as described (66). Error bars reflect technical variability. *P < 0.05 (Student t test). (F) The HiTox yeast strain was spotted onto plates containing uninducing media [SD −Leu; vector, crz1, PVIVIT-crz1 (high affinity to calcineurin) selective; Lower] and replica plated in threefold serial dilutions onto α-syn–inducing plates containing selective media and SGal −Leu (Upper) in the presence and absence of crz1 (crz1Δ). (G) β-Glactosidase luminescent assay for control and HiTox strains harboring a reporter for crz1p-dependent transcriptional activity in the presence and absence of 25 μg/mL FK506, calcineurin (cnb1Δ), and/or BSCaMIQ.
We focused further on the protective substrate Slm2p and the toxic substrate Crz1p, because Slm2p and Crz1p pathways are conserved from yeast to humans. Because CN is a phosphatase, we began by investigating the effect of α-syn on the phosphorylation status of Slm2p and Crz1p using selected reaction monitoring (SRM)-based mass spectrometry (MS) (40). This technique avoids the use of artificial protein tags, which can interfere with protein function, and more importantly, gives information about the nature of the phosphorylation sites in a highly specific and quantitative manner.
α-Syn expression increased Slm2p phosphorylation. Protective doses of FK506 further increased phosphorylation, whereas toxic doses blunted it (Fig. 4B). Although the role(s) of phosphorylation in regulating Slm2p activity is not clear, this result is consistant with an interaction between CN and Slm2p driving a protective response against α-syn toxicity. To test this, point mutations in Slm2p that selectively abrogate its interaction with CN (41) eliminated protection against α-syn toxicity, although having no effect on control strains (Fig. 4C and Fig. S6C).
Slm2p acts upon the TORC2 signaling pathway (38). To see whether this pathway is also affected by α-syn, we assayed the expression of two yeast genes that are classically up-regulated when TORC2 is inhibited (42). ylr194c and dia1 were both up-regulated in response to α-syn expression, indicating that α-syn does indeed inhibit the TORC2 pathway (Fig. 4D). As predicted from their effects on CN, overexpression of slm2 or moderate concentrations of FK506 both decreased expression of ylr194c and dia1, indicating a restoration of TORC2 function (Fig. 4D).
Activation of the transcription factor Crz1p is normally protective for yeast cells under stress (39). However, our results suggest that Crz1p drives a toxic response to α-syn. Phosphorylation of Crz1p inhibits its transcriptional activity. Using SRM to monitor Crz1p phosphorylation, we found that it was reduced in response to α-syn, whereas protective and toxic doses of FK506 increased it (Fig. 4E). A mutant of Crz1p with increased affinity for CN (43) had no effect on control cells but increased toxicity in cells expressing α-syn (Fig. 4F and Fig. S6D). This high-affinity allele of crz1 also reduced the protective effect of BSCaMIQ, confirming that CaM’s role in α-syn toxicity is mediated through the CN pathway (Fig. S6E). Further, deleting crz1 strongly rescued growth in HiTox cells (Fig. 4F and Fig. S6D).
If Crz1 activation is indeed driving a toxic transcriptional response to α-syn, Crz1p’s transcriptional activity should be up-regulated in the HiTox strain. Moreover, if this activity is connected to the Ca2+–CaM/CN pathway cascade, it should be down-regulated by BSCaMIQ, by protective concentrations of FK506 or by deletion of CN. Using a β-galactosidase reporter for Crz1p transcriptional activity (44), each of these predictions was fulfilled (Fig. 4G). Thus, α-syn activates a toxic Crz1p-regulated response to Ca2+ in yeast. Moderate inhibition of CN activity prevents activation of this toxic pathway while still allowing protective responses mediated through substrates such as Slm2p to remain intact.
Downstream Consequences of Calcineurin Activation by α-Syn in Mice.
To determine whether the effects of α-syn demonstrated in yeast are conserved in the central nervous system of mammals, we turned to an α-syn transgenic mouse model where α-syn is overexpressed in various regions of the brain, including areas affected in PD such as the olfactory bulb and cortex (45, 46). As they age, these mice display phenotypes reminiscent of PD, including neuroinflammation (increased numbers of astrocytes and microglia), as well as neuronal pathology. Calcineurin is highly expressed in both glia and neurons and, hence, could potentially contribute to protective or toxic responses in both.
First, we probed the status of the TORC2 pathway (activation was protective in yeast). In the brains of 12-mo-old mice, we monitored protein kinase C alpha (PKCα) phosphorylation at serine 657, a canonical downstream substrate of TORC2. PKCα phosphorylation was reduced in brain extracts from α-syn–overexpressing mice compared with controls (Fig. 5A). Thus, the inhibition of PKCα correlates with inhibition of the TORC2 pathway in mammalian brain in response to α-syn.
Fig. 5.

Calcineurin substrates in α-syn transgenic mice. (A) Western blot for phosphoserine 657, PKCα from brain lysates of 12-mo-old α-syn transgenic mice. PKCα serves as loading control. (B) Immunohistochemistry for NFATc4 in neurons from the mitral cell layer in the olfactory bulb of 13-mo-old α-syn transgenic mice and control (C) mice (Upper). Immunohistochemistry for NFATc3 in glia from the internal plexiform layer in the olfactory bulb of 13-mo-old α-syn transgenic. Immunohistochemistry for glial fibrillary acid protein (GFAP) for normal and reactive astrocytes in the internal plexiform layer of the olfactory bulb. Similarly, immunostaining for Iba-1 from normal and activated microglia in the nodules of the internal plexiform layer of the olfactory bulb of α-syn transgenic mice. Error bars reflect variability between two sections analyzed from a total of five animals in each group. *P < 0.05 (Student t test).
Although no direct homolog of crz1 exists in mammalian cells, nuclear factor of activated T cells (NFAT) is broadly accepted as its functional analog (41). Similar to Crz1p, calcineurin dephosphorylates various isoforms of NFAT, leading to their nuclear translocation. We monitored two isoforms, NFATc4 and NFATc3, which we found to be prominent in neurons and glia, respectively. In agreement with our yeast results, the neurons from the olfactory bulbs of the α-syn transgenic mice displayed increased nuclear neuronal localization of NFATc4, consistent with its activation (Fig. 5B). NFATc3 staining intensity and nuclear localization was increased in glia (Fig. 5B), indicating that it, too, was activated. Thus, a calcineurin-mediated response to α-syn was also conserved in the mammalian brain, in both neurons and glia.
NFAT Is Activated in Human Synucleinopathies.
Finally, we examined NFAT activation in fixed postmortem tissue from humans diagnosed with PD or with a more aggressive synucleinopathy, dementia with Lewy bodies (DLB). We could not detect NFATc3; however, staining of NFATc4 was highly reproducible and specific, as it was competed with a peptide that was used to raise the antibody (Fig. S7). Four of the five brains we analyzed from nondiseased controls exhibited cytoplasmic but no nuclear staining for NFATc4. In contrast, NFATc4 immunoreactivity was localized to nuclei in most of the eight cases of PD and DLB consistent with its activation (Fig. 6).
Fig. 6.

NFATc4 nuclear expression is increased in cases of PD and DLB. Immunohistochemistry for NFATc4 staining in neurons from the substantia nigra pars compacta, hippocampus area CA3, and layers 5 and 6 of the frontal cortex in human PD and DLB cases. An average of two sections from five control (C) and eight diseased cases were analyzed. Nuclear staining was scored by a neuropathologist. Scoring: 0, no nuclear staining; 1, scattered positive nuclei; 2, positive nuclear <30% of neurons; 3, positive nuclei focally >30% of neurons. n, neuronal nucleus; nm, neuromelanin; nuc, nucleolus. (Scale bar, 50 μm.) *P < 0.05 (Student t test).
The nuclear staining for NFATc4 in the diseased brains was evident across several different brain regions including the substantia nigra pars compacta (SNc), hippocampus, and frontal cortex, among others. In the frontal and cingulate cortices, nuclear NFATc4 staining was most prominent in the pyramidal neurons in layers 5 and 6. In DLB, these cortical layers bear the greatest burden of α-syn pathology (27). In the hippocampus, which is also critically involved in PD pathology (47), areas CA3 and CA4 stained more strongly than CA1 or CA2 or the subiculum. These results support an association between NFATc4 nuclear localization/activation and α-syn toxicity in human disease.
Discussion
We demonstrate that increasing levels of α-syn expression proportionally increase cytosolic Ca2+ concentrations, paralleling the extremely strong dosage dependence of α-syn toxicity previously described in our yeast models (18, 20) and in humans (23, 48). Sustained high cytosolic Ca2+ levels drive a Ca2+–CaM–calcineurin cascade that activates a toxic program regulated by the transcription factor Crz1p in yeast and NFAT in mammalian cells. However, calcineurin activation also triggers protective responses against α-syn involving TORC2. The balance between these responses and presumably other unidentified calcineurin substrates is critical in determining the ultimate outcome of activating the Ca2+–CaM–calcineurin pathway in response to α-syn (Fig. S8). Importantly, this multifaceted calcineurin-mediated response to α-syn, although first uncovered in yeast, is conserved in nematodes as well as mammalian neurons, glia, and brains, as, indeed, are the protective effects of moderate concentrations of FK506.
How does α-syn lead to increased cytosolic Ca2+? Our data clearly indicate that it is released from intracellular stores. Given the lipid-binding properties of α-syn, the protein might perturb Ca2+ homeostasis directly (through damage to the membrane or membrane channels) or indirectly [as a consequence of the profound block in endoplasmic reticulum (ER) to Golgi trafficking]. How might increased cytosolic Ca2+ determine calcineurin’s switch between protective and toxic substrates in a concentration-dependent manner? Differences in the sequence and accessibility of substrate docking sites, expression levels, and the number of dephosphorylations required to alter a substrate’s activity are all likely involved. These, in turn, will interface with differences in the degree of calcineurin activation, determined by cellular levels of CaM and Ca2+ in different compartments within the cell. Given the complexity of this problem, many strategies, including the use of genetically tractable organisms such as yeast, will be required to achieve a full understanding.
Because inhibition of calcineurin activity is routinely exploited clinically through the use of FK506 (tacrolimus) as an immunosuppressant, we now suggest that the repurposing of FK506, a compound that readily traverses the blood brain barrier merits investigation in the management of PD. Because it persists in the central nervous system long after systemic effects have resolved (49), intermittent dosing with this already FDA-approved drug could avoid systemic immunosuppression, while still providing a readily implemented, disease-modifying treatment strategy that targets a fundamental mechanism in the pathogenesis of α-synucleinopathies.
Indeed, FK506 was previously shown to have neuroprotective properties in mammalian PD models (50–53). Because FK506 impairs calcineurin function by locking it into a complex with FKBP12, FK506’s neuroprotective effects were thought to be mediated through FKBP12. Compounds that target FKBP12, without affecting calcineurin function, prevented neurodegeneration in specific animal models (54, 55) but failed to show reversal of PD motor symptoms in humans (56). Although we have yet to address any potential contributions of FKBP12 to the protective effects of FK506 that we have seen, our findings clearly establish the importance of reducing calcineurin activity to achieve neuroprotection.
Finally, our findings help explain why in PD, DA neurons in the SNc are particularly sensitive to α-syn–related dysfunction. Adult neurons in the SNc rely on voltage-dependent Ca2+ channels. These channels are regulated by CaM binding (57, 58) and drive autonomous pacemaking, which leads to relatively sustained elevations in cytosolic Ca2+ (9, 59). Intriguingly, CaM can be cross-linked to α-syn (60, 61), suggesting that α-syn could be locally sequestering CaM to further increase cytosolic Ca2+ flux through these channels. Moreover, SNc neurons have higher cytosolic dopamine levels than the less susceptible ventral tegmentum DA neurons (62). This neurotransmitter renders SNc neurons more susceptible to cell death and has also been linked to increased intracellular Ca2+ levels (62). The enhanced sensitivity of the SNc DA neurons to Ca2+ stress would exacerbate the defects in vesicle trafficking (18, 63, 64), mitochondrial dysfunction (21, 22), nitrosative stress (16, 17), and metal ion homeostasis (65) caused by α-syn. The combination is poised to create the “perfect storm” that devastates the SNc of patients with PD.
Materials and Methods
Primary Cells and Strains.
Yeast strains containing α-syn were generated and induced as previously described (18). Rat cortical and mouse dopaminergic neurons were infected with a lentivirus carrying α-syn as described in SI Materials and Methods. In vivo models for α-syn in Caenorhabditis elegans and mice where generated as described (32, 46). Human samples from PD and DLB patients were obtained through the Massachusetts Alzheimer Disease Research Center as described in SI Materials and Methods.
Imaging Techniques.
Ca2+ imaging was performed using aequorin (in yeast) and Fluo-4 (in neurons). CaM measurements were performed using FRET. MAP2 and TH staining in neurons were performed by immunofluorescence. NFAT staining in mice and human samples were performed by immunohistochemistry. Detailed description of all imaging techniques can be found in SI Materials and Methods.
Viability Assays.
For neurons we used ATP and MAP2 positive staining, whereas for yeast cells, spotting assays, growth curves using OD600, and propidium idodine incorporation by cell cytometry were performed as viability assays. Detailed description of all viability techniques can be found in SI Materials and Methods.
Biochemistry Assays.
To monitor phosphorylation of substrates selected reaction monitoring assays was performed. To monitor Crz1p activity in yeast we used the Crz1-dependent reporter element-luciferase reporter (44). Detailed description of these techniques can be found in SI Materials and Methods.
Molecular Biology.
RCN-1 overexpression and quantitation C. elegans was performed as described (34). RNA interference (RNAi) in C. elegans was performed as described (33). CaM mutants were performed by QuikChange XL site-directed mutagenesis as described in SI Materials and Methods.
Supplementary Material
Acknowledgments
Special thanks to Y. Schmitz, A. Mungenast, J. Heitman, M. Cyert, D. Petranovic, and members of the S.L. laboratory for discussions and/or critical reading of the manuscript. This work was supported by the Picower Foundation, the JPB Foundation, a Howard Hughes Medical Institute (HHMI) Collaborative Innovation Award, and the Eleanor Schwartz Charitable Foundation. C.Y.C. is supported by National Institutes of Health (NIH) Grant K01 AG038546, P.K.A., by a Beeson Career Development Award in Aging (NIH Grant K08 AG042774). E.V.M. and D.S. are supported by the JPB and Parkinson's Disease Foundation and the Udall Center of Excellence. E.V.M. is also supported by National Institute of Neurological Disorders and Stroke Grant R01 NS075222. S.L. is an HHMI Investigator.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1413201111/-/DCSupplemental.
References
- 1.Wojda U, Salinska E, Kuznicki J. Calcium ions in neuronal degeneration. IUBMB Life. 2008;60(9):575–590. doi: 10.1002/iub.91. [DOI] [PubMed] [Google Scholar]
- 2.Adamczyk A, Strosznajder JB. Alpha-synuclein potentiates Ca2+ influx through voltage-dependent Ca2+ channels. Neuroreport. 2006;17(18):1883–1886. doi: 10.1097/WNR.0b013e3280115185. [DOI] [PubMed] [Google Scholar]
- 3.Danzer KM, et al. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27(34):9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin ZS, et al. α-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: Relevance to human synucleopathic diseases. J Neurochem. 2012;120(3):440–452. doi: 10.1111/j.1471-4159.2011.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buttner S, et al. The Ca2+/Mn2+ ion-pump PMR1 links elevation of cytosolic Ca2+ levels to alpha-synuclein toxicity in Parkinson's disease models. Cell Death Differ. 2012;20(3):465–77. doi: 10.1038/cdd.2012.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurley MJ, Brandon B, Gentleman SM, Dexter DT. Parkinson’s disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain. 2013;136(Pt 7):2077–2097. doi: 10.1093/brain/awt134. [DOI] [PubMed] [Google Scholar]
- 7.Pasternak B, et al. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol. 2012;175(7):627–635. doi: 10.1093/aje/kwr362. [DOI] [PubMed] [Google Scholar]
- 8.Dryanovski DI, et al. Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci. 2013;33(24):10154–10164. doi: 10.1523/JNEUROSCI.5311-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan CS, et al. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447(7148):1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 10.Kupsch A, et al. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in non-human primates is antagonized by pretreatment with nimodipine at the nigral, but not at the striatal level. Brain Res. 1996;741(1-2):185–196. doi: 10.1016/s0006-8993(96)00917-1. [DOI] [PubMed] [Google Scholar]
- 11.Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011;21(2):91–103. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonilla M, Cunningham KW. Calcium release and influx in yeast: TRPC and VGCC rule another kingdom. Sci STKE. 2002;2002(127):pe17. doi: 10.1126/stke.2002.127.pe17. [DOI] [PubMed] [Google Scholar]
- 13.Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opin Genet Dev. 2001;11(5):505–512. doi: 10.1016/s0959-437x(00)00225-2. [DOI] [PubMed] [Google Scholar]
- 14.Rusnak F, Mertz P. Calcineurin: Form and function. Physiol Rev. 2000;80(4):1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 15.Auluck PK, Caraveo G, Lindquist S. α-Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Annu Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- 16.Chung CY, et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science. 2013;342(6161):983–987. doi: 10.1126/science.1245296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tardiff DF, et al. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates α-synuclein toxicity in neurons. Science. 2013;342(6161):979–983. doi: 10.1126/science.1245321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper AA, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313(5785):324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gitler AD, et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci USA. 2008;105(1):145–150. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302(5651):1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guzman JN, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468(7324):696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su LJ, et al. Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Dis Model Mech. 2010;3(3-4):194–208. doi: 10.1242/dmm.004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 24.Black DJ, Leonard J, Persechini A. Biphasic Ca2+-dependent switching in a calmodulin-IQ domain complex. Biochemistry. 2006;45(22):6987–6995. doi: 10.1021/bi052533w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Yang PS, Yang W, Yue DT. Enzyme-inhibitor-like tuning of Ca(2+) channel connectivity with calmodulin. Nature. 2010;463(7283):968–972. doi: 10.1038/nature08766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geiser JR, van Tuinen D, Brockerhoff SE, Neff MM, Davis TN. Can calmodulin function without binding calcium? Cell. 1991;65(6):949–959. doi: 10.1016/0092-8674(91)90547-c. [DOI] [PubMed] [Google Scholar]
- 27.Irwin DJ, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72(4):587–598. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polymeropoulos MH, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 29.Breuder T, Hemenway CS, Movva NR, Cardenas ME, Heitman J. Calcineurin is essential in cyclosporin A- and FK506-sensitive yeast strains. Proc Natl Acad Sci USA. 1994;91(12):5372–5376. doi: 10.1073/pnas.91.12.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hilioti Z, Cunningham KW. The RCN family of calcineurin regulators. Biochem Biophys Res Commun. 2003;311(4):1089–1093. doi: 10.1016/s0006-291x(03)01515-8. [DOI] [PubMed] [Google Scholar]
- 31.Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13(4):136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- 32.Cao S, Gelwix CC, Caldwell KA, Caldwell GA. Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci. 2005;25(15):3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrington AJ, Yacoubian TA, Slone SR, Caldwell KA, Caldwell GA. Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson’s disease. J Neurosci. 2012;32(6):2142–2153. doi: 10.1523/JNEUROSCI.2606-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JI, et al. The Caenorhabditis elegans homologue of Down syndrome critical region 1, RCN-1, inhibits multiple functions of the phosphatase calcineurin. J Mol Biol. 2003;328(1):147–156. doi: 10.1016/s0022-2836(03)00237-7. [DOI] [PubMed] [Google Scholar]
- 35.Piña FJ, et al. Hph1 and Hph2 are novel components of the Sec63/Sec62 posttranslational translocation complex that aid in vacuolar proton ATPase biogenesis. Eukaryot Cell. 2011;10(1):63–71. doi: 10.1128/EC.00241-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qureshi HY, Paudel HK. Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and alpha-synuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J Biol Chem. 2011;286(7):5055–5068. doi: 10.1074/jbc.M110.178905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fadri M, Daquinag A, Wang S, Xue T, Kunz J. The pleckstrin homology domain proteins Slm1 and Slm2 are required for actin cytoskeleton organization in yeast and bind phosphatidylinositol-4,5-bisphosphate and TORC2. Mol Biol Cell. 2005;16(4):1883–1900. doi: 10.1091/mbc.E04-07-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.deHart AK, Schnell JD, Allen DA, Tsai JY, Hicke L. Receptor internalization in yeast requires the Tor2-Rho1 signaling pathway. Mol Biol Cell. 2003;14(11):4676–4684. doi: 10.1091/mbc.E03-05-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cyert MS. Calcineurin signaling in Saccharomyces cerevisiae: How yeast go crazy in response to stress. Biochem Biophys Res Commun. 2003;311(4):1143–1150. doi: 10.1016/s0006-291x(03)01552-3. [DOI] [PubMed] [Google Scholar]
- 40.Picotti P, et al. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat Methods. 2010;7(1):43–46. doi: 10.1038/nmeth.1408. [DOI] [PubMed] [Google Scholar]
- 41.Roy J, Li H, Hogan PG, Cyert MS. A conserved docking site modulates substrate affinity for calcineurin, signaling output, and in vivo function. Mol Cell. 2007;25(6):889–901. doi: 10.1016/j.molcel.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Q, et al. Selective ATP-competitive inhibitors of TOR suppress rapamycin-insensitive function of TORC2 in Saccharomyces cerevisiae. ACS Chem Biol. 2012;7(6):982–987. doi: 10.1021/cb300058v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aramburu J, et al. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285(5436):2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- 44.Stathopoulos AM, Cyert MS. Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 1997;11(24):3432–3444. doi: 10.1101/gad.11.24.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doty RL. Olfactory dysfunction in Parkinson disease. Nat Rev Neurol. 2012;8(6):329–339. doi: 10.1038/nrneurol.2012.80. [DOI] [PubMed] [Google Scholar]
- 46.Lin X, et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64(6):807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Armstrong RA, et al. A quantitative study of α-synuclein pathology in fifteen cases of dementia associated with Parkinson disease. J Neural Transm. 2014;121(2):171–181. doi: 10.1007/s00702-013-1084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chartier-Harlin MC, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364(9440):1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 49.Butcher SP, Henshall DC, Teramura Y, Iwasaki K, Sharkey J. Neuroprotective actions of FK506 in experimental stroke: In vivo evidence against an antiexcitotoxic mechanism. J Neurosci. 1997;17(18):6939–6946. doi: 10.1523/JNEUROSCI.17-18-06939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kitamura Y, Itano Y, Kubo T, Nomura Y. Suppressive effect of FK-506, a novel immunosuppressant, against MPTP-induced dopamine depletion in the striatum of young C57BL/6 mice. J Neuroimmunol. 1994;50(2):221–224. doi: 10.1016/0165-5728(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 51.Costantini LC, et al. A novel immunophilin ligand: Distinct branching effects on dopaminergic neurons in culture and neurotrophic actions after oral administration in an animal model of Parkinson’s disease. Neurobiol Dis. 1998;5(2):97–106. doi: 10.1006/nbdi.1998.0185. [DOI] [PubMed] [Google Scholar]
- 52.Guo X, Dawson VL, Dawson TM. Neuroimmunophilin ligands exert neuroregeneration and neuroprotection in midbrain dopaminergic neurons. Eur J Neurosci. 2001;13(9):1683–1693. doi: 10.1046/j.0953-816x.2001.01542.x. [DOI] [PubMed] [Google Scholar]
- 53.Guo X, Dillman JF, 3rd, Dawson VL, Dawson TM. Neuroimmunophilins: Novel neuroprotective and neuroregenerative targets. Ann Neurol. 2001;50(1):6–16. doi: 10.1002/ana.1030. [DOI] [PubMed] [Google Scholar]
- 54.Armistead DM, et al. Design, synthesis and structure of non-macrocyclic inhibitors of FKBP12, the major binding protein for the immunosuppressant FK506. Acta Crystallogr D Biol Crystallogr. 1995;51(Pt 4):522–528. doi: 10.1107/S0907444994014502. [DOI] [PubMed] [Google Scholar]
- 55.Steiner JP, et al. Neurotrophic actions of nonimmunosuppressive analogues of immunosuppressive drugs FK506, rapamycin and cyclosporin A. Nat Med. 1997;3(4):421–428. doi: 10.1038/nm0497-421. [DOI] [PubMed] [Google Scholar]
- 56.Meissner W, Hill MP, Tison F, Gross CE, Bezard E. Neuroprotective strategies for Parkinson’s disease: Conceptual limits of animal models and clinical trials. Trends Pharmacol Sci. 2004;25(5):249–253. doi: 10.1016/j.tips.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 57.Tadross MR, Ben Johny M, Yue DT. Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels. J Gen Physiol. 2010;135(3):197–215. doi: 10.1085/jgp.200910308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dick IE, et al. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451(7180):830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bean BP. Neurophysiology: Stressful pacemaking. Nature. 2007;447(7148):1059–1060. doi: 10.1038/4471059a. [DOI] [PubMed] [Google Scholar]
- 60.Martinez J, Moeller I, Erdjument-Bromage H, Tempst P, Lauring B. Parkinson’s disease-associated alpha-synuclein is a calmodulin substrate. J Biol Chem. 2003;278(19):17379–17387. doi: 10.1074/jbc.M209020200. [DOI] [PubMed] [Google Scholar]
- 61.Lee D, Lee SY, Lee EN, Chang CS, Paik SR. alpha-Synuclein exhibits competitive interaction between calmodulin and synthetic membranes. J Neurochem. 2002;82(5):1007–1017. doi: 10.1046/j.1471-4159.2002.01024.x. [DOI] [PubMed] [Google Scholar]
- 62.Mosharov EV, et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62(2):218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thayanidhi N, et al. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell. 2010;21(11):1850–1863. doi: 10.1091/mbc.E09-09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nemani VM, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65(1):66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gitler AD, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41(3):308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bodenmiller B, et al. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci Signal. 2010;3(153):rs4. doi: 10.1126/scisignal.2001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.