

Abstract
New reliable and cost-effective antimalarial drug screening assays are urgently needed to identify drugs acting on different stages of the parasite Plasmodium falciparum, and particularly those responsible for human-to-mosquito transmission, that is, the P. falciparum gametocytes. Low Z′ factors, narrow dynamic ranges, and/or extended assay times are commonly reported in current gametocyte assays measuring gametocyte-expressed fluorescent or luciferase reporters, endogenous ATP levels, activity of gametocyte enzymes, or redox-dependent dye fluorescence. We hereby report on a dual-luciferase gametocyte assay with immature and mature P. falciparum gametocyte stages expressing red and green-emitting luciferases from Pyrophorus plagiophthalamus under the control of the parasite sexual stage-specific pfs16 gene promoter. The assay was validated with reference antimalarial drugs and allowed to quantitatively and simultaneously measure stage-specific drug effects on parasites at different developmental stages. The optimized assay, requiring only 48 h incubation with drugs and using a cost-effective luminogenic substrate, significantly reduces assay cost and time in comparison to state-of-the-art analogous assays. The assay had a Z′ factor of 0.71 ± 0.03, and it is suitable for implementation in 96- and 384-well microplate formats. Moreover, the use of a nonlysing d-luciferin substrate significantly improved the reliability of the assay and allowed one to perform, for the first time, P. falciparum bioluminescence imaging at single-cell level.
Malaria still represents the deadliest parasitic infection afflicting humans worldwide, with Plasmodium falciparum causing the most severe form of the disease. In the goal to globally eliminate malaria, it is increasingly recognized that antiparasite interventions need to target not only the pathogenic asexual forms of the parasite but also the Plasmodium developmental stages responsible for transmission between the human and the Anopheles hosts. Such reinvigorated efforts include the challenge of revising high throughput (HTS) drug screening approaches, currently tailored against the Plasmodium replicative asexual blood stages, to identify compounds active against multiple stages of the malarial parasite life cycle. This is particularly important at a time where no safe drug is available against P. falciparum transmission stages and reports have established the emergence of parasite resistance to the frontline artemisinin drugs.
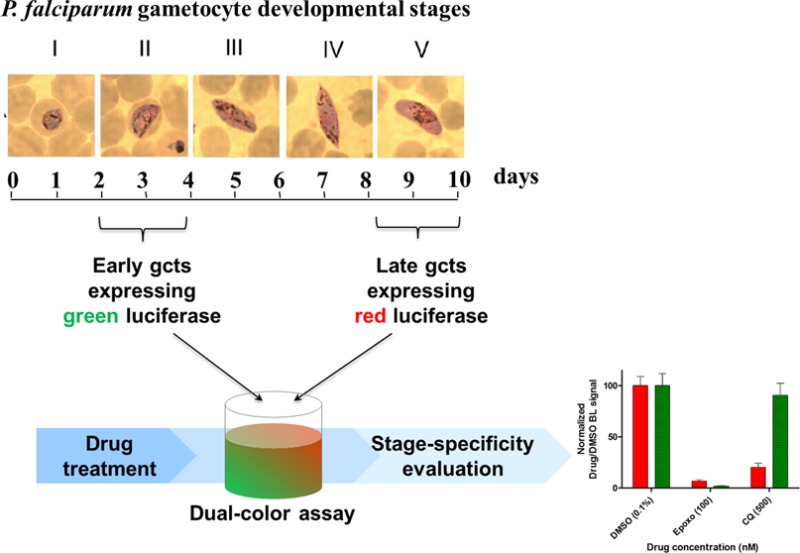
P. falciparum gametocytes are the parasite sexual stages responsible for the human-to-mosquito transmission. These are formed in the human bloodstream where they undergo a 10-day multistage development showing remarkable morphological1 and physiological2 differences that distinguish early and mature stages. The nonreplicative nature of these sexual blood stages and their long maturation time constrained so far the ability of the recently developed gametocyte assays to sensitively and reliably monitor compound effects on gametocyte viability, mainly because activity of fluorescent reporters or endogenous parasite enzymes tends to persist in the unhealthy drug treated gametocytes.3 Current cell-based reporter assays against these parasite stages generally show suboptimal robustness, require long assay time and expensive reagents, and do not provide information about stage-specificity of target drugs.
Bioluminescence (BL) is the emission of light in living organisms in which an enzyme, generally called luciferase, catalyzes the oxidation of a specific substrate, luciferin, with a release of photons in the visible spectrum. Luciferase genes cloned from different organisms are used in several bioanalytical applications thanks to peculiar characteristic of BL reactions such as high quantum yield, high signal-to-noise ratio, and the possibility to multiplex assays using luciferases emitting at different wavelengths. As luciferases do not require post-translational modifications for activity and are not toxic to cells even at high concentrations, these enzymes have been successfully exploited as reporters in a variety of ultrasensitive cell-based assays.4,5 The possibility to tune luciferase emission properties such as emission wavelength and kinetics or thermo- and pH-stability6 via random or site-directed mutagenesis opens the avenue to significantly improve P. falciparum HTS luciferase assays, so far restricted to the use of the Photinus pyralis wild-type enzyme.7−11 Improvements can be achieved by combining luciferases emitting at different wavelength under the control of different regulatory sequences to monitor multiple molecular targets or signaling events, resulting in increased information from the same cell/well and reduced assay cost/time. Also, the introduction of a second, constitutively expressed luciferase in the same cell can provide an internal viability control to correct the analytical signal, improving assay reliability and robustness. Moreover, the possibility to monitor in real-time the BL signal emitted by a single parasite by non invasive BL imaging offers tremendous potential for clarifying mechanisms of action of drugs.
In this work, the combination of multicolor bioluminescence and use of an optimized luminogenic substrate are reported for the first time in P. falciparum to quantitatively and simultaneously assess the viability of parasites at different developmental stages in a format scalable to HTS and to introduce single-cell imaging methodology in the study of this parasite.
Experimental Section
Parasite Cultures and Transfection
The P. falciparum 3D7A line12 was cultured in human 0+ erythrocytes, kindly provided by Prof. G. Girelli, Dipartimento di Biopatologia Umana, University of Rome ‘‘La Sapienza”, at 5% hematocrit under 5% CO2, 2% O2, 93% N2.13 Cultures were grown in medium containing RPMI 1640 medium (Gibco) supplemented with 25 mM Hepes, 50 μg/mL hypoxanthine, 0.25 mM NaHCO3, 50 μg/mL gentamicin sulfate, and 10% pooled heat-inactivated 0+ human serum. Ring stage parasites at 3–5% parasitaemia were transfected by electroporation with 80–100 μg of transfection vectors using the following conditions: voltage, 0.31 kV; capacitance, 960 μF; resistance to infinity.14 Following transfection, parasites were maintained in drug free medium for 24 h; at this time, positive selection was initiated by the addition of 1.2 μg/mL of blasticidin S to select the parasites stably maintaining the episomal constructs.
Production of transgenic lines with stably integrated luciferase cassettes in the pfelo1 locus was attempted equipping the luciferase cassettes with pfelo1 homology regions for Zinc Finger Nuclease (ZFN)-mediated genome editing.15 The episome-containing transgenic parasites were transfected with ZFN expression plasmid and double selection started after 24 h by adding 1.2 μg/mL of BSD and 2.5 nM WR99210. After 7 days of selection, parasites were allowed to recover in the absence of any drug. Southern blot analysis of the resulting parasites with pfelo1- and bsd-specific probes on parental parasites and on parasites containing the episomal plasmids before and after transfection of the ZFN plasmids revealed, however, that successful disruption of the pfelo1 locus was mediated by integration of the entire plasmid via homologous recombination through the pfelo1 3′ homology region.
Plasmid Construction
Plasmids containing the myc-tagged luciferase expression cassettes, equipped with Bbx1 attB sites under the expression control of the pfs16 regulatory regions into pCR2.1 vector (Life Technologies), were obtained through a multistep strategy described in details in the Supporting Information experimental section. To generate constructs able to mediate the chromosomal integration of the above luciferase cassettes, each luciferase expression cassette was excised from its pCR2.1 vector as a HindIII–HindIII fragment and cloned into a pDC2-based vector previously modified as described in detail in the Supporting Information experimental section. The resulting expression plasmids contained the blasticidine-S-deaminase (BSD) for selection of transgenic parasites. ZFNs (Sangamo) were engineered for expression in P. falciparum as described in the Supporting Information experimental section.
Plasmid Copy Number Determination by SYBR-Green Real-Time PCR
The procedure to determine luciferase cassette plasmid copy number in the transgenic parasite lines was based on amplification of sequences of the bsd selectable marker contained in the episomal plasmids and of the chromosomal parasite gene pfeba175 as a control for genome numbers. Details of the methodology are described in the Supporting Information experimental section.
Southern Blot Analysis
Procedures for digestion, blotting, and hybridization of parasite genomic DNA with specific pfelo1 and bsd probes are described in detail in the Supporting Information experimental section.
P. falciparum Gametocyte Drug Treatments
Drug assays were performed on gametocytes at different stages of maturation. For early (stage II) gametocytes, induced cultures were treated 48 h with 50 mM N-acetyl-glucosamine (NAG) to eliminate asexual stages before drug treatment. Late (IV–V) stage gametocytes had been NAG-treated 96 h at the onset of gametocytogenesis and then allowed to mature. Drug treatments were performed in 100 μL final volume in 96-well culture plates at a final hematocrit of 1%. In these cultures, gametocytemias were routinely ranging from 1% to 2.5% for episomal transgenic parasites and from 2% to 3.5% for 3D7elo1-pfs16-CBG99. Drugs were dissolved in dimethyl sulfoxide (DMSO), except for chloroquine, which is soluble in water. Control samples were treated with DMSO at the highest concentration present in treated samples, which never exceeded 0.1%.
Immunofluorescence Analysis
Mixed stage gametocyte smears were fixed with acetone for 5 min at room temperature, blocked with PBS/3% BSA for 30 min, and incubated with a rabbit anti-Pfg27 antibody16 (1:500 in 1xPBS 2% BSA) to label all gametocytes and with a mouse antimyc antibody (ab32, Abcam) (1:200 in 1xPBS 2% BSA). After incubation and washes in 1XPBS, slides were incubated with a 1:200 dilution of affinity purified, rhodamine-conjugated, and FITC-conjugate secondary antibody against rabbit and mouse IgG, respectively.
Luciferase Assays
Comparison of luciferase activities from the six episome-expressing transgenic parasites was performed on stage III gametocytes after Percoll purification.17 Frozen aliquots of equal numbers of gametocytes were resuspended in ice with 100 μL of PBS just prior to luminometric measurements, transferred to 96-well white plates, and 100 μL of Britelite plus Reporter Gene Assay System (PerkinElmer) was added. Total light outputs were recorded using a Microplate Scintillation and Luminescence CounterTopCount NXT (PerkinElmer) over a 20-min period in 3-s intervals. Equivalent samples were also read using a Varioskan Flash multimode reader (Thermo Fisher). Luminescence measurements were expressed as signal-to-noise ratio with respect to untransfected 3D7 control parasite samples.
Commercial substrates Britelite, Neolite, and Steadylite were from PerkinElmer; One-Glo, Steady-Glo, and Bright-Glo were from Promega. Frozen pellets of gametocyte expressing CBG99 luciferase were used to compare bioluminescence emission kinetics. Briefly, 100 μL of the same aliquot of resuspended gametocytes was transferred to a 96-well white microtiter plate, and 100 μL of each substrate was added simultaneously with a multichannel pipette. Bioluminescence emissions were acquired for 45 min with 300 ms integration time using a Varioskan reader. All samples were tested in triplicate at least three times. Luciferase assays after drug-treatment experiments were performed after transferring samples to 96-well white microplates. Different d-luciferin concentrations and buffer compositions were tested, and the optimal substrate was 0.5 mM d-luciferin (final concentration) in citrate buffer 0.1 M, pH 5.5. Substrate (1 mM d-luciferin) was added directly to the samples at a 1:1 ratio, and plates were read within 2 min after addition. Luminescence measurements were performed as described above.
Bioluminescence Single-Cell Imaging
Single-cell bioluminescence imaging was performed on control (DMSO 0.1%) or drug treated gametocytes (epoxomicin 100 nM, chloroquine 500 nM). After 48 h of incubation, gametocytes (100 μL at 0.1% HCT) were transferred to a clear bottom 96-well microplate (ibidi GmbH) coated with Cell-Tak cell and tissue adhesive (BD Biosciences) according to the manufacturer’s instructions and allowed to adhere for 30 min at 37 °C. BL imaging was performed using an inverted microscope (Olympus CK40) connected to an electron multiplying charge coupled device (EM-CCD) camera (ImagEM-X2, Hamamatsu). BL images were acquired for 10 min with 40× objective (UApo, Olympus) after addition of 100 μL of d-luciferin 1 mM. The setup was enclosed in a custom-built dark box to shield from ambient light. Images were processed with HCImage software (v4.1.5.12, Hamamatsu) applying a cosmic ray removal option (threshold 10 000) and brightness/contrast adjusted with normal linear function.
Dual-Luciferase Gametocyte Assays
The dual-reporter assays have been performed using CBG99 and CBR expressing parasites (1% hematocrit), at stage II and stage V of development, mixed in a 1:10 ratio, respectively, to compensate for the different BL signals from the two enzymes. Gametocyte mixed populations were seeded in a 96-well plate and treated for 48 h with 500 nM chloroquine (CQ), 100 nM epoxomicin (Epoxo), or DMSO 0.1% (control). By adding BL, substrate emissions kinetics were acquired for 15 min (300 ms integration time) with Varioskan Flash using both F545 (510–580 nm) and F615 (590–640 nm) high transmittance band-pass emission filters. Raw BL intensities taken from 5 to 10 min were elaborated with Chroma-Luc Calculator22 to unmix BL emission (corrected BL) of the green- and red-emitting gametocytes. The mean value of each corrected BL kinetic is plotted and normalized with respect to DMSO control.
Results and Discussion
Expression of Green- and Red-Emitting Luciferases in P. falciparum
A panel of six ATP-dependent luciferases derived from different bioluminescent species or obtained by rational mutagenesis were selected according to their enzymatic properties (Table 1) and expressed for the first time in the malaria parasite. The repertoire of luciferases used so far as reporters in malaria parasites is to our knowledge restricted to the Renilla and the P. pyralis enzymes, with only the latter luciferase being used in HTS and live imaging applications. Selected luciferases were the green wild-type Photinus pyralis enzyme (PpyWT18), a red-shifted emission variant (PpyRE1019), a mutant with green-shifted emission and increased thermal stability (PpyGRTS18), a red-emitting variant of the firefly Luciola italica (LitRE620), and the green- and red-emitting luciferases from the click beetle Pyrophorus plagiophthalamus (CBG99 and CBR21).
Table 1. Properties of Luciferases Selected for Expression in Plasmodium falciparum.
| reporter gene (organism) | in vitro BL emissiona (λmax, nm) | half-life (37 °C, h) | pH-dependent emission | half bandwidth (nm)b |
|---|---|---|---|---|
| PpyWT (Photinus pyralis) | 557 | 0.26 | yes | 66 |
| PpyGRTS (Photinus pyralis) | 548 | 10.5 | no | 62 |
| PpyRE10 (Photinus pyralis) | 617 | 3.6 | no | 42 |
| LitRE6 (Luciola italica) | 610 | 9.6 | no | 70 |
| CBG99 (Pyrophorus plagiophthalamus) | 537 | >5 | no | 65 |
| CBR (Pyrophorus plagiophthalamus) | 613 | >5 | no | 62 |
Bioluminescence emission spectra measured in Hek293 cell lines.
Bandwidths (nm) of emission spectra were measured at 50% of the intensity at the maximum wavelength.
P. falciparum parasites were transfected with plasmid constructs in which the promoter and the 3′ untranslated region (UTR) of the gametocyte specific gene pfs16 (Figure S1, Supporting Information) were used to specifically drive cytoplasmic expression of the above luciferases from the onset of sexual differentiation (stage I) through the 10-day long maturation to stage V gametocytes.1 Six transgenic parasite lines were selected for blasticidin resistance, and real-time PCR experiments determined that plasmid copy number differed at most 2-fold between these lines (Figure S2, Supporting Information).
To identify the most active luciferases in view of the development of a dual-color assay, BL emissions were measured from an equal number of purified stage III gametocytes of the six transgenic lines. The comparison of the BL signals from the different luciferase-expressing gametocytes revealed in our hands that the green- and the red-emitting P. plagiophthalamus enzymes provide BL signals significantly higher than those of the other tested luciferases and display stable emission kinetics (Figure 1a). Although the emission spectra of these luciferases show a remarkable overlap, the distance between the two λmax of approximately 76 nm, combined with the use of appropriate emission filters and a spectral unmixing algorithm,22 are adequate to successfully establish a dual-luciferase gametocyte assay in which contribution of each luciferase can be sensitively and robustly determined. In addition, these reporters exhibit pH-independent emission and glow-type emission kinetics, making them the best candidates for the implementation of the dual-color assay.
Figure 1.
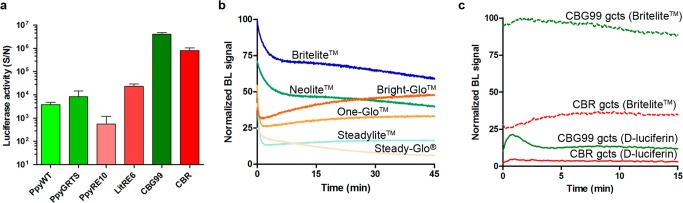
Luciferase selection and characterization: (a) BL emission intensities of the same amount (2.5 × 105) of purified stage III transgenic gametocytes expressing the luciferases indicated (with Britelite substrate). (b) Normalized emission kinetics obtained with CBG99 expressing gametocytes (4 × 104 gametocytes/well) using different commercial substrates. (c) Comparison of BL intensities and kinetic profiles obtained with transgenic gametocytes expressing CBG99 or CBR luciferase using Britelite or d-luciferin substrates. In (b) and (c), the highest BL signal is set as 100%.
Improvement in Luciferase Assay Performance on the Nonproliferating Parasite Sexual Stages
The need to identify novel drugs active against P. falciparum gametocytes recently prompted much work to establish cell-based assays against such parasite stages. To improve current assays, the effect of several BL commercial substrates on the assay analytical performance was evaluated. Emission kinetics of the gametocyte-expressed CBG99 luciferase in a 45 min window showed that the highest and most stable BL signal was obtained using Britelite (Figure 1b). However, as commercial substrates generally contain additives (luciferase inhibitors, lysing components, ATP) to enhance and stabilize the bioluminescent signal, a homemade d-luciferin solution was developed as an alternative substrate to better reflect the viability of the gametocytes in drug screening assays, avoiding artifactual BL emission in the treated cells. Gametocytes expressing the CBG99 or the CBR luciferase were incubated with Britelite or with a formulation of 0.5 mM d-luciferin dissolved in 0.1 M citrate buffer pH 5.5, optimized to enter cells in other eukaryotic and prokaryotic systems.23 Although the nonlysing d-luciferin substrate produced a lower BL signal than Britelite, a stable signal was obtained from 5 to 15 min after substrate addition, with only 5% variability over 10 min (Figure 1c). To directly compare the performance of Britelite and d-luciferin substrates in faithfully monitoring parasite viability exposed to drug treatment, synchronous unpurified cultures (1% hematocrit, 2% gametocytemia) of stage II and stage IV gametocytes expressing CBG99 or CBR were exposed for 48 h to a 100 nM concentration of the reference gametocytocidal drug epoxomicin.24 Results showed that the decline in luciferase activity in the drug-treated gametocytes was significantly more pronounced using the d-luciferin substrate than Britelite in both gametocyte stages for both parasite lines (Figure 2a,b). At 48 h, with d-luciferin, residual luciferase activity was virtually absent or <1% in early and late gametocytes, respectively, whereas with Britelite it was 37 ± 5% in early gametocytes and 57 ± 4% and 72 ± 6%, respectively, in the CBR- and CBG99-late stages. The robustness of this determination was indicated by a cumulative assay Z′ factor25 of 0.92 ± 0.09 and was confirmed by the comparable epoxomicin IC50 values on early and late gametocyte stages obtained with the commercial and the d-luciferin substrates (12.5 ± 0.4 and 14.3 ± 0.6 nM with d-luciferin and Britelite, respectively, on early and 10.7 ± 0.8 and 10.1 ± 0.5 nM with d-luciferin and Britelite, respectively, on late stages) (Figure 2c).
Figure 2.

Bioluminescence in control and drug treated P. falciparum gametocytes: (a) d-Luciferin and Britelite performances in drug treatment assay. Bioluminescence from early stage gametocytes expressing CBG99 (left) and CBR (right) measured with d-luciferin (●) and Britelite (▲) at t = 0, t = 24 h, and t = 48 h. (b) Same experiment on stage IV gametocytes. Results are expressed as drug/DMSO ratio. Statistics are performed with the GraphPad Prism software. (c) Comparison of d-luciferin and Britelite performances in dose–response gametocytocidal assays. Early (left) and late (right) stage gametocytes expressing the CBG99 luciferase were treated for 72 h with increasing doses of epoxomicin (0–90 nM) before luciferase activity was measured with 0.5 mM d-luciferin and Britelite. IC50 values were calculated using the GraphPad Prism software.
As compared to the assays traditionally used to identify compounds blocking the multiplication of the parasite asexual stages, gametocyte assays face the challenge to reliably measure the ability of compounds to inhibit development or metabolism of such nonproliferating parasite stages. Low Z′ factors, narrow dynamic ranges, and/or extended assay times are commonly reported in current gametocyte cell-based assays measuring gametocyte-expressed fluorescent or luciferase reporters,9,10,26,27 endogenous ATP levels,28 activity of gametocyte enzymes,3 or redox-dependent dye fluorescence.29,30 Such suboptimal performances are largely due to the persistence or slow decay of the above reporters or signals in the nondividing gametocytes, even when they are affected by the compound treatment.
The replacement of commonly used cell-lysing BL substrates with a formulation of nontoxic, nonlysing d-luciferin substrate solution resulted in assays where the BL signal relies both on the expression of the luciferase reporter and, importantly, on the use of the parasite endogenous ATP, thus reliably reflecting the viability of the treated and untreated gametocytes. This improvement results in a cost-effective and less time-consuming protocol able to measure the drop to background level in gametocyte viability produced by epoxomicin in only 48 h, whereas as long as 144 h is required in similar assays based on the gametocyte endogenous lactate dehydrogenase activity.3 Such an improvement was observed not only on the immature gametocytes but also on the late sexual stages, the ones freely circulating in the bloodstream ready for uptake by the mosquito bite, whose apparently quiescent metabolic state makes the identification of inhibitory compounds particularly challenging.
Single-Cell Bioluminescence Imaging in Live P. falciparum Parasites
The possibility to use a nonlysing d-luciferin substrate to produce a BL signal from whole living parasites was here exploited to introduce BL imaging at the single-cell level, unprecedented in malaria parasites, to visualize live gametocytes of different stages. To perform these experiments, a parasite line stably expressing the CBG99 luciferase from an integrated chromosomal locus was produced. A derivative of the P. falciparum 3D7 line was generated in which diagnostic Southern blot analysis (Figure S3, Supporting Information) confirmed the integration of the pfs16-CBG99 cassette in the P. falciparum locus encoding the fatty acid elongase-1, pfelo1, dispensable for gametocyte, mosquito, and liver stage development (Santha Kumar, T. R. and Fidock, D. A., unpublished observations). Immunofluorescence experiments on gametocytes of this line, named 3D7elo1-pfs16-CBG99, confirmed that the Myc-tagged luciferase reporter can be readily detected in >80% of the gametocytes (Figure S4, Supporting Information).
Stage IV gametocytes from the 3D7elo1-pfs16-CBG99 line, immobilized in 96-well plates, were incubated with 0.5 mM d-luciferin and imaged with an optical microscope connected to an EM-CCD camera. Individual bioluminescent live gametocytes could be readily imaged and clearly distinguished from uninfected red blood cells (Figure 3a), representing to our knowledge the first report of single-cell BL imaging in a protozoan species. Stage IV gametocytes were then treated for 48 h with 100 nM epoxomicin, with 500 nM chloroquine, which on such stages has a limited activity at high concentration, or with the DMSO vehicle. In this experiment, no bioluminescent cells were detectable in the epoxomicin treated wells, weak BL signals were seen from gametocytes treated with chloroquine, whereas strong signals were detected on the metabolically active control parasites (Figure 3b). The failure to detect BL signals from morphologically recognizable gametocytes after drug treatment clearly indicates that the BL imaging signal is diagnostic of the viability of such cells. These results further confirmed that the use of a nonlysing d-luciferin substrate greatly improves the reliability of measuring luciferase reporter activity in the assessment of parasite viability.
Figure 3.

(a) Single gametocyte bioluminescence imaging of 3D7elo1-pfs16-CBG99 DMSO-treated gametocytes (60× objective, 10 min acquisition). Magnification bar is 10 μm. (b) BL imaging of control (0.1% DMSO) and drug-treated stage IV gametocytes (500 nm chloroquine (CQ) and 100 nM epoxomicin) (40× objective, 10 min acquisition). Parts of the photographs showing representative gametocytes in the bright field and in the BL images are enlarged. Magnification bar is 10 μm.
Imaging approaches based on the detection of transgenic BL cells are widely used in several biological systems, and have been exploited also in protozoan unicellular parasites such as P. falciparum, P. berghei, Trypanosome cruzi, and Leishmania to detect parasite infections in whole animals. The availability of several luciferase variants whose emissions are optimized for detection from deep tissues is currently improving the sensitivity of such approaches to describe the patterns of sequestration in natural or engrafted mouse tissues by populations of parasites.31−34
Development and Validation of a Dual-Color Stage-Specific Luciferase Assay
To fully exploit the potential of multicolor bioluminescence, the selected green and red luciferases were combined to develop a dual-color assay to quantitatively measure stage-specific effects of drugs on gametocytes at different developmental stages. Only one report on the rodent P. berghei parasites describes a dual luciferase assay exploiting the fact that the two enzymes require different luminogenic substrates,35 whereas our approach is based on the use of the same BL substrate and on the ability to quantitatively distinguish the simultaneous emissions of two reporters at different wavelengths.
A sensitive and robust dual-color assay ideally requires two luciferases with comparable BL intensities and virtually non-overlapping emission spectra. Our test of different novel luciferases in P. falciparum was motivated by the fact that the significant red-shift emission, caused by slight pH and temperature changes, of the green PpyWT luciferase23 makes this reporter unsuitable for dual-color assays. Although the firefly red-emitting mutant PpyRE10, with the longest emission wavelength (λmax = 617 nm) and the narrowest emission spectrum (half bandwidth of 42 nm), would have represented the ideal red-emitting partner of the green-emitting CBG luciferase (λmax = 537 nm), the better performance of the CBR luciferase led us to develop the dual-color BL assay with the P. plagiophthalamus enzymes.
Experiments were preliminarily performed to achieve an efficient spectral unmixing of the CBG99 and the CBR luciferase BL emissions. Green F545 (510–580 nm) and red F615 (590–640 nm) high-transmittance bandpass emission filters were used for the simultaneous BL acquisition. The F545 filter allowed acquisition of about 65% of the CBG and only 5% of the CBR luciferase emission, whereas with the F615 filter about 70% of the red luciferase and 16% of the green one were detected (Figure S5, Supporting Information). Thanks to a spectral unmixing algorithm (Chroma-Luc calculator,)22 the overlap between BL emissions can be reliably calculated to achieve a quantitative determination of the specific BL contributions of the individual luciferases. Dual-color BL assays were then performed, with the above filter pair, on samples containing different proportions of CBG99- and CBR-expressing gametocytes. Results confirmed that the activities of the two luciferases within mixed gametocyte populations can be accurately quantified from the corrected green and red light emissions simultaneously recorded from the same well (Figure S6, Supporting Information).
The ability of the dual-reporter assay to measure stage-specific drug effects was validated treating for 48 h mixtures of stage II and of stage V gametocyte cultures with 500 nM chloroquine, virtually inactive on mature gametocytes,20 or 100 nM epoxomicin, killing all sexual stages.24
The dual assays calculated the specific emissions of the CBG99 and the CBR luciferases, which were respectively produced by early and by late gametocytes in one experiment (Figure 4a) and by late and by early stages in the reciprocal experiment (Figure 4b). Results were that neither luciferase showed any activity after epoxomicin treatment, confirming that this drug efficiently killed both gametocyte stages. By contrast, in chloroquine-treated parasites, only a minor decrease in activity was measured for either the red or the green luciferase when these were produced by the chloroquine-insensitive mature gametocytes, whereas a dramatic drop in activity of both luciferases was observed when these were expressed by the early gametocytes. The dual assay was also performed comparing the d-luciferin and the Britelite substrates, further supporting that assays using the nonlysing d-luciferin substrate formulation more faithfully reflect the differential stage-specific activity of these drugs and assess parasite viability (Figure 4c). Drugs killing all gametocyte stages or differentially active against immature and mature sexual stages validated the ability of the dual-color assay to quantitatively measure such stage-specific effects. The calculated assay Z′ factor of 0.71 ± 0.03 indicated an excellent robustness for scaling up to HTS formats. The use of a dual-color BL assay to monitor different parasite sexual stages provides a proof of principle that this approach can be used on other parasite stages, which has relevant implications for future strategies in the frame of malaria eradication.
Figure 4.

Dual-color gametocyte assay validation. (a) Dual-luciferase assay with early and mature gametocytes, respectively, expressing the CBG99 (green bar) and the CBR luciferase (red bar). BL intensities were acquired with a Varioskan luminometer using F545 and F615 optical filters. Raw BL measurements were spectrally unmixed with the Chroma-Luc calculator and normalized with respect to DMSO control. (b) Reciprocal dual-color assay with mature and early stage gametocytes expressing the CBG99 and the CBR luciferase, respectively. (c) Comparison of Britelite and d-luciferin performance in an independent dual-color assay using CBG99 mature stage and CBR early stage gametocytes.
Conclusion
In this work, we developed a robust, cost-effective, and reliable dual-color gametocyte assay that exploits, for the first time in malaria, the potentiality of multicolor bioluminescence. The assay provided superior analytical performance in comparison to previously reported assays with the potential of improving current drug screenings in terms of cost, time, and sensitivity. We envisage that the same approach could be easily applied to develop new screening assays for identifying antimalarial drugs targeting different parasite stages. Besides, this methodology can be used, for instance, to simultaneously compare expression of stage-specific gene products, measure distinct cellular pathways, or evaluate the activity of an inducible or treatment-responsive promoter as compared to a constitutive internal viability control, respectively driving the expression of the two luciferases in the same parasite.
Moreover, the single-cell BL imaging of the human malaria parasite P. falciparum opens the possibility to monitor in real-time individual luciferase-expressing parasites in their stage-specific functional interactions with host tissues and cells and to assess how distinct cell types affect viability of specific parasite stages in in vitro and in ex vivo settings.
The concomitant significant development of affordable plate readers with customizable technical modules dedicated to multiplexing and BL imaging will greatly facilitate the applicability of the approaches presented here both in the study of and in the fight against this deadly parasite.
Acknowledgments
This work was supported by Grants OPP1040394 and OPP1040399 from the Global Health Program of the Bill & Melinda Gates Foundation to P.A. and D.A.F., respectively, and Grant R01 AI085584 from the NIH to D.A.F. We gratefully acknowledge Drs. G. Girelli, Blood Center of University of Rome “La Sapienza”, for the gift of human erythrocytes; Mauro Andreotti, ISS, for help in qPCR; Zuleika Michelini, ISS, and Carolina Scagnolari, University of Rome “La Sapienza”, for help in luciferase assays; and Marcus C. S. Lee and Judith Straimer, Columbia University, for advice on plasmid construction.
Supporting Information Available
Production of the plasmids used in this work; determination of luciferase plasmid copy number in P. falciparum transgenic lines; molecular characterization of the P. falciparum line containing the chromosomally integrated pfs16CBG99 cassette; immunofluorescence analysis of 3D7elo1-pfs16-CBG99 gametocytes; bioluminescence spectra of the CBG99 and the CBR luciferases; spectral unmixing of CBG99- and CBR-luciferase expressing gametocytes; list and sequence of the oligonucleotide primers used in the work. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
○ Liverpool School of Tropical Medicine, Pembroke Place, Liverpool, L35QA, United Kingdom.
Author Contributions
L.C.: Imaging and dual assays design and experiments, data analysis, manuscript preparation. G.C.: Plasmid constructs, transgenic lines, luciferase drug assays, data analysis, manuscript preparation. E.M.: Dual luciferase assay design, manuscript preparation. G.S.: Plasmid constructs, transgenic lines, Southern blot analysis. M.M.C.: Luciferase characterization and imaging experiments. R.B.: qPCR experiments. T.R.S.K.: pfelo1 genotypic/phenotypic analysis. A.C.: Plasmid design, project supervision. B.R.B.: Luciferase characterization. D.A.F.: Transgenic line work supervision. A.R.: Dual luciferase assay design, project supervision. P.A.: Project design and supervision, manuscript preparation.
Author Contributions
◆ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hawking F.; Wilson M. E.; Gammage K. Trans. R. Soc. Trop. Med. Hyg. 1971, 65, 549–559. [DOI] [PubMed] [Google Scholar]
- Silvestrini F.; Lasonder E.; Olivieri A.; Camarda G.; van Schaijk B.; Sanchez M.; Younis Younis S.; Sauerwein R.; Alano P. Mol. Cell. Proteomics 2010, 9, 1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro S.; Silvestrini F.; Dechering K.; Corbett Y.; Parapini S.; Timmerman M.; Galastri L.; Basilico N.; Sauerwein R.; Alano P.; Taramelli D. J. Antimicrob. Chemother. 2013, 68, 2048–2058. [DOI] [PubMed] [Google Scholar]
- Cevenini L.; Michelini E.; D’Elia M.; Guardigli M.; Roda A. Anal. Bioanal. Chem. 2013, 405, 1035–1045. [DOI] [PubMed] [Google Scholar]
- Ekström L.; Cevenini L.; Michelini E.; Schulze J.; Thörngren J. O.; Belanger A.; Guillemette C.; Garle M.; Roda A.; Rane A. Eur. J. Clin. Invest. 2013, 43, 248–255. [DOI] [PubMed] [Google Scholar]
- Michelini E.; Cevenini L.; Mezzanotte L.; Roda A. Methods Mol. Biol. 2009, 574, 1–13. [DOI] [PubMed] [Google Scholar]
- Che P.; Cui L.; Kutsch O.; Cui L.; Li Q. Assay Drug Dev. Technol. 2012, 10, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenkamp S.; Wong E. H.; Horrocks P. Malar. J. 2012, 10, 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adjalley S. H.; Johnston G. L.; Li T.; Eastman R. T.; Ekland E. H.; Eappen A. G.; Richman A.; Sim B. K.; Lee M. C.; Hoffman S. L.; Fidock D. A. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, E1214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucantoni L.; Duffy S.; Adjalley S. H.; Fidock D. A.; Avery V. M. Antimicrob. Agents Chemother. 2013, 57, 6050–6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T. Q.; Dehdashti S. J.; Nguyen D. T.; McKew J. C.; Zheng W.; Williamson K. C. Mol. Biochem. Parasitol. 2013, 188, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walliker D.; Quakyi I. A.; Wellems T. E.; McCutchan T. F.; Szarfman A.; London W. T.; Corcoran L. M.; Burkot T. R.; Carter R. Science 1987, 236, 1661–1666. [DOI] [PubMed] [Google Scholar]
- Trager W.; Jensen J. B. Science 1976, 193, 673–675. [DOI] [PubMed] [Google Scholar]
- Fidock D. A.; Wellems T. E. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 10931–10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straimer J.; Lee M. C.; Lee A. H.; Zeitler B.; Williams A. E.; Pearl J. R.; Zhang L.; Rebar E. J.; Gregory P. D.; Llinás M.; Urnov F. D.; Fidock D. A. Nat. Methods 2012, 9, 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri A.; Camarda G.; Bertuccini L.; van de Vegte-Bolmer M.; Luty A. J.; Sauerwein R.; Alano P. Mol. Microbiol. 2009, 73, 180–193. [DOI] [PubMed] [Google Scholar]
- Carter R.; Graves P. M.; Creasey A.; Byrne K.; Read D.; Alano P.; Fenton B. Exp. Parasitol. 1989, 69, 140–149. [DOI] [PubMed] [Google Scholar]
- Branchini B. R.; Ablamsky D. M.; Murtiashaw M. H.; Uzasci L.; Fraga H.; Southworth T. L. Anal. Biochem. 2007, 361, 253–262. [DOI] [PubMed] [Google Scholar]
- Branchini B. R.; Ablamsky D. M.; Rosenberg J. C. Bioconjugate Chem. 2010, 21, 2023–2030. [DOI] [PubMed] [Google Scholar]
- Maguire C. A.; van der Mijn J. C.; Degeling M. H.; Morse D.; Tannous B. A. Mol. Imaging 2012, 11, 13–21. [PubMed] [Google Scholar]
- Wood K. V.; Lam Y. A.; McElroy W. D. J. Biolumin. Chemilumin. 1989, 1, 289–301. [DOI] [PubMed] [Google Scholar]
- Almond B.; Hawkins E.; Stecha P.; Garvin D.; Paguio A.; Butler B.; Beck M.; Wood M.; Wood K. Promega Notes 2003, 85, 11–14. [Google Scholar]
- Michelini E.; Cevenini L.; Mezzanotte L.; Ablamsky D.; Southworth T.; Branchini B. R.; Roda A. Photochem. Photobiol. Sci. 2008, 2, 212–217. [DOI] [PubMed] [Google Scholar]
- Czesny B.; Goshu S.; Cook J. L.; Williamson K. C. Antimicrob. Agents Chemother. 2009, 53, 4080–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. H.; Chung T. D.; Oldenburg K. R. J. Biomol. Screening 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- Buchholz K.; Burke T. A.; Williamson K. C.; Wiegand R. C.; Wirth D. F.; Marti M. J. Infect. Dis. 2011, 203, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Liu M.; Liang X.; Siriwat S.; Li X.; Chen X.; Parker D. M.; Miao J.; Cui L. PLoS One 2014, 9, e93825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelièvre J.; Almela M. J.; Lozano S.; Miguel C.; Franco V.; Leroy D.; Herreros E. PLoS One 2012, 7, e35019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T. Q.; Williamson K. C. Mol. Biochem. Parasitol. 2011, 177, 160–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S.; Avery V. M. Malar. J. 2013, 12, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claser C.; Malleret B.; Peng K.; Bakocevic N.; Gun S. Y.; Russell B.; Ng L. G.; Rénia L. Parasitol. Int. 2014, 63, 187–194. [DOI] [PubMed] [Google Scholar]
- Vaughan A. M.; Mikolajczak S. A.; Wilson E. M.; Grompe M.; Kaushansky A.; Camargo N.; Bial J.; Ploss A.; Kappe S. H. J. Clin. Invest. 2012, 122, 3618–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis M. D.; Fortes Francisco A.; Taylor M. C.; Burrell-Saward H.; McLatchie A. P.; Miles M. A.; Kelly J. M. Cell. Microbiol. 2014, 10.1111/cmi.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M. C.; Kelly J. M. Trends Parasitol. 2014, 30, 161–162. [DOI] [PubMed] [Google Scholar]
- Helm S.; Lehmann C.; Nagel A.; Stanway R. R.; Horstmann S.; Llinas M.; Heussler V. T. PLoS One 2010, 5, e13653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.