

Summary
Ataxia-telangiectasia (A-T) is a rare autosomal recessive disorder caused by mutations in the ATM gene, resulting in faulty repair of breakages in double-stranded DNA. The clinical phenotype is complex, and is characterized by neurologic abnormalities, immunodeficiencies, susceptibility to malignancies, recurrent sinopulmonary infections, and cutaneous abnormalities. Lung disease is common in patients with A-T and often progresses with age and neurological decline. Diseases of the respiratory system cause significant morbidity and are a frequent cause of death in the A-T population. Lung disease in this population is thought to exhibit features of one or more of the following phenotypes: recurrent sinopulmonary infections with bronchiectasis, interstitial lung disease, and lung disease associated with neurological abnormalities. Here, we review available evidence and present expert opinion on the diagnosis, evaluation, and management of lung disease in A-T, as discussed in a recent multidisciplinary workshop. Although more data are emerging on this unique population, many recommendations are made based on similarities to other more well-studied diseases. Gaps in current knowledge and areas for future research in the field of pulmonary disease in A-T are also outlined.
Background
Ataxia-telangiectasia (A-T) is an autosomal recessive disorder caused by mutations in the gene ATM, which encodes a serine/threonine kinase responsible for the cellular response to double stranded DNA breaks (4; 53). The clinical features of A-T include neurologic abnormalities (cerebellar ataxia, dysarthria, drooling, ocular apraxia and choreoathetosis), immunodeficiency (antibody deficiency and lymphopenia), recurrent sinopulmonary infections, susceptibility to malignancies (especially leukemia and lymphoma), and cutaneous abnormalities (telangiectasia and progeric changes in the hair and skin). A-T is a heterogeneous disorder, however, and not all people with A-T exhibit the same constellation of features. In addition, whereas some features are progressive (ataxia, oculomotor apraxia), others are often static (immunodeficiency) or variable in their appearance (cancer) (22; 49). Estimates place the incidence of A-T between 1 to 2.5 per 100,000 (63). Lung disease is common in patients with A-T and often progresses with age and neurological decline. (11). Diseases of the respiratory system cause significant morbidity and are a frequent factor or cause of death in the A-T population (11; 13; 24).
A multidisciplinary workshop on “Pulmonary Disease in Ataxia-Telangiectasia” was held in Baltimore, MD on April 23-24, 2009 to review available data, discuss diagnostic and management strategies and propose topics for future research. The discussions generated at this meeting provided the impetus for this paper. This document is not intended as a set of evidence-based guidelines or consensus panel recommendations, given that the data are simply too sparse to attempt such statements. Rather, this document should be considered a reflection of current approaches to A-T care. These approaches generally reflect best available evidence in A-T, or when data are unavailable, are drawn from approaches used in other more common disorders that share features with A-T lung disease. Many topics for future investigations are suggested and discussed as well. Research to address the unanswered questions about lung disease in A-T will help establish more evidence-based care in the future.
Mechanisms and Features of Lung Disease in A-T
Pathophysiology of lung disease in A-T
The mechanisms that underlie the particular susceptibility of people with A-T to develop lung disease are thought to be related to a constellation of factors. In A-T, abnormal injury repair, premature aging, systemic inflammation and oxidative stress are believed to contribute to the pathophysiology of lung disease (61). Several of these mechanisms contribute to disease progression in pulmonary disorders such as emphysema and pulmonary fibrosis (2; 39), and are thus thought to play a role in A-T. Clinical factors that contribute to lung disease in A-T include susceptibility to infections, abnormal immune response, recurrent aspiration, and impaired clearance of respiratory tract secretions. Clarifying the impact of cellular and clinical factors on lung disease and its progression may help with discovery and development of therapeutic interventions. The identification of strategies that limit progression and development of pulmonary disease may also improve quality of life (QOL) and increase life expectancy until a cure for the disease becomes available.
Lung disease phenotypes in A-T
Three major types of lung disease are generally recognized in A-T patients: recurrent sinopulmonary infections and bronchiectasis, interstitial lung disease (ILD)/pulmonary fibrosis, and lung disease associated with neuromuscular deficits resulting in bulbar and spinal system impairment (11; 42; 57; 73). In A-T, the clinical picture is often a mixture of two or more of these phenotypes. Nonetheless, for the purposes of this discussion, we will focus on each as a separate entity. Because data are limited on lung disease in A-T, parallels are often drawn to other more well-characterized forms of chronic lung disease when formulating general diagnostic and management approaches (Table 1). Although the three types of lung disease discussed here are thought to be directly related to the pathophysiology of A-T, having A-T does not preclude children and adults from having other respiratory conditions, not necessarily related to their underlying diagnosis. Asthma is common in the general population and reversible small airways obstruction has been demonstrated in a cohort of A-T patients from Israel (6).
Table 1. Respiratory Disease Patterns in Ataxia-Telangiectasia.
Common symptoms, presentations and treatment considerations for lung disease in A-T
| Category | Common Symptoms/Presentations | Treatment Considerations |
|---|---|---|
| Sinopulmonary Disease and Bronchiectasis |
|
|
|
| ||
| Interstitial Lung Disease |
|
|
|
| ||
| Neuromuscular and Bulbar abnormalities |
|
|
Recurrent sinopulmonary disease and bronchiectasis
Summary of current knowledge
Recurrent sinopulmonary disease was one of three cardinal features noted in the first descriptions of A-T by Boder and Sedgwick (9; 10). Recurrent lower airway infections are common in A-T and can be associated with the development of bronchiectasis (52; 57) (Figure 1). In a population of A-T patients in Iran, 54% had a history of pneumonia while 64% had a history of recurrent upper respiratory infections (52). A chart review of 100 consecutive A-T patients in the United States revealed that 38% of those over 20 years of age had a history of recurrent lower respiratory tract infections (57). In a report from France, 7 of 15 A-T patients had bronchiectasis by imaging studies (11). Another study from Japan found that four A-T patients who died of respiratory failure with a tissue diagnosis of bronchiolitis obliterans had pulmonary infections with mycoplasma pneumonia, cytomegalovirus pneumonia and Pseudomonas aeruginosa (37).
Figure 1.
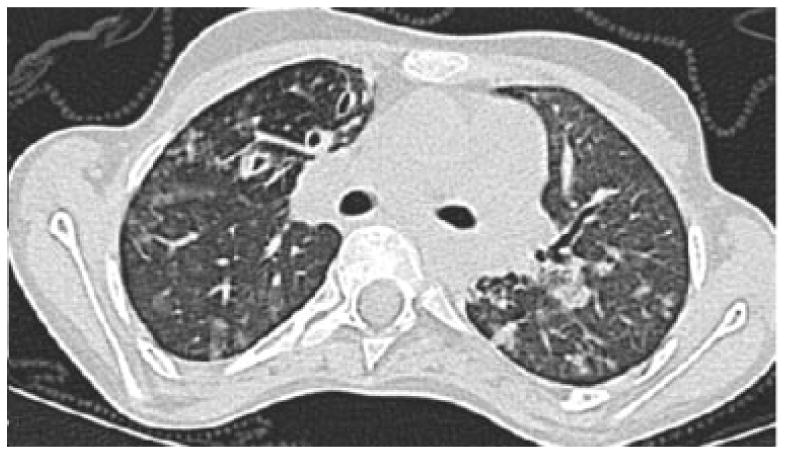
CT image of 17 year old female with A-T and history of recurrent pneumonias, dysphagia and bronchiectasis. This single CT image demonstrates right middle lobe bronchiectasis and bronchial wall thickening, peripheral tree in bud opacities and scattered areas of ground glass opacity.
Although sinopulmonary infections and bronchiectasis have been commonly reported in patients with A-T, little has been published regarding the organisms that cause these pulmonary infections. The incidence of chronic bacterial colonization in individuals with A-T is also unknown. Bacterial colonization with organisms including non-typeable Haemophilus influenza, Streptococcus pneumonia and Moraxhella caterrhalis have been reported in non A-T patients with bronchiectasis and persistent bacterial bronchitis (18) while colonization with Pseudomonas aeruginosa is commonly associated with bronchiectasis in cystic fibrosis (CF) and primary ciliary dyskinesia (PCD) (31; 56). Upper airway infections including chronic sinusitis are common in patients with A-T but as with other primary immune deficiency conditions, sinusitis is often refractory to conventional therapies (14).
Selective IgA deficiency, hypogammaglobulinemia, deficiency of antibody production particularly to polysaccharide antigens, IgG subclass deficiency, and T cell lymphopenia and dysfunction are present to varying degrees in people with A-T (13) and likely contribute to the increase in lower airway infections found in this population. In a study by Novak-Wegrzyn and colleagues, IgG4 deficiency was found in 65%, IgA deficiency in 63%, IgG2 deficiency in 48%, IgE deficiency in 23%, and IgG deficiency in 18% of patients with A-T (57). The majority of patients were also lymphopenic (71%), with leukopenia present in 37%. Interestingly, despite low CD4 T lymphocytic counts in many patients, infections with pneumocystis and severe varicella infections were found to be rare (57). In addition to immune defects, a chronically heightened inflammatory state, poor pulmonary clearance, and recurrent aspiration may also contribute to recurrent and chronic lung infections in A-T (42).
Current approaches based on best available evidence
Symptoms of lower respiratory tract infections include chronic or intermittent wet cough and chest congestion, though some people with A-T appear asymptomatic because of a diminished ability to cough. Since little has been published regarding pulmonary organisms in A-T, antibiotic treatment of upper and lower airway infections is often based on common organisms found in patients with other disorders associated with recurrent sinopulmonary disease and bronchiectasis. Identification of specific organisms from sputum, throat or bronchoalveolar lavage (BAL) should be pursued in guiding treatment of A-T patients with an inadequate or poor response to therapy. This approach mirrors paradigms used in CF, PCD and chronic obstructive pulmonary disease (COPD) (27; 30; 58). During acute and chronic infections, chest clearance techniques may help augment clearance of bronchial secretions.
Gaps in knowledge and future directions
Future research should address the identification of common organisms associated with sinopulmonary infections in A-T and the incidence of bacterial airway colonization in these patients. Studies should be done to determine if aggressive antimicrobial treatment can improve pulmonary outcomes. The role for long term prophylactic antibiotic should be examined in those patients with recurrent pulmonary exacerbations or bronchiectasis with and without proven bacterial colonization. The utility of inhaled antibiotics in A-T patients with chronic airway colonization is unclear and should also be studied. Although inhaled antibiotics are commonly used in CF patients with bacterial colonization (30), it is not known if the risk of antibiotic resistance outweighs their potential beneficial effect.
Given that elevated serum levels of IL-8 are relatively common in people with A-T (47), the role of macrolide antibiotics and anti-inflammatory agents, with regard to their anti IL-8 effects (5; 32; 44) should be examined in this population. Chronic administration of macrolides have also been shown to slow decline in lung function in patients with CF and diffuse panbronchiolitis and should be studied in A-T patients with chronic sinopulmonary disease and bronchiectasis (25; 30; 41).
ILD/Pulmonary Fibrosis
Summary of current knowledge
ILD has been described in individuals with A-T (73), although the exact incidence is unknown. A study in non-A-T adult patients with symptomatic IgG deficiency reported an ILD prevalence of 19.6% (66). In A-T patients, Schroeder and colleagues have published the most complete description of ILD to date. They retrospectively reviewed 437 charts of patients with A-T and found 25 patients diagnosed with ILD, with a mean onset of 17.5 years (73). Non-productive cough, dyspnea, and fever were the most common symptoms preceding the radiographic appearance of interstitial infiltrates. Radiographic findings in A-T patients with ILD were varied but generally included bilateral interstitial changes with interlobular opacities on chest radiograph and septal thickening on chest CT (73). Schroeder and colleagues presented data suggesting that steroids may enhance survival in A-T patients with ILD. Although observational and based on small numbers, these are the only data published on treatment of ILD in individuals with A-T.
The histological pattern described by Schroeder and colleagues in 13 A-T patients with ILD included lung fibrosis and chronic inflammation with lymphocytic or lymphohistiocytic cells, and all specimens demonstrated “atypical epithelial and interstitial cells with large hyperchromatic and pleiomorphic nuclei.” (73). This constellation of histological findings was believed to be unique to A-T since it did not fit any previous classification of ILD (Figure 2). Interestingly, nuclear changes such as these have been reported in other organs of patients with A-T (36).
Figure 2.
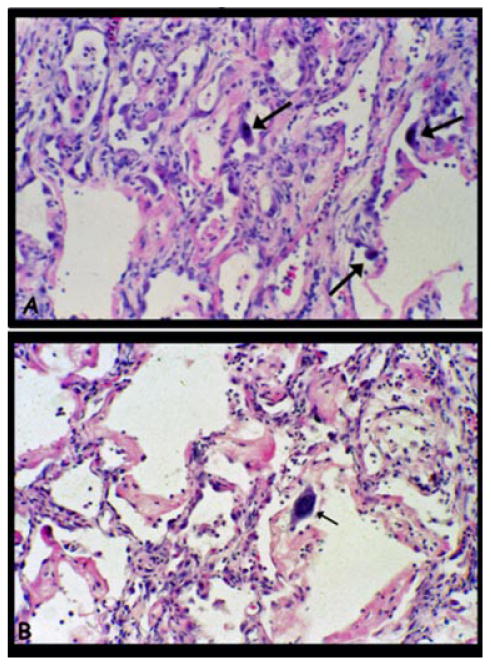
Lung biopsy of a 19 year old male with A-T and chronic pulmonary symptoms. A. Lung biopsy showed a diffuse interstitial process characteristic of early organizing diffuse alveolar damage with mild alveolar wall fibrosis and lobular remodeling, a mild interstitial infiltrate of predominantly lymphocytes, and patchy alveolar epithelial hyperplasia with scattered atypical cells having large hyperchromatic nuclei (arrows). B. In other regions there were more superimposed acute changes with hyaline membrane formation and mild mixed inflammation.
Individuals with A-T are at increased risk for the development of malignancies (49). Pulmonary parenchymal involvement secondary to lymphoma (15; 82) has been reported among children and young adults with A-T, and may mimic ILD clinically and radiographically. Chemotherapy for the treatment of a malignancy is also a known risk factor for the development of pulmonary fibrosis in people with A-T (20) (Figure 3). The risk of rapid pulmonary decline following the use of chemotherapeutic agents is well-described in A-T, and may be due to an underlying defect in injury repair (20; 70; 71). Of note, none of the patients in Schroeder's study were reported as having been exposed to chemotherapy (73), suggesting that their cases might represent a different type of ILD from that associated with cytotoxic medications.
Figure 3.
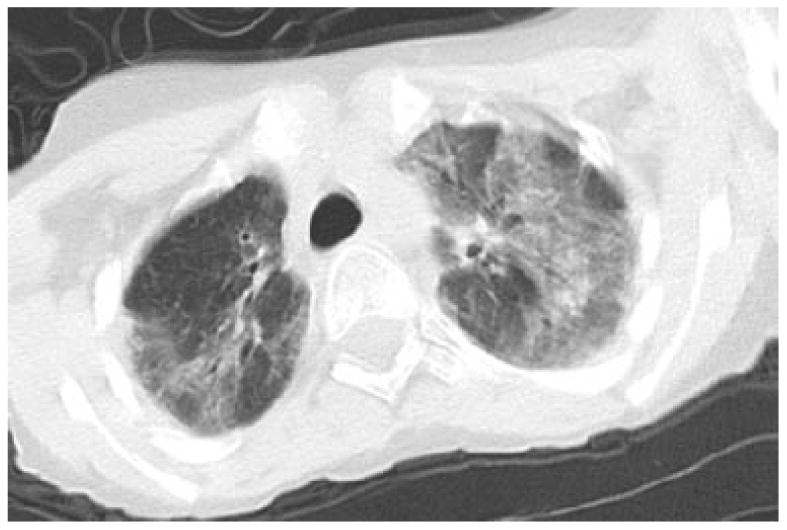
CT image of 14 year old boy with respiratory failure following chemotherapy for lymphoma. This CT image, through the upper lungs demonstrates mosaic attenuation with bilateral geographic areas of predominantly ground glass opacity. Several dilated bronchi are seen in the right upper lobe. Note that this image shows some blurring from motion, to decrease radiation exposure, images that are diagnostic but not ideal should not be repeated in people with A-T.
Current approaches based on best available evidence
ILD should be considered in patients with clinical symptoms of chronic cough, tachypnea, dyspnea on exertion, hypoxemia, and crackles, in the absence of detectable viral or bacterial infections. Restrictive lung disease has been reported in patients with A-T (11; 46), and may suggest the presence of an interstitial process. However, decreased forced vital capacity (FVC) by spirometry may be due to abnormalities in respiratory muscle coordination or scoliosis. Helium dilution measurements can help discriminate between true restriction and an inability to expire to residual volume (RV) (46). While a lung biopsy may be obtained to confirm the diagnosis of ILD in patients with A-T, the diagnostic benefits of such a procedure should be weighed against the risks associated with anesthesia and surgery. A trial of steroid therapy may be considered, although optimal time for initiation, dosing and length of therapy are not known. Pulmonary involvement of an oncologic process should be considered in patients with diffuse infiltrates and an ILD-like clinical picture.
Gaps in knowledge and future directions
Studies are needed to further characterize the unique forms and risk factors of ILD in A-T. Further assessment of steroid responsiveness and determination of optimal dosing regimens including strategies for tapering and cessation of therapy are needed. Hydroxychloroquine and immunomodulatory doses of gammaglobulin have been shown to be beneficial in non-A-T ILD (51; 62; 72), and warrant further evaluation in A-T associated ILD. Early identification of ILD is likely to improve outcomes. Serum levels of KL-6 and surfactant proteins A and D have been shown to be useful as biomarkers in other forms of ILD (1; 40; 59) and may help identify presymptomatic A-T patients who have or are at risk for developing ILD. In patients with malignancies, affecting between 10-30% of individuals with A-T (49; 76), further study is needed regarding the least pulmonary toxic regimens for treating these patients.
Lung disease associated with neuromuscular and bulbar abnormalities
Summary of current knowledge
Some aspects of lung disease seen in A-T may be attributable to progressive neurologic decline. Impairment in bulbar muscle function can lead to swallowing dysfunction and chronic aspiration, which can cause and/or exacerbate lung disease (42). In A-T, dysphagic problems commonly emerge in the second decade of life (23; 42). Abnormal respiratory muscle function from neurologic decline can lead to decreased tidal volumes and ineffective cough (46). From this standpoint, A-T patients may be approached in much the same way as those with other diseases causing neuromuscular impairment such as Duchene's Muscular Dystrophy (DMD) and Spinal Muscular Atrophy (SMA). Unlike individuals with these disorders, however, patients with A-T often become wheelchair-bound due to severe ataxia rather than primary muscle weakness. Lack of conditioning in patients with A-T who are wheelchair-bound may contribute to deterioration in lung function. Clinical indicators of lung disease due to impairment of upper airway and respiratory muscle systems may include chronic cough and prolonged or difficult recovery from respiratory illnesses. Cough or congestion with meals may suggest dysfunctional swallow and aspiration. Poor weight gain may be an indicator of excessive caloric expenditure due to increased work of breathing or difficulty taking in adequate calories.
Current approaches based on best available evidence
Measures to improve pulmonary clearance and prevent atelectasis may be beneficial in A-T patients, especially those with more advanced neurologic decline. Such interventions may include the use of chest physiotherapy, flutter valve therapy, and cough assist devices. As in other neuromuscular disease disorders, regimens may be increased during episodes of acute illness (28). Evaluation of maximum inspiratory and expiratory pressures (MIP and MEP, respectively) are well-established markers of respiratory muscle strength (68). Measurements of MIP and MEP in A-T patients should be considered in deciding when to initiate aggressive pulmonary toilet measures or non-invasive ventilation (38; 75). Dietary changes or gastrostomy tube (GT) placement should be considered in patients at risk for pulmonary aspiration due to bulbar dysfunction. In A-T patients with severe bulbar dysfunction and gastroesophageal reflux refractory to medical therapy, fundoplication may be indicated in addition to gastrostomy.
Gaps in knowledge and future directions
Future research should address techniques to assess lung-related neuromuscular weakness in A-T patients, as well as the optimal time to institute regular pulmonary clearance measures, and how these regimens should be designed. The use of devices to increase expiratory muscle strength and improve swallowing has been shown to be useful in Parkinson disease (64; 69) and should be studied in patients with A-T. Non-invasive ventilation (NIV) has been shown to increase long term survival in patients with DMD who develop chronic respiratory failure (28; 45). As chronic and acute respiratory failure have been reported in patients with A-T (37; 81), the use of NIV in such situations merits further investigations.
Evaluation of Lung Disease in A-T
Clinical Assessment
Summary of current knowledge
Lung disease is a major cause of morbidity and mortality in this population, and that risk increases with advancing age. Assessment of respiratory symptoms may be difficult in patients with A-T, so close attention to subtle signs and symptoms of pulmonary limitation is needed, since they may indicate early signs of respiratory deterioration. Due to progressive ataxia, many patients are wheelchair-bound by 12 years of age and physical activity is limited. Children and adults with A-T therefore, may not have a history of dyspnea on exertion, even in the face of significant pulmonary limitation. Disease severity during an acute respiratory illness may be underestimated due to a weak or ineffective cough.
Current approaches based on best available evidence
The natural course of pulmonary function decline is unknown in A-T. It is currently recommended that the pulmonary status of children with A-T should be evaluated by two years of age, and at least annually thereafter. Patients should undergo a complete pulmonary evaluation with emphasis on a history of dyspnea with or without exertion, prolonged respiratory symptoms after an acute respiratory illness and clinical symptoms of chronic cough or chest and nasal congestion. Because recurrent lower respiratory tract infections during childhood, even among otherwise healthy children, have been associated with a more rapid decline in FEV1 (79), timely identification and treatment of respiratory infections may help preserve lung function in children with A-T. As with any childhood lung disease, environmental exposure to cigarette smoke, air pollution, and other potential respiratory irritants should be avoided since these exposures can exacerbate respiratory symptoms and have been linked to decreased lung function in adulthood (16; 17).
Gaps in knowledge and future directions
Longitudinal studies of the nature and course of lung disease in A-T will be crucial for the development of more effective means of assessing and treating these patients. Standardized assessment tools and more formal management guidelines are needed to assure high quality, evidence-based care for patients with A-T.
Pulmonary Function Testing
Summary of current knowledge
Standard spirometry may be used in the evaluation of children and adolescents with A-T (46). The reproducibility of testing is increased by stabilizing the patient's head and holding the cheeks while the patient performs the forced expiratory maneuver (Figure 4). Inability to expire fully to RV may result in a decreased FVC and the appearance of restrictive lung disease. Static lung volumes can be useful to confirm the presence of restriction. However, as many patients are dependent on wheelchairs and may be unable to perform the required respiratory maneuvers, body plethysmography is often not a practical option. If available, helium dilution provides an effort-independent alternative for measuring lung volumes.
Figure 4.
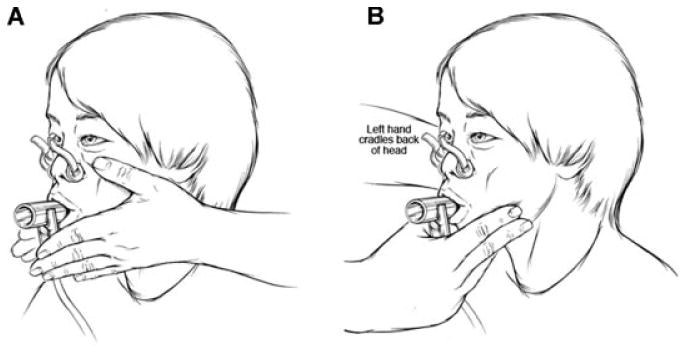
Diagram of set-up for performing PFTs on patient with A-T. Panel A illustrates the use of both hands to stabilize the patient's cheeks during the forced expiratory maneuver. Panel B shows the use of one hand to stabilize the cheeks and mandible while using the opposite hand to stabilize the head.
Current approaches based on best available evidence
Regular assessment of lung function in children who are old enough to complete the maneuver can provide valuable information about subtle progression of lung disease, even in the absence of clinical symptoms. Establishing baseline spirometry values at an early age can allow for longitudinal assessment and early interventions. To assess the degree of lung decline with age and to assess risk during respiratory illnesses and operative procedures, yearly spirometry should be performed on all able patients with A-T. Reversible small airways obstruction has been reported in children with A-T (6) and therefore, bronchodilator response should be assessed, particularly if an obstructive pattern is seen on baseline flow-volume loops. Although the incidence of obstructive lung disease is not known in the A-T population, patients with evidence of small airways obstruction and bronchodilator reversibility may benefit from inhaled steroids and β-agonist therapy (6).
Gaps in knowledge and future directions
Peak cough flow may have a role in the evaluation of the patient's ability to clear and protect the airway, especially in those patients with an observed or reported weak cough (8). A number of well-studied, effort-independent tests of pulmonary function are also available for use in young children (7), and may be useful in identifying early lung dysfunction in children with A-T unable to perform spirometry. The ability to recognize children who are at risk for poor respiratory outcomes, and to have a better understanding of pulmonary decline with age, may allow for earlier and more aggressive interventions.
Imaging Studies
Summary of current knowledge
Due to the heightened sensitivity to ionizing radiation in patients with A-T (65), radiographic studies are often avoided. Nonetheless, the risk of developing cancer has not been clearly linked to exposure to diagnostic imaging in A-T. Although the need to minimize radiation exposure in patients with A-T should be recognized, not all X-ray studies require a high dose of radiation. Chest radiographs use very little radiation, typically 0.02 mSv for frontal and lateral views in a five-year old child. This dose can be reduced by two-thirds by obtaining only the frontal view, which, in most situations is an appropriate first imaging study. The dose for a frontal view is approximately the amount of background radiation received from natural sources in the United States every two days (12) .
CT scanning provides excellent evaluation of the lungs, but the radiation dose is often 100 times that of chest radiographs. The dose can be reduced by over 70% using an interval technique that images 1mm of every 10mm of lung (60). This technique (high resolution CT) is the method of choice for imaging ILD and can provide an overall evaluation of the lungs. Because radiation dose is directly proportional to the amount of the body that is imaged, limiting CT scanning to a specific area of concern further reduces dose. Magnetic resonance imaging (MRI) does not provide the same lung detail as CT imaging, but can be useful. Areas of collapse and consolidation are similarly demonstrated by CT and MRI. Bronchiectasis and bronchial wall thickening can be demonstrated by MRI as well (67).
Radiographic confirmation of sinus disease and swallowing dysfunction is often necessary for guiding treatment in the patient with A-T. The paranasal sinuses can be imaged with plain radiographs, CT, and MRI. The radiation dose for sinus X-rays is approximately 0.05 mSv, and 0.1 mSv for CT (54). Sinus radiographs are limited in both sensitivity and specificity in the diagnosis of acute sinusitis. Both CT and MRI provide similar evaluation of the sinuses and are limited in specificity, with soft tissue in the sinuses reported in 30% of asymptomatic children (26; 43). A video fluoroscopic swallow study (VFSS) protocol has been developed that captures dysphagic patterns while limiting radiation exposure (42).
Current approaches based on best available evidence
Imaging studies should be performed in patients with A-T when clinically indicated since avoidance of their use could result in delayed or missed diagnoses in a patient with pulmonary pathology. The clinician and imager should consider the risks and benefits of each study and minimize radiation exposure as much as possible.
Gaps in knowledge and future directions
Further investigations better quantifying the risk of routine imaging studies are needed. Investigations into the use of minimal-dose protocols and non-ionizing radiation to evaluate the lungs, paranasal sinuses, and swallowing mechanics are needed in patients with A-T. Since dysphagia is common in this population and may significantly contribute to lung disease, future studies that evaluate the utility of non-radiographic techniques that detect dysphagia are warranted.
Polysomnography
Summary of current knowledge
Data suggest that sleep disordered breathing is not common among stable adolescents with A-T (48). This finding is in contrast to other diseases characterized by bulbar muscle weakness and decreased pulmonary reserve, such as DMD and SMA, in which airway tone is significantly compromised, obstructive sleep apnea syndrome is found regularly and respiratory failure is progressively more common with age (78; 80). Sleep efficiency, however, was found to be decreased in A-T patients (48). Decreased sleep efficiency is common in children with chronic illnesses and may negatively impact QOL (3; 55). No data have been published on the use of polysomnography in A-T patients with severe lung disease.
Current approaches based on best available evidence
Polysomnography should be considered in A-T patients with signs or symptoms of excessive daytime sleepiness, snoring, disrupted sleep, and/or witnessed apnea to evaluate for sleep disordered breathing. In patients with severe lung disease, polysomnography can be useful in evaluating for hypoxemia or hypercapnea, and may support the need for supplemental oxygen or NIV support.
Gaps in knowledge and future directions
Assessment of the impact of decreased sleep efficiency on QOL and evaluation of interventions to improve sleep efficiency in this population should be pursued. The use of polysomnography in the assessment of A-T patients with severe lung disease and possible respiratory failure warrants formal evaluation.
General Care Issues
Immunizations
Summary of current knowledge
Immunizations may be less effective in this population (77). People with A-T often have a suboptimal response to the 23-valent pneumococcal polysaccharide vaccine (34), as well as to other vaccines. Limited data suggest that the response to the 7-valent pneumococcal conjugate vaccine may be more robust (74).
Current approaches based on best available evidence
Despite the limitations described above, immunizations to common respiratory pathogens should be given to individuals with A-T, and those with impaired immune responses should be referred to an immunologist. As with all individuals with chronic illness, those with A-T and their household contacts, should receive annual influenza immunization (21). Because people with A-T may not generate a protective antibody response, current management strategies include the measurement of post-immunization antibody levels to guide the need for additional immunizations or to initiate other preventive measures (Table 2). Gammaglobulin replacement therapy, delivered via an intravenous or subcutaneous route, should be considered in people with deficient antibody responses to vaccines. Hypogammaglobulinemia or deficiency of an IgG subclass is not in itself, however, an indication for gammaglobulin replacement therapy unless there is an accompanying deficiency of antibody production.
Table 2. Recommendations for immunizations in A-T patients.
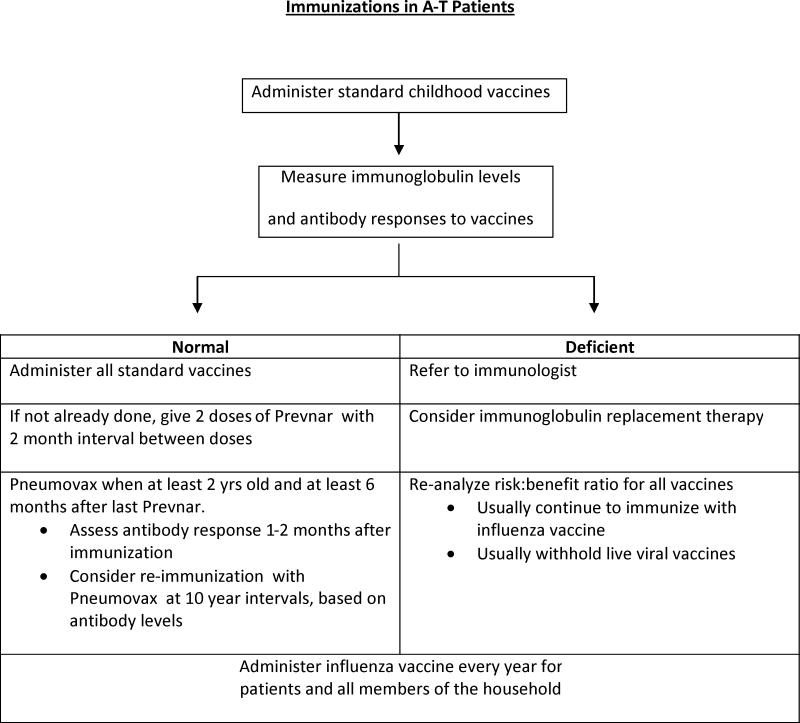
|
Gaps in knowledge and future directions
Further investigations into the optimal use of vaccines for the prevention of recurrent respiratory tract infections are necessary and ongoing.
Nutritional Support
Summary of current knowledge
Pulmonary function is related to nutritional status in patients with CF and COPD (19; 50). Whether this paradigm holds true for A-T is unknown, although poor growth is a common feature of A-T and may be associated with a general decline in overall health, poor caloric intake or endocrine abnormalities (49). Nutrition via GT has been used in patients with A-T but no data exist to confirm the optimal time for GT placement or whether nutritional intervention provides any benefit to lung health. Furthermore, targets for optimal nutritional management are not known in the A-T population.
Current approaches based on best available evidence
Although no published data on this population exists, use of GT should be considered in A-T patients with failure to thrive, or in those at risk for aspiration. It is currently recommended that GT placement be performed only where intensive care services are available. Young adult A-T patients with impaired lung function may be at increased risk for pulmonary complications following GT placement and a trial of nasogastric feedings should be considered to determine the utility of this intervention.
Gaps in knowledge and future recommendations
Prospective longitudinal studies will be needed to address the optimal timing and modes of nutritional intervention, such as GT placement and use, and whether such interventions afford improved pulmonary health in patients with A-T.
Peri-Operative and Critical Care Management
Summary of current knowledge
The peri-operative and intensive care management of patients with A-T is often challenging. Case reports have described difficulties with mechanical ventilation due to restrictive lung disease (81). Recommendations have been made in the post-operative and intensive care management of individuals with DMD (28; 29).
Current approaches based on best available evidence
A full pre-operative evaluation should be considered prior to any planned procedures involving general anesthesia or sedation to verify stability of neurologic, pulmonary, immunologic and oncologic conditions. Diagnostic and operative procedures requiring anesthesia or sedation should be performed at facilities where personnel are experienced with medically complex patients, and where intensive care services are readily available. Ongoing discussion of advance directives should take place while the patient is clinically stable, and written directives should be put in place whenever possible prior to major surgical procedures. Close attention to providing adequate respiratory support, augmenting pulmonary clearance measures, optimizing pain control, and supporting nutrition are likely to be beneficial in individuals with A-T requiring intensive or post-operative care.
Gaps in knowledge and future directions
Further research is needed to help determine the optimal modes and strategies for ventilation in patients with A-T and lung disease. Additionally, determination of the most safe and effective medications and approaches for providing anesthesia and sedation should help reduce peri- and post-operative morbidity and mortality.
Multidisciplinary Care
Summary of current knowledge
Comprehensive coordinated care has become the standard for many complex chronic diseases, and has been particularly successful in the care of pediatric patients with CF and hemophilia through development of disease-specific, multidisciplinary specialty clinics throughout the United States (33). These centers provide a “medical home” for patients and their families and ensure standardized and evidence-based care (35). By contrast, patients with A-T are often seen by multiple specialists including neurologists, immunologists, oncologists, speech-language pathologists and pulmonologists. How their care is directed and provided depends largely on geographical location and resources available.
Current approaches based on best available evidence
Establishment of dedicated multidisciplinary care centers at multiple sites in the United States is not practical for A-T due to its rarity, the geographical dispersion of patients, and the lack of care providers with expertise in the care of A-T patients. Nonetheless, providing multidisciplinary care whenever possible should be a goal of providers. Ideally, this approach would include active participation by primary care providers, multiple medical specialists (immunology, pulmonology, neurology), genetic counseling, as well as nursing, nutrition, and social/psychological support. Such a model exists at the Johns Hopkins A-T Clinical Center, and has been successful in providing consultative services to other sites.
Gaps in knowledge and future directions
Future research should address the question of whether centralized and multidisciplinary care of A-T is practical and whether it improves outcomes, or if patients with A-T could receive adequate standardized care at centers dedicated to other chronic disease such as CF, ILD, or neuromuscular disorders. A comprehensive registry of patients with A-T should be established. The use of the registry would allow for following patient outcomes, identifying best practice guidelines and would allow for the development of comprehensive clinical trials to assess treatment modalities.
Conclusions
A-T is a rare and extremely complex lifelong disease. Lung disease is a significant source of morbidity and mortality among patients with A-T. Current knowledge about this disorder and its pulmonary complications is limited by the lack of prospective data. Lessons must therefore be drawn from other diseases with similar features. A great need exists for multicenter collaboration and prospective studies to better characterize the lung disease seen in children with A-T and to more effectively address its evaluation and management.
Acknowledgments
Funding for the Workshop on “Pulmonary Disease in Ataxia-Telangiectasia” was provided by The A-T Children's Project.
Support: Ataxia-Telangiectasia Children's Project, 5300 W. Hillsboro Blvd. Ste 105, Coconut Creek, FL 33073
Footnotes
The authors express gratitude to all workshop attendees: David Paydarfar, Allan H. Ropper, Gerald M. Loughlin, John L. Carroll, Leland L. Fan, Ronald B. Easley, Michael P. Boyle, Michelle Troche, Asbjorg Stray-Pedersen, Shyam Biswal, Andrew R. Exley, John T. Sandlund, Stefan Zielen, Jonathan Finder, Howard M. Lederman, Maureen A. Lefton-Greif, Paul G. Auwaerter, Cynthia J. Rothblum-Oviatt, Thomas Crawford, Karla Au Yeung, Laura Sterni, Sharon McGrath-Morrow, C.M. R. Weemaes, Peter J. Mogayzel, Sande Okelo, Danilo A. Tagle, Karen Rosquist, Amber Kelly, W. Adam Gower, Scott A. Schroeder, Brad A. Margus, Jayesh Bhatt, Jennifer Thornton
Reference List
- 1.Al Salmi QA, Walter JN, Colasurdo GN, Sockrider MM, Smith EO, Takahashi H, Fan LL. Serum KL-6 and surfactant proteins A and D in pediatric interstitial lung disease. Chest. 2005;127:403–407. doi: 10.1378/chest.127.1.403. [DOI] [PubMed] [Google Scholar]
- 2.Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, Phillips JA, III, Lansdorp PM, Loyd JE, Armanios MY. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amin R, Bean J, Burklow K, Jeffries J. The relationship between sleep disturbance and pulmonary function in stable pediatric cystic fibrosis patients. Chest. 2005;128:1357–1363. doi: 10.1378/chest.128.3.1357. [DOI] [PubMed] [Google Scholar]
- 4.Barzilai A, Rotman G, Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst) 2002;1:3–25. doi: 10.1016/s1568-7864(01)00007-6. [DOI] [PubMed] [Google Scholar]
- 5.Benden C, Boehler A. Long-term clarithromycin therapy in the management of lung transplant recipients. Transplantation. 2009;87:1538–1540. doi: 10.1097/TP.0b013e3181a492b2. [DOI] [PubMed] [Google Scholar]
- 6.Berkun Y, Vilozni D, Levi Y, Borik S, Waldman D, Somech R, Nissenkorn A, Efrati O. Reversible airway obstruction in children with ataxia telangiectasia. Pediatr Pulmonol. 2010;45:230–235. doi: 10.1002/ppul.21095. [DOI] [PubMed] [Google Scholar]
- 7.Beydon N, Davis SD, Lombardi E, Allen JL, Arets HG, Aurora P, Bisgaard H, Davis GM, Ducharme FM, Eigen H, Gappa M, Gaultier C, Gustafsson PM, Hall GL, Hantos Z, Healy MJ, Jones MH, Klug B, Lodrup Carlsen KC, McKenzie SA, Marchal F, Mayer OH, Merkus PJ, Morris MG, Oostveen E, Pillow JJ, Seddon PC, Silverman M, Sly PD, Stocks J, Tepper RS, Vilozni D, Wilson NM. An official American Thoracic Society/European Respiratory Society statement: pulmonary function testing in preschool children. Am J Respir Crit Care Med. 2007;175:1304–1345. doi: 10.1164/rccm.200605-642ST. [DOI] [PubMed] [Google Scholar]
- 8.Bianchi C, Baiardi P. Cough peak flows: standard values for children and adolescents. Am J Phys Med Rehabil. 2008;87:461–467. doi: 10.1097/PHM.0b013e318174e4c7. [DOI] [PubMed] [Google Scholar]
- 9.Boder E. Ataxia-telangiectasia: some historic, clinical and pathologic observations. Birth Defects Orig Artic Ser. 1975;11:255–270. [PubMed] [Google Scholar]
- 10.Boder E, Sedgwick RP. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics. 1958;21:526–554. [PubMed] [Google Scholar]
- 11.Bott L, Lebreton J, Thumerelle C, Cuvellier J, Deschildre A, Sardet A. Lung disease in ataxia-telangiectasia. Acta Paediatr. 2007;96:1021–1024. doi: 10.1111/j.1651-2227.2007.00338.x. [DOI] [PubMed] [Google Scholar]
- 12.Brody AS, Frush DP, Huda W, Brent RL. Radiation risk to children from computed tomography. Pediatrics. 2007;120:677–682. doi: 10.1542/peds.2007-1910. [DOI] [PubMed] [Google Scholar]
- 13.Buckley RH. Pulmonary complications of primary immunodeficiencies. Paediatr Respir Rev. 2004;5(Suppl A):S225–S233. doi: 10.1016/s1526-0542(04)90043-7. [DOI] [PubMed] [Google Scholar]
- 14.Buehring I, Friedrich B, Schaaf J, Schmidt H, Ahrens P, Zielen S. Chronic sinusitis refractory to standard management in patients with humoral immunodeficiencies. Clin Exp Immunol. 1997;109:468–472. doi: 10.1046/j.1365-2249.1997.4831379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canny GJ, Roifman C, Weitzman S, Braudo M, Levison H. A pulmonary infiltrate in a child with ataxia telangiectasia. Ann Allergy. 1988;61:422–428. [PubMed] [Google Scholar]
- 16.Carlsen KC, Haland G, Carlsen KH. Natural history of lung function in health and diseases. Curr Opin Allergy Clin Immunol. 2009;9:146–150. doi: 10.1097/ACI.0b013e3283292243. [DOI] [PubMed] [Google Scholar]
- 17.Carlsen KH, Carlsen KC. Respiratory effects of tobacco smoking on infants and young children. Paediatr Respir Rev. 2008;9:11–19. doi: 10.1016/j.prrv.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Chang AB, Redding GJ, Everard ML. Chronic wet cough: Protracted bronchitis, chronic suppurative lung disease and bronchiectasis. Pediatr Pulmonol. 2008;43:519–531. doi: 10.1002/ppul.20821. [DOI] [PubMed] [Google Scholar]
- 19.Chapman KM, Winter L. COPD: using nutrition to prevent respiratory function decline. Geriatrics. 1996;51:37–42. [PubMed] [Google Scholar]
- 20.Chen RL, Wang PJ, Hsu YH, Chang PY, Fang JS. Severe lung fibrosis after chemotherapy in a child with ataxia-telangiectasia. J Pediatr Hematol Oncol. 2002;24:77–79. doi: 10.1097/00043426-200201000-00021. [DOI] [PubMed] [Google Scholar]
- 21.Committe on Infectious Diseases. Influenza. In: Pickering L, Baker C, Kimberlin D, Long S, editors. 2009 Report of the Committee on Infectious Diseases. Elk Grove Village: American Academy of Pediatrics; 2009. [Google Scholar]
- 22.Crawford TO. Ataxia telangiectasia. Semin Pediatr Neurol. 1998;5:287–294. doi: 10.1016/s1071-9091(98)80007-7. [DOI] [PubMed] [Google Scholar]
- 23.Crawford TO, Mandir AS, Lefton-Greif MA, Goodman SN, Goodman BK, Sengul H, Lederman HM. Quantitative neurologic assessment of ataxia-telangiectasia. Neurology. 2000;54:1505–1509. doi: 10.1212/wnl.54.7.1505. [DOI] [PubMed] [Google Scholar]
- 24.Crawford TO, Skolasky RL, Fernandez R, Rosquist KJ, Lederman HM. Survival probability in ataxia telangiectasia. Arch Dis Child. 2006;91:610–611. doi: 10.1136/adc.2006.094268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crosbie PA, Woodhead MA. Long-term macrolide therapy in chronic inflammatory airway diseases. Eur Respir J. 2009;33:171–181. doi: 10.1183/09031936.00042208. [DOI] [PubMed] [Google Scholar]
- 26.Duerinckx AJ, Hall TR, Whyte AM, Lufkin R, Kangarloo H. Paranasal sinuses in pediatric patients by MRI: normal development and preliminary findings in disease. Eur J Radiol. 1991;13:107–112. doi: 10.1016/0720-048x(91)90090-i. [DOI] [PubMed] [Google Scholar]
- 27.Fauroux B, Tamalet A, Clement A. Management of primary ciliary dyskinesia: the lower airways. Paediatr Respir Rev. 2009;10:55–57. doi: 10.1016/j.prrv.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Finder JD. A 2009 perspective on the 2004 American Thoracic Society statement, “respiratory care of the patient with Duchenne muscular dystrophy”. Pediatrics. 2009;123(Suppl 4):S239–S241. doi: 10.1542/peds.2008-2952I. [DOI] [PubMed] [Google Scholar]
- 29.Finder JD, Birnkrant D, Carl J, Farber HJ, Gozal D, Iannaccone ST, Kovesi T, Kravitz RM, Panitch H, Schramm C, Schroth M, Sharma G, Sievers L, Silvestri JM, Sterni L. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med. 2004;170:456–465. doi: 10.1164/rccm.200307-885ST. [DOI] [PubMed] [Google Scholar]
- 30.Flume PA, O'Sullivan BP, Robinson KA, Goss CH, Mogayzel PJ, Jr, Willey-Courand DB, Bujan J, Finder J, Lester M, Quittell L, Rosenblatt R, Vender RL, Hazle L, Sabadosa K, Marshall B. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176:957–969. doi: 10.1164/rccm.200705-664OC. [DOI] [PubMed] [Google Scholar]
- 31.George AM, Jones PM, Middleton PG. Cystic fibrosis infections: treatment strategies and prospects. FEMS Microbiol Lett. 2009;300:153–164. doi: 10.1111/j.1574-6968.2009.01704.x. [DOI] [PubMed] [Google Scholar]
- 32.Gottlieb J, Szangolies J, Koehnlein T, Golpon H, Simon A, Welte T. Long-term azithromycin for bronchiolitis obliterans syndrome after lung transplantation. Transplantation. 2008;85:36–41. doi: 10.1097/01.tp.0000295981.84633.bc. [DOI] [PubMed] [Google Scholar]
- 33.Grosse SD, Schechter MS, Kulkarni R, Lloyd-Puryear MA, Strickland B, Trevathan E. Models of comprehensive multidisciplinary care for individuals in the United States with genetic disorders. Pediatrics. 2009;123:407–412. doi: 10.1542/peds.2007-2875. [DOI] [PubMed] [Google Scholar]
- 34.Guerra-Maranhao MC, Costa-Carvalho BT, Nudelman V, Barros-Nunes P, Carneiro-Sampaio MM, Arslanian C, Nagao-Dias AT, Sole D. Response to polysaccharide antigens in patients with ataxia-telangiectasia. J Pediatr (Rio J) 2006;82:132–136. doi: 10.2223/JPED.1460. [DOI] [PubMed] [Google Scholar]
- 35.Homer CJ, Klatka K, Romm D, Kuhlthau K, Bloom S, Newacheck P, Van Cleave J, Perrin JM. A review of the evidence for the medical home for children with special health care needs. Pediatrics. 2008;122:e922–e937. doi: 10.1542/peds.2007-3762. [DOI] [PubMed] [Google Scholar]
- 36.Huber J, Zegers BJ, Schuurman HJ. Pathology of congenital immunodeficiencies. Semin Diagn Pathol. 1992;9:31–62. [PubMed] [Google Scholar]
- 37.Ito M, Nakagawa A, Hirabayashi N, Asai J. Bronchiolitis obliterans in ataxia-telangiectasia. Virchows Arch. 1997;430:131–137. doi: 10.1007/BF01008034. [DOI] [PubMed] [Google Scholar]
- 38.Jackson CE, Rosenfeld J, Moore DH, Bryan WW, Barohn RJ, Wrench M, Myers D, Heberlin L, King R, Smith J, Gelinas D, Miller RG. A preliminary evaluation of a prospective study of pulmonary function studies and symptoms of hypoventilation in ALS/MND patients. J Neurol Sci. 2001;191:75–78. doi: 10.1016/s0022-510x(01)00617-7. [DOI] [PubMed] [Google Scholar]
- 39.Karrasch S, Holz O, Jorres RA. Aging and induced senescence as factors in the pathogenesis of lung emphysema. Respir Med. 2008;102:1215–1230. doi: 10.1016/j.rmed.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 40.Kohno N. Serum marker KL-6/MUC1 for the diagnosis and management of interstitial pneumonitis. J Med Invest. 1999;46:151–158. [PubMed] [Google Scholar]
- 41.Koyama H, Geddes DM. Erythromycin and diffuse panbronchiolitis. Thorax. 1997;52:915–918. doi: 10.1136/thx.52.10.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lefton-Greif MA, Crawford TO, Winkelstein JA, Loughlin GM, Koerner CB, Zahurak M, Lederman HM. Oropharyngeal dysphagia and aspiration in patients with ataxia-telangiectasia. J Pediatr. 2000;136:225–231. doi: 10.1016/s0022-3476(00)70106-5. [DOI] [PubMed] [Google Scholar]
- 43.Lim WK, Ram B, Fasulakis S, Kane KJ. Incidental magnetic resonance image sinus abnormalities in asymptomatic Australian children. J Laryngol Otol. 2003;117:969–972. doi: 10.1258/002221503322683858. [DOI] [PubMed] [Google Scholar]
- 44.Lopez-Boado YS, Rubin BK. Macrolides as immunomodulatory medications for the therapy of chronic lung diseases. Curr Opin Pharmacol. 2008;8:286–291. doi: 10.1016/j.coph.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 45.Manzur AY, Kinali M, Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch Dis Child. 2008;93:986–990. doi: 10.1136/adc.2007.118141. [DOI] [PubMed] [Google Scholar]
- 46.McGrath-Morrow S, Lefton-Greif M, Rosquist K, Crawford T, Kelly A, Zeitlin P, Carson KA, Lederman HM. Pulmonary function in adolescents with ataxia telangiectasia. Pediatr Pulmonol. 2008;43:59–66. doi: 10.1002/ppul.20738. [DOI] [PubMed] [Google Scholar]
- 47.McGrath-Morrow SA, Collaco JM, Crawford TO, Carson KA, Lefton-Greif MA, Zeitlin P, Lederman HM. Elevated serum IL-8 levels in ataxia telangiectasia. J Pediatr. 2010;156:682–684. doi: 10.1016/j.jpeds.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 48.McGrath-Morrow SA, Sterni L, McGinley B, Lefton-Greif MA, Rosquist K, Lederman H. Polysomnographic values in adolescents with ataxia telangiectasia. Pediatr Pulmonol. 2008;43:674–679. doi: 10.1002/ppul.20838. [DOI] [PubMed] [Google Scholar]
- 49.McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep. 2004;5:772–776. doi: 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milla CE. Nutrition and lung disease in cystic fibrosis. Clin Chest Med. 2007;28:319–330. doi: 10.1016/j.ccm.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 51.Miyazaki E, Ando M, Muramatsu T, Fukami T, Matsuno O, Nureki S, Ueno T, Tsuda T, Kumamoto T. Early assessment of rapidly progressive interstitial pneumonia associated with amyopathic dermatomyositis. Clin Rheumatol. 2007;26:436–439. doi: 10.1007/s10067-005-0147-4. [DOI] [PubMed] [Google Scholar]
- 52.Moin M, Aghamohammadi A, Kouhi A, Tavassoli S, Rezaei N, Ghaffari SR, Gharagozlou M, Movahedi M, Purpak Z, Mirsaeid GB, Mahmoudi M, Farhoudi A. Ataxia-telangiectasia in Iran: clinical and laboratory features of 104 patients. Pediatr Neurol. 2007;37:21–28. doi: 10.1016/j.pediatrneurol.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 53.Morio T, Kim H. Ku, Artemis, and ataxia-telangiectasia-mutated: signalling networks in DNA damage. Int J Biochem Cell Biol. 2008;40:598–603. doi: 10.1016/j.biocel.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 54.Mulkens TH, Broers C, Fieuws S, Termote JL, Bellnick P. Comparison of effective doses for low-dose MDCT and radiographic examination of sinuses in children. AJR Am J Roentgenol. 2005;184:1611–1618. doi: 10.2214/ajr.184.5.01841611. [DOI] [PubMed] [Google Scholar]
- 55.Naqvi SK, Sotelo C, Murry L, Simakajornboon N. Sleep architecture in children and adolescents with cystic fibrosis and the association with severity of lung disease. Sleep Breath. 2008;12:77–83. doi: 10.1007/s11325-007-0123-0. [DOI] [PubMed] [Google Scholar]
- 56.Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, Zariwala MA, Knowles MR. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med. 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 57.Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr. 2004;144:505–511. doi: 10.1016/j.jpeds.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 58.O'Sullivan BP, Flume P. The clinical approach to lung disease in patients with cystic fibrosis. Semin Respir Crit Care Med. 2009;30:505–513. doi: 10.1055/s-0029-1238909. [DOI] [PubMed] [Google Scholar]
- 59.Ohnishi H, Yokoyama A, Kondo K, Hamada H, Abe M, Nishimura K, Hiwada K, Kohno N. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med. 2002;165:378–381. doi: 10.1164/ajrccm.165.3.2107134. [DOI] [PubMed] [Google Scholar]
- 60.Owens C. Pearls and pitfalls in HRCT in children. Paediatr Respir Rev. 2006;7(Suppl 1):S44–S49. doi: 10.1016/j.prrv.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 61.Pagano G, Korkina LG, Brunk UT, Chessa L, Degan P, del Principe D, Kelly FJ, Malorni W, Pallardo F, Pasquier C, Scovassi I, Zatterale A, Franceschi C. Congenital disorders sharing oxidative stress and cancer proneness as phenotypic hallmarks: prospects for joint research in pharmacology. Med Hypotheses. 1998;51:253–266. doi: 10.1016/s0306-9877(98)90084-6. [DOI] [PubMed] [Google Scholar]
- 62.Paiva MA, Amaral SM. Chronic interstitial lung diseases in children. J Bras Pneumol. 2009;35:792–803. doi: 10.1590/s1806-37132009000800012. [DOI] [PubMed] [Google Scholar]
- 63.Palau F, Espinos C. Autosomal recessive cerebellar ataxias. Orphanet J Rare Dis. 2006;1:47. doi: 10.1186/1750-1172-1-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pitts T, Bolser D, Rosenbek J, Troche M, Okun MS, Sapienza C. Impact of expiratory muscle strength training on voluntary cough and swallow function in Parkinson disease. Chest. 2009;135:1301–1308. doi: 10.1378/chest.08-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pollard JM, Gatti RA. Clinical radiation sensitivity with DNA repair disorders: an overview. Int J Radiat Oncol Biol Phys. 2009;74:1323–1331. doi: 10.1016/j.ijrobp.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Popa V, Colby TV, Reich SB. Pulmonary interstitial disease in Ig deficiency. Chest. 2002;122:1594–1603. doi: 10.1378/chest.122.5.1594. [DOI] [PubMed] [Google Scholar]
- 67.Puderbach M, Eichinger M, Gahr J, Ley S, Tuengerthal S, Schmahl A, Fink C, Plathow C, Wiebel M, Muller FM, Kauczor HU. Proton MRI appearance of cystic fibrosis: comparison to CT. Eur Radiol. 2007;17:716–724. doi: 10.1007/s00330-006-0373-4. [DOI] [PubMed] [Google Scholar]
- 68.Rochester DF, Esau SA. Assessment of ventilatory function in patients with neuromuscular disease. Clin Chest Med. 1994;15:751–763. [PubMed] [Google Scholar]
- 69.Saleem AF, Sapienza CM, Okun MS. Respiratory muscle strength training: treatment and response duration in a patient with early idiopathic Parkinson's disease. NeuroRehabilitation. 2005;20:323–333. [PubMed] [Google Scholar]
- 70.Sandoval C, Swift M. Treatment of lymphoid malignancies in patients with ataxia-telangiectasia. Med Pediatr Oncol. 1998;31:491–497. doi: 10.1002/(sici)1096-911x(199812)31:6<491::aid-mpo5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 71.Sandoval C, Swift M. Hodgkin disease in ataxia-telangiectasia patients with poor outcomes. Med Pediatr Oncol. 2003;40:162–166. doi: 10.1002/mpo.10251. [DOI] [PubMed] [Google Scholar]
- 72.Sapir T, Blank M, Shoenfeld Y. Immunomodulatory effects of intravenous immunoglobulins as a treatment for autoimmune diseases, cancer, and recurrent pregnancy loss. Ann N Y Acad Sci. 2005;1051:743–778. doi: 10.1196/annals.1361.118. [DOI] [PubMed] [Google Scholar]
- 73.Schroeder SA, Swift M, Sandoval C, Langston C. Interstitial lung disease in patients with ataxia-telangiectasia. Pediatr Pulmonol. 2005;39:537–543. doi: 10.1002/ppul.20209. [DOI] [PubMed] [Google Scholar]
- 74.Schubert R, Reichenbach J, Rose M, Zielen S. Immunogenicity of the seven valent pneumococcal conjugate vaccine in patients with ataxia-telangiectasia. Pediatr Infect Dis J. 2004;23:269–270. doi: 10.1097/01.inf.0000115737.35353.55. [DOI] [PubMed] [Google Scholar]
- 75.Soliman MG, Higgins SE, El Kabir DR, Davidson AC, Williams AJ, Howard RS. Non-invasive assessment of respiratory muscle strength in patients with previous poliomyelitis. Respir Med. 2005;99:1217–1222. doi: 10.1016/j.rmed.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 76.Spector BD, Filipovich AH, Perry GS, Kersey JH. Epidemiology of cancer in ataxia-telangiectasia. In: Bridges BA, Harnden DG, editors. Ataxia-telangiectasia: A Cellular and Molecular Ling Between Cancer, Neuropathology, and Immune Deficiency. New York: Wiley; 1982. pp. 103–138. [Google Scholar]
- 77.Stray-Pedersen A, Aaberge IS, Fruh A, Abrahamsen TG. Pneumococcal conjugate vaccine followed by pneumococcal polysaccharide vaccine; immunogenicity in patients with ataxia-telangiectasia. Clin Exp Immunol. 2005;140:507–516. doi: 10.1111/j.1365-2249.2005.02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suresh S, Wales P, Dakin C, Harris MA, Cooper DG. Sleep-related breathing disorder in Duchenne muscular dystrophy: disease spectrum in the paediatric population. J Paediatr Child Health. 2005;41:500–503. doi: 10.1111/j.1440-1754.2005.00691.x. [DOI] [PubMed] [Google Scholar]
- 79.Svanes C, Sunyer J, Plana E, Dharmage S, Heinrich J, Jarvis D, Demarco R, Norback D, Raherison C, Villani S, Wjst M, Svanes K, Anto J. Early life origins of chronic obstructive pulmonary disease. Thorax. 2009 doi: 10.1136/thx.2008.112136. [DOI] [PubMed] [Google Scholar]
- 80.Testa MB, Pavone M, Bertini E, Petrone A, Pagani M, Cutrera R. Sleep-disordered breathing in spinal muscular atrophy types 1 and 2. Am J Phys Med Rehabil. 2005;84:666–670. doi: 10.1097/01.phm.0000176362.24957.77. [DOI] [PubMed] [Google Scholar]
- 81.Verhagen MM, van Deuren M, Willemsen MA, Van der Hoeven HJ, Heijdra YF, Yntema JB, Weemaes CM, Neeleman C. Ataxia-Telangiectasia and mechanical ventilation: a word of caution. Pediatr Pulmonol. 2009;44:101–102. doi: 10.1002/ppul.20957. [DOI] [PubMed] [Google Scholar]
- 82.Yalcin B, Kutluk MT, Sanal O, Akyuz C, Anadol D, Caglar M, Gocmen A, Buyukpamukcu M. Hodgkin's disease and ataxia telangiectasia with pulmonary cavities. Pediatr Pulmonol. 2002;33:399–403. doi: 10.1002/ppul.10057. [DOI] [PubMed] [Google Scholar]