

Abstract
We describe SILIRID (Simple Ligand–Receptor Interaction Descriptor), a novel fixed size descriptor characterizing protein–ligand interactions. SILIRID can be obtained from the binary interaction fingerprints (IFPs) by summing up the bits corresponding to identical amino acids. This results in a vector of 168 integer numbers corresponding to the product of the number of entries (20 amino acids and one cofactor) and 8 interaction types per amino acid (hydrophobic, aromatic face to face, aromatic edge to face, H-bond donated by the protein, H-bond donated by the ligand, ionic bond with protein cation and protein anion, and interaction with metal ion). Efficiency of SILIRID to distinguish different protein binding sites has been examined in similarity search in sc-PDB database, a druggable portion of the Protein Data Bank, using various protein–ligand complexes as queries. The performance of retrieval of structurally and evolutionary related classes of proteins was comparable to that of state-of-the-art approaches (ROC AUC ≈ 0.91). SILIRID can efficiently be used to visualize chemogenomic space covered by sc-PDB using Generative Topographic Mapping (GTM): sc-PDB SILIRID data form clusters corresponding to different protein types.
Keywords: Protein–ligand interactions, Interaction fingerprints, Protein similarity, Protein classification, Chemogenomics, Generative Topographic Mapping
1. Introduction
Nowadays, a comparison of protein–ligand binding sites is widely used to predict new targets or new ligands using protein–ligand (PL) complexes as search queries [1–3]. Efficiency of this procedure clearly depends on computer representation of the binding sites (BSs). The simplest BS representation as amino acid sequence is, unfortunately, insufficient because protein families with identical folds may have very low sequence similarity. More appropriate approaches encode BSs by surface [4,5], mesh [6], cloud-of-atoms [7] or graphs [8–13].
The interaction fingerprint (IFP) approach [14] represents an alternative way to encode protein–ligand complexes. Generally, an IFP encodes a presence (1) or an absence (0) of interactions of the ligand with specified amino acids of the binding site, thus forming a binary string (bitstring). Each amino acid of the binding site is described by one same number of interaction types (hydrophobic, hydrogen donor, hydrogen acceptor, etc.), thus all complexes of the given protein could be described by IFPs of the same length. Therefore, they can be easily compared using similarity measures. IFPs directly characterize the binding modes rather than the ligand structure and, hence, they may be similar for the PL-complexes containing ligands with different scaffolds.
Typically, the IFP length depends on the binding site size which limits their application to one protein's family. Therefore, some efforts have been made to construct binding site independent IFP [15]. The most common approach consists in the binning of geometrical patterns of interactions resulting in either a fixed size vector or a vector which size depends on the training set. For example, atom-pair based interaction fingerprint (APIF) by Pérez-Nueno et al. [16] encodes a quadruple forming by interacting atom pairs of the ligand and the protein. APIF has a fixed size of 294 bits for any protein corresponding to six possible combinations of PL interaction types and seven distance bins [16]. In FuzCav cavity fingerprint by Weill and Rognan [17], a BS is represented by the 4833-integer vector in which each component registers the count of unique pharmacophoric triplets (three properties and three related distances) occurring at binned inter-feature distances. Recently, it has been shown [18] that IFP for a given class of protein can be predicted directly from 2D structure of ligands.
Here, we describe a new type of binding site size independent IFP — Simple Ligand–Receptor Interaction Descriptor (SILIRID) which is a vector constructed from binding site dependent IFP. It has been demonstrated that this approach could efficiently be used for the binding site comparison and analysis of PL-interactions.
2. Method
SILIRIDs are calculated from the IFP described by Marcou and Rognan [19] and stored in the sc-PDB database (version 2011) [20]. Every IFP consists of 8 bits per amino acid: hydrophobic, aromatic face to face, aromatic edge to face, hydrogen bond donated by the protein, hydrogen bond donated by the ligand, ionic bond with protein cation and protein anion, and metal ion. The length of this IFP depends on the size of the biding site.
Preparation of SILIRID vectors from IFP by merging bitstrings for a given type of amino acid is shown in Fig. 1. Here, two bitstrings corresponding to VAL18 and VAL64 are transformed into a vector based on integer numbers; similar operation is performed for PHE80 and PHE82. Amino acids which do not interact with the ligand (all bits are zero) must also be taken into account. Co-factors are also considered as an additional 8 bit entry. At the second step, bitstrings of 21 entries (20 amino acids and cofactor) are concatenated into one vector. For any protein, the order of the amino acids in SILIRID is fixed according to the lipophilicity and pKa of the amino acids. Thus, any binding site is described by a fixed length 21 ∗ 8 = 168 dimensional vector. SILIRIDs for the whole sc-PDB were generated using in-house script.
Fig. 1.

SILIRID generation workflow: IFP bits corresponding to the same amino acids are concatenated resulting in a numerical vector with fixed size (168), which corresponds to the product of the number of amino acids plus cofactor (21) and the number of interaction types (8).
Similarity between SILIRIDs was calculated using the Jaccard index using R-package vegdist [21]:
Here, , where xik are i-th components of SILIRIDs describing, respectively, PL complexes j and k.
The ROCR package [22] for R statistical environment [23] was used to plot ROC curves and to perform ROC AUC calculation.
Notice that obtaining SILIRID from 3D structure and comparison of SILIRIDs corresponding to different binding sites are very fast. Thus, calculations of SILIRID based pairwise similarities for ~ 9000 sc-PDB entries take around 15 min on standard Linux station, 64 bit, single core, Intel i5, using standard 64 bit R statistical environment.
SILIRID vectors extracted from the sc-PDB database are available for download at https://github.com/chupvl/silirid.
3. Results and discussion
3.1. Ability of SILIRID to detect similar binding sites
SILIRID efficiency in alignment-free binding site comparison has been investigated for three protein classes: kinases, serine-proteases and nuclear receptors. Every studied protein class was treated as class 1 and all other PL-complexes in sc-PDB as class 2. Within each class, sub-classes 1a and 1b have been selected using either EC number (enzyme classification) or Structural Classification of Proteins (SCOP) or both (Table 1) and additionally manually cleaned. Protein family 1a is a sub-class of 1b, which, in turn, is a sub-class of 1 (see Fig. 2). This setup allows us to study the ability of SILIRID to retrieve proteins of the given class and sub-classes in similarity search using PL complexes of 1a proteins as query. Thus, the ability of a CDK2 binding site encoded by SILIRID has been tested to retrieve binding sites of other CDK2 (class 1a), similar binding sites of serine–threonine protein kinases (class 1b), and those of protein kinases (class 1).
Table 1.
Classes and subclasses of proteins used for similarity search studies. The number of entries from the sc-PDB database is shown in parenthesis.
| Class 1a | CDK2 kinase (123) | Androgen receptor (29) | Trypsin (78) |
| Class 1b | Serine–threonine kinase (488) | NA | Trypsin-like fold (378) |
| Class 1 | Protein kinase (754) | Nuclear receptor (282) | Serine protease (417) |
Fig. 2.
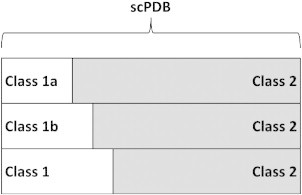
Setup of protein classes and subclasses used for SILIRID comparison in similarity search experiments. See Table 1 for details.
For a given protein family 1a, each representative has been used as query. Therefore, in order to characterize the results of similarity search, the average ROC curves have been plotted and corresponding ROC AUC values have been calculated.
Similarity search results reported in Fig. 3 and Table 2 show that SILIRID efficiency to compare protein binding sites is similar to that of the state-of-the-art approaches. Thus, SILIRID-based similarity search with trypsin as queries to retrieve trypsin-like fold proteins among all sc-PDB entries resulted in average ROC AUC = 0.95 which is similar to the values obtained with SiteAlign [12] (ROC AUC = 0.88) and BSAlign [9] (0.91). Similarly, with CDK2 as queries, we achieved average ROC AUC = 0.81 to retrieve protein kinases, which is similar to the value obtained by SiteAlign (ROC AUC = 0.76). Androgen receptor queries retrieve nuclear receptor entries with average ROC AUC = 0.92 that is also similar to the SiteAlign results (0.98).
Fig. 3.
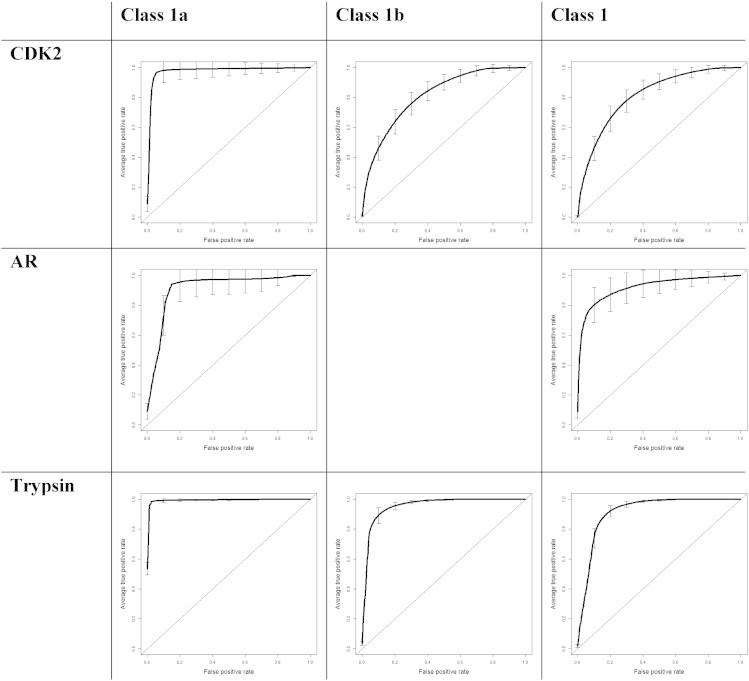
ROC for classification results. CDK2 was used as query to retrieve various protein families, such as class 1a — CDK2 entries itself, class 1b — serine–threonine protein kinases, and class 1 — protein kinases. Androgen receptor (AR) entries were used to retrieve just two classes: class 1a — androgen receptor themselves and class 1 — all nuclear receptors. Trypsin: class 1a — trypsin, class 1b — trypsin-like fold, class 1 — serine proteases. ROC AUC averaged according to true positive rate and the standard deviation boxplot was added for every curve.
Table 2.
Average ROC AUC for similarity search corresponding to setup described in Fig. 2 and Table 1. In the brackets minimum and maximum ROC AUC values are given.
| Query | Class 1a | Class 1b | Class 1 |
|---|---|---|---|
| CDK2 | 0.98 (0.58–0.99) | 0.81 (0.54–0.88) | 0.81 (0.54–0.88) |
| Androgen receptor | 0.91 (0.58–0.95) | – | 0.92 (0.68–0.96) |
| Trypsin | 0.99 (0.95–1.00) | 0.95 (0.93–0.97) | 0.92 (0.89–0.94) |
Some PL-complexes were found dissimilar to the query. Most of them represent a case of allosteric binding. For example, 2PIV (androgen receptor) as query poorly retrieves androgen receptors (ROC AUC = 0.56), because the ligand (3,5,3′-triiodothyronine) is bound not to the steroid-binding site of the receptor, but to the periphery co-activator binding site. Similar situation was detected for 2QPY, also an androgen receptor complex. Weak retrieval rate (ROC AUC = 0.58) with 3QHW used to query CDK2 and protein kinase space can be explained by errors of the semi-automatic algorithm of sc-PDB construction which mistakenly treats a small part of the protein disconnected from its main part as a ligand, thus leading to erroneous IFP and SILIRID calculations.
Discrimination power of SILIRID can be related to the difference in the binding patterns for different protein families. Fig. 4 is a median frequency distribution of the SILIRID components for trypsin and thrombin — functionally and structurally similar protein families and CDK2 — as an example of the distinct protein family both structurally and functionally. Significant difference of the component occurrences corresponding to particular interactions reflects the fact that one contribution of the same amino acid in PL binding varies as a function of protein family. For example, according to median bit count, serine is important as hydrogen bond donor for thrombin only (Fig. 4), but not for trypsin and CDK2. On the other hand, serine as H-bond acceptor is equally important for trypsin and CDK2, but not for thrombin. Leucine in turn is equally important as H-bond acceptor and H-bond donor for CDK2, but it doesn't exhibit these binding models in cases of thrombin and trypsin. CDK2 realizes hydrophobic interactions with ligands mostly via leucine, phenylalanine, lysine and isoleucine, whereas trypsin via cysteine and serine.
Fig. 4.
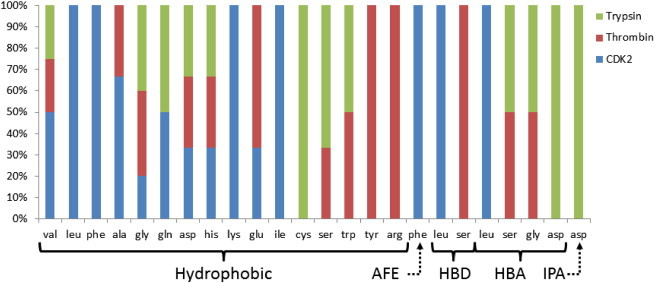
SILIRID: median vector component frequency for trypsin, thrombin and CDK2 in the form of cumulative histogram with percent. Components with zero values for all entries were removed. Components were grouped according to interaction type: hydrophobic, AFE — aromatic face to edge, HBD — hydrogen bond (protein donor), HBA — hydrogen bond (protein acceptor), IPA — ionic interaction (protein anion).
3.2. Visualization of SILIRID-based chemogenomic space using Generative Topographic Mapping
SILIRIDs can efficiently be used to visualize chemogenomic space of studied PL-complexes. For this purpose we used Generative Topographic Mapping (GTM), a probabilistic variant of the self-organizing Kohonen map, which projects the objects in N-dimensional vector space onto two-dimensional space [24,25]. GTM is an unsupervised method describing the hidden structure of data represented by SILIRID vectors. This type of study has a special interest for structure-based drug design helping to find similarities and relationships between protein families [26].
In this study, GTM has been built on the raw SILIRID descriptors for 417 serine proteases, 304 asparagine proteases, 253 phosphorylases, 241 tyrosine kinases, 488 serine/threonine kinases and 282 nuclear receptors. On the map shown in Fig. 5 nuclear receptors and proteases form distinct and non-overlapping clusters. Tyrosine and serine/threonine kinases produce highly overlapping clusters which also overlap with phosphorylases clusters; these protein families share similar functional property of transferring of phosphate group to the protein or chemical substrate. Fig. 5 clearly reveals an efficacy of SILIRIDs to encode major functional properties of different protein families.
Fig. 5.
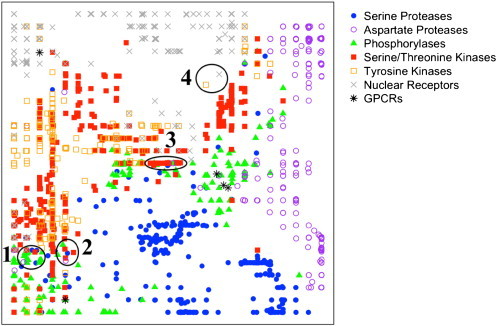
Generative Topographic Mappings of various protein families: serine and aspartate proteases, serine/threonine and tyrosine protein kinases, phosphorylases and nuclear receptors extracted from sc-PDB. Composition of zones 1–4 is given in Supplementary material and discussed in Section 3.2. The following parameters have been used: map resolution 25 × 25, radial basis function network with grid 5 × 5 and width factor 1.0, regularization coefficient 0.1.
There are, nevertheless, several zones where some of these main clusters do overlap (e.g., zones 1–4, Fig. 5). Typically, they gather PL-complexes either containing similar ligands, or representing examples of allosteric binding, or characterizing atypical binding modes (see details in Supplementary material).
3.3. Pose retrieval
The question arises whether SILIRIDs are able to retrieve correct binding poses in docking experiments. In order to investigate this question one PL-complex per protein for CDK2, androgen receptor and trypsin families (PDB IDs: 1W0X, 2AX8 and 2UUK2AX82UUK, correspondingly) was considered. In each complex, extracted ligand was re-docked with GOLD v5.1 [27] to generate 100 conformations with diverse orientation within binding site. Both IFP and SILIRID calculated for X-ray structure were compared with those calculated for each generated conformer in order to retrieve the best pose. RMSD less than 2.0 Å for a top-one scored conformer was considered as success. In these calculations, IFP displayed good retrieval rate for all proteins. On the other hand, with SILIRID the correct poses have been retrieved only for the androgen receptor. Thus, SILIRID can hardly be used to retrieve the correct binding pose, and, therefore, cannot be recommended for postprocessing of the virtual screening results, a common practice of IFP usage. This drawback of SILIRID can be explained by the fact that they, unlike IFP, implicitly describe protein–ligand interactions which, in most cases, is not sufficient to determine exact ligand binding mode.
4. Conclusions
Here, we introduced SILIRID (Simple Ligand–Receptor Interaction Descriptor), a novel fixed vector characterizing protein–ligand interactions. It can be produced from 3D structure of PL complex through the step of generation of binary interaction fingerprints. It has been demonstrated that SILIRIDs well distinguish different protein binding sites and, therefore, they can be particularly useful to map protein–ligand complexes to the functional family using similarity search or data analysis methods. SILIRID can also be used as a fast method to detect groups within collections of binding sites. The short length of SILIRID allows easy to percept visualization of the ligand–protein interactions for specific protein families as well as individual PL-complexes.
Acknowledgments
We thank Dr D. Rognan for fruitful discussions. Dr N. Kireeva is acknowledged for the help with GTM calculations.
Appendix A. Supplementary data
Supplementary material.
References
- 1.Defranchi E., De Franchi E., Schalon C., Messa M., Onofri F. Binding of protein kinase inhibitors to synapsin I inferred from pair-wise binding site similarity measurements. PLoS One. 2010;5:e12214. doi: 10.1371/journal.pone.0012214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan L., Batista J., Bajorath J. Computational methodologies for compound database searching that utilize experimental protein–ligand interaction information. Chem Biol Drug Des. 2010;76:191–200. doi: 10.1111/j.1747-0285.2010.01007.x. [DOI] [PubMed] [Google Scholar]
- 3.Rognan D. Structure-based approaches to target fishing and ligand profiling. Mol Inform. 2010;29:176–187. doi: 10.1002/minf.200900081. [DOI] [PubMed] [Google Scholar]
- 4.Das S., Kokardekar A., Breneman C.M. Rapid comparison of protein binding site surfaces with property encoded shape distributions. J Chem Inf Model. 2009;49:2863–2872. doi: 10.1021/ci900317x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das S., Krein M.P., Breneman C.M. PESDserv: a server for high-throughput comparison of protein binding site surfaces. Bioinformatics. 2010;26:1913–1914. doi: 10.1093/bioinformatics/btq288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pang B., Zhao N., Korkin D., Shyu C.-R. Fast protein binding site comparisons using visual words representation. Bioinformatics. 2012;28:1345–1352. doi: 10.1093/bioinformatics/bts138. [DOI] [PubMed] [Google Scholar]
- 7.Hoffmann B., Zaslavskiy M., Vert J.-P., Stoven V. A new protein binding pocket similarity measure based on comparison of clouds of atoms in 3D: application to ligand prediction. BMC Bioinforma. 2010;11:99. doi: 10.1186/1471-2105-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurumatani N, Monji H, Ohkawa T. Binding site extraction by similar subgraphs mining from protein molecular surfaces. Bioinformatics Bioengineering (BIBE), 2012 IEEE 12th International Conference on. (n.d.), pp. 255–259.
- 9.Aung Z., Tong J.C. BSAlign: a rapid graph-based algorithm for detecting ligand-binding sites in protein structures. Genome Inform. 2008;21:65–76. [PubMed] [Google Scholar]
- 10.Van Voorst J.R., Tong Y., Kuhn L.A. Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine. ACM; New York, NY, USA: 2012. ArtSurf: a method for deformable partial matching of protein small-molecule binding sites; pp. 36–43. [Google Scholar]
- 11.Konc J., Janežič D. ProBiS-2012: web server and web services for detection of structurally similar binding sites in proteins. Nucleic Acids Res. 2012;40:W214–W221. doi: 10.1093/nar/gks435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schalon C., Surgand J.-S., Kellenberger E., Rognan D. A simple and fuzzy method to align and compare druggable ligand-binding sites. Proteins. 2008;71:1755–1778. doi: 10.1002/prot.21858. [DOI] [PubMed] [Google Scholar]
- 13.Reisen F., Weisel M., Kriegl J.M., Schneider G. Self-organizing fuzzy graphs for structure-based comparison of protein pockets. J Proteome Res. 2010;9:6498–6510. doi: 10.1021/pr100719n. [DOI] [PubMed] [Google Scholar]
- 14.Deng Z., Chuaqui C., Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein–ligand binding interactions. J Med Chem. 2004;47:337–344. doi: 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- 15.Sato T., Honma T., Yokoyama S. Combining machine learning and pharmacophore-based interaction fingerprint for in silico screening. J Chem Inf Model. 2010;50:170–185. doi: 10.1021/ci900382e. [DOI] [PubMed] [Google Scholar]
- 16.Pérez-Nueno V.I., Rabal O., Borrell J.I., Teixidó J. APIF: a new interaction fingerprint based on atom pairs and its application to virtual screening. J Chem Inf Model. 2009;49:1245–1260. doi: 10.1021/ci900043r. [DOI] [PubMed] [Google Scholar]
- 17.Weill N., Rognan D. Alignment-free ultra-high-throughput comparison of druggable protein–ligand binding sites. J Chem Inf Model. 2010;50:123–135. doi: 10.1021/ci900349y. [DOI] [PubMed] [Google Scholar]
- 18.Chupakhin V., Marcou G., Baskin I., Varnek A., Rognan D. Predicting ligand binding modes from neural networks trained on protein–ligand interaction fingerprints. J Chem Inf Model. 2013;53:763–772. doi: 10.1021/ci300200r. [DOI] [PubMed] [Google Scholar]
- 19.Marcou G., Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J Chem Inf Model. 2007;47:195–207. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- 20.Meslamani J., Rognan D., Kellenberger E. sc-PDB: a database for identifying variations and multiplicity of “druggable” binding sites in proteins. Bioinformatics. 2011;27:1324–1326. doi: 10.1093/bioinformatics/btr120. [DOI] [PubMed] [Google Scholar]
- 21.Oksanen J., Blanchet F.G., Kindt R., Legendre P., Minchin P.R. 2012. Vegan: community ecology package. [Google Scholar]
- 22.Sing T., Sander O., Beerenwinkel N., Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 23.R Development Core Team R: a language and environment for statistical computing. 2012. http://www.r-project.org Available:
- 24.Kireeva N., Baskin I.I., Gaspar H.A., Horvath D., Marcou G. Generative Topographic Mapping (GTM): universal tool for data visualization, structure–activity modeling and dataset comparison. Mol Inform. 2012;31:301–312. doi: 10.1002/minf.201100163. [DOI] [PubMed] [Google Scholar]
- 25.Gaspar H.A., Marcou G., Arault A., Lozano S., Vayer P., Varnek A. GTM-based classification models and their applicability domain: application to the Biopharmaceutics Drug Disposition Classification System (BDDCS) J Chem Inf Model. 2013;53:763–772. doi: 10.1021/ci400423c. [DOI] [PubMed] [Google Scholar]
- 26.Lounnas V., Ritschel T., Kelder J., McGuire R., Bywater R.P. Current progress in structure-based rational drug design marks a new mindset in drug discovery. Comput Struct Biotechnol J. 2013;5 doi: 10.5936/csbj.201302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones G., Willett P., Glen R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.