

Abstract
Background
Early and accurate identification of adverse drug reactions (ADRs) is critically important for drug development and clinical safety. Computer-aided prediction of ADRs has attracted increasing attention in recent years, and many computational models have been proposed. However, because of the lack of systematic analysis and comparison of the different computational models, there remain limitations in designing more effective algorithms and selecting more useful features. There is therefore an urgent need to review and analyze previous computation models to obtain general conclusions that can provide useful guidance to construct more effective computational models to predict ADRs.
Principal Findings
In the current study, the main work is to compare and analyze the performance of existing computational methods to predict ADRs, by implementing and evaluating additional algorithms that have been earlier used for predicting drug targets. Our results indicated that topological and intrinsic features were complementary to an extent and the Jaccard coefficient had an important and general effect on the prediction of drug-ADR associations. By comparing the structure of each algorithm, final formulas of these algorithms were all converted to linear model in form, based on this finding we propose a new algorithm called the general weighted profile method and it yielded the best overall performance among the algorithms investigated in this paper.
Conclusion
Several meaningful conclusions and useful findings regarding the prediction of ADRs are provided for selecting optimal features and algorithms.
Introduction
Early and accurate identification of ADRs is critically important for drug development and clinical safety. Traditional clinical trials to recognize ADRs are expensive and time-consuming. Conversely, computer-aided methods for predicting ADRs are much cheaper and quicker than clinical trials and highly reliable [1]–[3].
Constructing machine learning models by combining intrinsic features of drugs and ADRs with topological features of drug-ADR association networks has been one of typical computer-aided methods for predicting ADRs [4], [5]. However, many other state-of-the-art methods have been proposed to predict drug targets [6]–[15]. Computer-aided prediction of drug targets is similar to prediction of ADRs: there are close relationships between ADRs and drug targets that have been identified in biological systems [16], [17]. In addition, in terms of mathematics, the prediction of ADRs and drug targets can both be abstracted into link prediction models on a bipartite network; therefore most of the computational processing steps are similar between these two systems. We therefore hypothesize these series of state-of-the-art methods, which have been successfully applied in the prediction of drug targets, could also achieve excellent performance in the prediction of ADRs. Our results also support this hypothesis indirectly. Hence, in recent years, many computational methods have been proposed to predict ADRs or drug targets, whereas less attention has been paid to compare and analyze existing computational methods and features. Here, we summarize the existing computation methods and features that have been proposed, extract classical methods and features to construct different representative computational models for predicting ADRs, and compare and analyze these methods and features. Finally, useful findings are provided for searching optimal features, appropriate algorithms for predicting ADRs. A brief illustration of the main workflow in this paper is shown in Figure 1 .
Figure 1. Overview of the main workflow in this paper.

First, data were integrated from multiple sources, including network data (drug-ADR associations) and intrinsic data (chemical structures and ATC taxonomies of drugs and MedDRA taxonomies of ADRs). Next, topological features and intrinsic features were constructed based on network data and intrinsic data, respectively, and then integrated features were constructed by integrating topological features with intrinsic features, Finally different algorithms were selected to construct models to predict ADR, and comparative analyses were performed for features, algorithms and prediction results based on modeling experiments.
Materials and Methods
Materials
In this paper, two drug-ADR association networks were constructed; one was called the training network, and the other was called the testing network.
To construct the training network, drug data were collected from the following databases: DrugBank [18], Kegg [19], FDA Adverse Event Reporting System (FAERS, website: www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/default.htm) of 2005, and SIDER [20]. To reduce the proportion of false positives in drug-ADR associations from SIDER and FAERS (in 2005), an interacting drug-ADR pair was taken only when the drug-ADR pair was recorded in both databases. In addition, according to the Medical Dictionary for Regulatory Activities (MedDRA) [21], ADRs can be divided into five different levels: the System outraged Class (SOC), the High Level Group Term (HLGT), the High Level Term (HLT), the Preferred Term (PT), and the Lowest Level Term (LLT). Here, only ADRs in the HLT Level were considered; therefore, ADRs recorded in FAERS and SIDER that belonged to PT or LLT were first mapped to HLT.
We obtained the testing network by adding drug-ADR associations recorded in both FAERS and SIDER from 2006 to 2011 to the training network. Finally, the network node sets (consisting of drug nodes and ADR nodes) were identical in the training and testing networks, whereas the network edge sets (interacting drug-ADR pairs) were different. The related quantitative statistics of the drug-ADR networks are provided in Table 1 and Figure 2 .
Table 1. Statistics for the drug-ADE networks.
| Statistics | Train drug-ADE network | Test drug-ADE network |
| Number of drugs | 404 | 404 |
| Number of ADEs | 461 | 461 |
| Number of drug-ADE associations | 9180 | 19182 |
| Average degree of drugs | 22.7 | 47.5 |
| Average degree of ADEs | 19.9 | 41.6 |
Figure 2. Degree distributions of drugs and ADRs.

The left panel depicts the histograms of the degrees of drugs. The right panel depicts the histograms of the degrees of ADRs.
Problem formalization
The problem of predicting ADRs of drugs can be abstracted to the problem of predicting new interactions in a drug-ADR association network. Formally,  and
and  represent a set of the drug nodes and ADRs nodes in a drug-ADR association network, respectively, and the edges in the network represent interacting drug-ADR pairs. Furthermore, this bipartite network can be characterized as an
represent a set of the drug nodes and ADRs nodes in a drug-ADR association network, respectively, and the edges in the network represent interacting drug-ADR pairs. Furthermore, this bipartite network can be characterized as an  adjacency matrix Y. That is,
adjacency matrix Y. That is,  if an existing association is previously known between
if an existing association is previously known between  and
and  , and
, and  otherwise. In addition, to make it more convenient for later description, the set of prediction scores for each drug-ADR pair are characterized as an
otherwise. In addition, to make it more convenient for later description, the set of prediction scores for each drug-ADR pair are characterized as an  matrix
matrix  , where the element
, where the element  represents the prediction score of the drug-ADR pair
represents the prediction score of the drug-ADR pair  . The set of similarity scores of drugs and similarity scores of ADRs are characterized as an
. The set of similarity scores of drugs and similarity scores of ADRs are characterized as an  similarity matrix
similarity matrix  and an
and an  similarity matrix
similarity matrix  , respectively. The elements
, respectively. The elements  and
and  represent the similarities of the drug-drug pair
represent the similarities of the drug-drug pair  and the ADR-ADR pair
and the ADR-ADR pair  , respectively. One of main tasks in this paper was to compute the prediction score of each non-interacting drug-ADR pair
, respectively. One of main tasks in this paper was to compute the prediction score of each non-interacting drug-ADR pair  and then to determine whether an association between
and then to determine whether an association between  and
and  existed using the prediction score of the drug-ADR pair
existed using the prediction score of the drug-ADR pair  .
.
Model features
Features of drugs or ADRs in this paper were used to characterize the similarity of drugs or ADRs. Here, intrinsic features and topological features of drugs' and ADRs were employed.
Topological feature
To extensively investigate the effect of topological features on computational models for predicting ADRs, six common topological features and a new topological feature designed by us were employed to characterize the similarity of drugs or ADRs.
Jaccard coefficient (denoted
 ):
):  . Here,
. Here,  and
and  represent the neighborhood set of homology nodes x and y, respectively. In drug-ADR association network, there are two classes of nodes (drug nodes or ADR nodes). Therefore, the relationship of any two drug (ADR) nodes is homologous, while, the relationship between a drug node and an ADR node is heterologous, here, if two nodes both belong to drug or ADR nodes, we call them as homology nodes. In addition, the symbol
represent the neighborhood set of homology nodes x and y, respectively. In drug-ADR association network, there are two classes of nodes (drug nodes or ADR nodes). Therefore, the relationship of any two drug (ADR) nodes is homologous, while, the relationship between a drug node and an ADR node is heterologous, here, if two nodes both belong to drug or ADR nodes, we call them as homology nodes. In addition, the symbol  represents the number of elements in a set.
represents the number of elements in a set.Gaussian interaction profile kernel (denoted
 ): this feature is proposed in by the scholar Laarhoven and has been successfully applied to predict drug-target interactions [12].
): this feature is proposed in by the scholar Laarhoven and has been successfully applied to predict drug-target interactions [12].A topological feature proposed by the scholar Allali [22] (denoted
 ):
):  ;
;Neighbors Product (denoted
 ):
):  ;
;Common Neighbors (denoted
 ):
):  ;
;A feature proposed by the scholar L.A.Adamic [23] (denoted
 ):
):  ;
;A new feature is designed by us, which is similar to
 (denoted
(denoted  ):
):  ;
;
Intrinsic features
The intrinsic features were obtained from chemical structures or biological functions of drugs or ADRs. The intrinsic features of drugs were based on chemical structures and the ATC taxonomy of drugs [24], [25], and the intrinsic features of ADRs were based on the MedDRA taxonomy of ADRs. The chemical similarities between drugs were computed using SIMCOMP [26], and the ATC taxonomy similarities between drugs and the MedDRA taxonomy similarities between ADRs were both computed using the semantic similarity algorithm [5], [11], [27].
Classification algorithm
There are many state-of-the-art methods to predict drug targets. In this study, we selected the regularized least – squares classifier, semi-supervised link prediction classifier and the nearest – neighbor classifier from these existing methods to predict ADRs. There are several justifications for this selection. The performance of methods [7], [8], [9], [12], [13], [14] have been tested on a same dataset [7], the performance of method [12] based on the regularized least – squares and method [9] based on the semi-supervised link prediction was competitive with others, especially, method [12] yielded the highest performance among these methods. On the other hand, regularized least – squares classifier, semi-supervised link prediction classifier and the nearest – neighbor classifier belong to supervised learning, semi-supervised learning and memory-based algorithm, respectively, therefore, these three classifiers were representative of different classes of algorithms among existing methods. We briefly discuss these algorithms below.
RLS
The Regularized Least-Squares classifier (denoted RLS) [12], [28] is a basic supervised learning algorithm. If an appropriate kernel has been chosen for RLS, the accuracy of RLS will be similar to support vector machine (SVM), whereas the computation complexity of RLS is much less than SVM. The RLS algorithm can be divided into three separate sub algorithms for defining the kernel matrix: RLS-KP, RLS-KS and RLS-avg. Here, KP and KS are short for Kronecker Product [25], [29] and Kronecker Sum [29], respectively.
SLP
Semi-supervised Link Prediction classifier (denoted SLP) is a semi-supervised learning algorithm [9], [30], and the basic assumption of SLP is “Two node pairs that are similar to each other are likely to have the same link strength” [30]. Based on this assumption, the objective function is defined as:
where  is a regularization parameter and
is a regularization parameter and  is a Laplacian matrix. SLP also can be divided into three independent sub algorithms for defining
is a Laplacian matrix. SLP also can be divided into three independent sub algorithms for defining  : SLP-KP, SLP-KS and SLP-avg.
: SLP-KP, SLP-KS and SLP-avg.
NN
The Nearest-Neighbor classifier (denoted NN) is a simple memory-based algorithm (more detailed descriptions regarding algorithms are provided in File S1).
Results and Discussion
Evaluation
Ten-fold cross validation and prospective evaluation were used to evaluate the performance of each model. For ten-fold cross validation, interacting drug-ADR pairs and non-interacting drug-ADR pairs were each randomly divided into ten folds of roughly equal size; in each run of the method, one fold of interacting drug-ADR pairs and one fold of non-interacting drug-ADR pairs were left out by setting their entries in adjacency matrix Y to 0. We then attempted to recover their true labels using the remaining data. Note that the Y matrix corresponds to the training network. For prospective evaluation, the training data consisted of the training network and the validation data consisted of all the testing network drug-ADR pairs that were non-edges in the training network. We attempted to recover the true labels of the validation using the training network.
We assessed the model performance with the following two common quantitative indexes: AUC [31] and AUPR [32]. The value of AUC is determined from the area below a curve relating the proportion of true positives versus the proportion of false positives, whereas the value of AUPR is determined from the area below a curve relating precision versus recall.
Feature analysis
In this paper, two types of features (topological features and intrinsic features) were employed in the modeling experiment. To comprehensively analyze these features, associations between features were first investigated, and then the performances of models constructed using only intrinsic features or topological features were tested, and lastly, the performances of models constructed with integrated features were evaluated.
Associations between features
Here, Pearson correlation coefficients among drug or ADR features were calculated separately. The detailed results are listed in Table S1 and Table S2. The Pearson coefficients among drug and ADR features were consistent. The intrinsic features of drugs or ADRs were not significantly correlated with topological features of drugs or ADRs, indicating that the information of topological features and intrinsic features may be complementarily used in a prediction model.
Modeling with intrinsic features
Within the intrinsic features, the chemical similarity of drugs, ATC similarity of drugs and MedDRA similarity of ADRs were denoted by  ,
,  and
and  , respectively. The Pearson coefficient between
, respectively. The Pearson coefficient between  and
and  was 0.0905, indicating no significant association between
was 0.0905, indicating no significant association between  and
and  . Here,
. Here,  is defined by integrating
is defined by integrating  with
with  as follows:
as follows:  , where
, where  ; and
; and  . In the modeling experiments, ten-fold cross validation and the Grid Search Method [33] were used to obtain the optimal value of
. In the modeling experiments, ten-fold cross validation and the Grid Search Method [33] were used to obtain the optimal value of  . The detailed results are listed in Table S3 and Table S4: when
. The detailed results are listed in Table S3 and Table S4: when  , the model achieved slight better overall performance than other models.
, the model achieved slight better overall performance than other models.
Modeling with topological features
The process of modeling with topological features was similar to as with intrinsic features. Here, seven topological features were respectively used to construct models. The detailed results are listed in
Table 2
and
Table 3
. Almost all models built with the topological feature  yielded good performance (except SLP-KP). Hence, compared with the other six topological features,
yielded good performance (except SLP-KP). Hence, compared with the other six topological features,  has the most important and general effect on predicting drug-ADR associations.
has the most important and general effect on predicting drug-ADR associations.
Table 2. AUC scores of the models built with different topological features.
| AUC | |||||||

|

|

|

|

|

|

|
|
| RLS-KP | 91.1(0.1) | 92.7(0.1) | 80.9(0.1) | 90.2(0.1) | 50.8(0.3) | 48.8(0.5) | 50.5(0.7) |
| RLS-KS | 92.1(0.1) | 93.3(0.1) | 89(0.1) | 91.2(0.1) | 68.4(0.2) | 54.4(0.2) | 50.4(1.0) |
| RLS-avg | 93.1(0.2) | 92.7(0.1) | 91.9(0.2) | 91.1(<0.1) | 72.6(0.2) | 63.5(0.4) | 52.2(3.0) |
| SLP-KP | 43.2(2.4) | 48.6(1.4) | 48.3(5.6) | 9.8(0.1) | 49.5(1.1) | 49.7(0.8) | 49.5(0.4) |
| SLP-KS | 91.1(<0.1) | 34.4(0.1) | 30.9(4.5) | 8.7(0.1) | 49(3.7) | 49.7(2.1) | 85.9(3.0) |
| SLP-avg | 93.1(<0.1) | 90.7(<0.1) | 93.2(<0.1) | 91.9(<0.1) | 92.7(<0.1) | 92.6(<0.1) | 93.1(0.1) |
| NN | 92.1(0.1) | 36.2(0.1) | 90.9(0.1) | 89.4(0.1) | 89.9(<0.1) | 89.7(<0.1) | 91.3(0.8) |
| GWPM | 93.0(0.1) | 90.7(<0.1) | 93.0(<0.1) | 91.0(0.1) | 92.9(0.1) | 92.8(0.1) | 93.6(<0.1) |
Determined from ten-fold cross validation experiments. The AUC scores are normalized to 100.
Table 3. AUPR scores of the models built with different topological features.
| AUPR | |||||||

|

|

|

|

|

|

|
|
| RLS-KP | 63.5(0.2) | 60.4(0.2) | 38.5(0.3) | 53.1(0.1) | 5.9(0.1) | 4.7(<0.1) | 5.3(0.3) |
| RLS-KS | 63.6(0.1) | 58.7(0.1) | 45.6(0.2) | 52.8(0.1) | 10.5(0.1) | 6(0.1) | 5.8(0.2) |
| RLS-avg | 63.4(0.1) | 58.4(0.1) | 59(0.2) | 53.2(<0.1) | 14.2(0.1) | 7.3(0.1) | 7.2(0.9) |
| SLP-KP | 4.9(0.5) | 4.1(0.5) | 20.5(5.3) | 3.6(2.2) | 6.3(0.2) | 5.5(0.2) | 4.9(0.4) |
| SLP-KS | 51.7(0.1) | 3.9(0.7) | 10(0.4) | 2.6(<0.1) | 10.1(1.5) | 7.3(0.6) | 26.9(5.4) |
| SLP-avg | 57.7(0.1) | 30.1(<0.1) | 57.4(0.1) | 53.5(<0.1) | 56.1(0.1) | 55.9(0.1) | 58.1(0.1) |
| NN | 52.1(0.2) | 4.1(<0.1) | 41.7(0.1) | 36.4(0.1) | 39.5(0.1) | 39.1(0.2) | 51.9(0.2) |
| GWPM | 60.0(0.1) | 30.9(<0.1) | 59.5(0.1) | 53.2(0.1) | 58.1(0.1) | 57.4(0.1) | 65.4(0.2) |
Determined from ten-fold cross validation experiments. The AUPR scores are normalized to 100.
Modeling with integrated features
Here, the features that integrate topological features with intrinsic features were further investigated. The intrinsic similarity matrices of drugs and ADRs were defined as  and
and  , respectively (
, respectively ( ;
;  ). The integrated features were as follows:
). The integrated features were as follows: 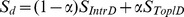 ;
;  ; where
; where  ,
,  , and the topological features of drugs and ADRs were denoted as
, and the topological features of drugs and ADRs were denoted as  and
and  , respectively. In the modeling experiments, ten-fold cross validation and the Grid Search Method were used to obtain the optimal values of
, respectively. In the modeling experiments, ten-fold cross validation and the Grid Search Method were used to obtain the optimal values of  and
and  for each integrated feature. The detailed results are delineated in Table S5, Table S6, Table S7 and Table S8. Compared with models constructed with intrinsic or topological features separately, models constructed with integrated features yielded better performance; that is, the information of intrinsic features and topological features was complementary.
for each integrated feature. The detailed results are delineated in Table S5, Table S6, Table S7 and Table S8. Compared with models constructed with intrinsic or topological features separately, models constructed with integrated features yielded better performance; that is, the information of intrinsic features and topological features was complementary.
Algorithm analysis
According to the above results, the best performance of models was obtained from RLS-avg with an optimal integrated feature that integrated  with intrinsic features of drugs and ADRs ([AUC,AUPR] = [0.933,0.635]). While, models constructed using SLP-avg with either intrinsic features or topological features of drugs and ADRs all yielded excellent performance. Therefore, among these seven sub algorithms, models constructed using SLP-avg yielded the best overall performance, demonstrating that SLP-avg is a more general algorithm for predicting drug-ADR associations.
with intrinsic features of drugs and ADRs ([AUC,AUPR] = [0.933,0.635]). While, models constructed using SLP-avg with either intrinsic features or topological features of drugs and ADRs all yielded excellent performance. Therefore, among these seven sub algorithms, models constructed using SLP-avg yielded the best overall performance, demonstrating that SLP-avg is a more general algorithm for predicting drug-ADR associations.
By comparative analysis of the structure of each algorithms, final formulas of these algorithms could be unified as:  or
or  ; here,
; here,  , where
, where  is a function of the similarity matrices
is a function of the similarity matrices  and
and  , and
, and  is a symmetric matrix. More detailed descriptions of the unified formulas are provided in the File S2. For unify formulas,
is a symmetric matrix. More detailed descriptions of the unified formulas are provided in the File S2. For unify formulas,  was considered as a similarity matrix of drug-ADR pairs, therefore, all models in this paper can be converted to simple linear models, and the major difference between these models occurs in methods regarding the construction of
was considered as a similarity matrix of drug-ADR pairs, therefore, all models in this paper can be converted to simple linear models, and the major difference between these models occurs in methods regarding the construction of  . Based on the above analysis, we attempted a simple general linear method to construct
. Based on the above analysis, we attempted a simple general linear method to construct  and then designed a simple algorithm called general weighted profile method (denoted GWPM, a more detail description of GWPM is provided in File S1) And the performance of this algorithm of prediction ADRs by ten-fold cross validation was shown in
Table 2
,
Table 3
, Tables S5, Tables S6, Tables S7 and Tables S8. Although the computation complexity of GWPM is relatively lower than other algorithms (except NN), the overall performances of models constructed using GWPM was even better than SLP-avg, especially, the model constructed using GWPM with the optimal integrated feature integrating
and then designed a simple algorithm called general weighted profile method (denoted GWPM, a more detail description of GWPM is provided in File S1) And the performance of this algorithm of prediction ADRs by ten-fold cross validation was shown in
Table 2
,
Table 3
, Tables S5, Tables S6, Tables S7 and Tables S8. Although the computation complexity of GWPM is relatively lower than other algorithms (except NN), the overall performances of models constructed using GWPM was even better than SLP-avg, especially, the model constructed using GWPM with the optimal integrated feature integrating  with intrinsic features yielded the best performance ([AUC, AUPR] = [0.942, 0.657]) among all test models in this paper. Hence, finding a good method for constructing
with intrinsic features yielded the best performance ([AUC, AUPR] = [0.942, 0.657]) among all test models in this paper. Hence, finding a good method for constructing  (which is equivalent to finding a proper mapping function from drug and ADR space to drug-ADR pair space) is the key to predicting of drug-ADR associations.
(which is equivalent to finding a proper mapping function from drug and ADR space to drug-ADR pair space) is the key to predicting of drug-ADR associations.
Statistical analysis of model predictions
According to the above results regarding model performance based on ten-fold cross validation, models were rebuilt by each algorithm with the optimal feature and then validated by prospective evaluation. For RLS and SLP, we selected one sub algorithm among the three sub algorithms (RLS-avg and SLP-avg, respectively), and the detailed results are presented in Table 4 . The associations between prediction scores of drug-ADR pairs and degrees of drugs or ADRs were also investigated. If the degree of drug or ADR was more than 40 in the training network, then the drug or ADR was considered as a high degree drug or ADR; otherwise, was considered as a low degree drug or ADR. Hence, all drug-ADR pairs were divided into four types: low degree drug- low degree ADR pair, high degree drug- low degree ADR pair, low degree drug- high degree ADR and high degree drug- high degree ADR. The prediction score distribution of these four type drug-ADR pairs is shown in Figure 3 . Drug-ADR pairs that had known interactions in the training network were not recorded in the prediction score distribution. According to Figure 3 , the prediction scores of drug-ADR pairs and degrees of drugs or ADRs displayed positive correlations, indicating that the interaction between drug-ADR pairs containing high degree drugs or ADRs were more likely to be predicted correctly by models. Each model has limited ability to predict low degree drug- low degree ADR associations. On one hand, this result demonstrated the limitation of topological features; on the other hand, although integrated features have integrated topological and intrinsic features, the limitation of topological features was not compensated sufficiently well by intrinsic features. Therefore, more effective intrinsic features of drugs and ADRs still require further investigation to improve the model prediction performance.
Table 4. The performances of the optimal models validated by prospective evaluation.
| Weight coefficient | Model performance | ||||
| Algorithm | feature |

|

|
AUC | AUPR |
| RLS-avg |

|
0.9 | 1 | 79.2 | 25.5 |
| SLP-avg |

|
0.9 | 0.7 | 85.1 | 26.4 |
| NN |

|
0.7 | 1 | 83.1 | 26.1 |
| GWPM |

|
0.8 | 0.8 | 82.7 | 26.9 |
Here,  indicates the weight coefficient of drug topological features,
indicates the weight coefficient of drug topological features,  indicates the weight coefficient of ADR topological features; The AUC and AUPR scores are normalized to 100.
indicates the weight coefficient of ADR topological features; The AUC and AUPR scores are normalized to 100.
Figure 3. Distribution of prediction scores for different types of drug-ADR pairs.

The histograms of distributions of prediction scores of models built by four algorithms are shown. In each sub panel, the blue, green, yellow and red histograms represent the distributions of prediction scores for low degree drug- low degree ADRs, high degree drug- low degree ADRs, low degree drug- high degree ADRs and high degree drug- high degree ADRs, respectively.
Comparative with other existing ADR prediction literature
We are aware of only a few other studies that attempts to predict unknown likely ADRs through combining intrinsic and topological features methods [4], [5]. The study [5] and the current study are similar in that they both integrate various types of information to predict unknown likely ADRs, and conclusions about various features are consistent. The data and methods used by the two studies differ in several ways. In current study, drug-ADR associations were extracted from following databases: FAERS and SIDER, and to reduce false positives in drug-ADR associations, a drug-ADR pair was taken only when it was recorded in both databases. While, in study [5], drug-ADR associations were mainly extracted from a proprietary commercial database widely used in hospitals today, provided by Lexicomp (http://www.lexi.com). Perhaps the most important distinction between these two studies lies in computational methods for predicting ADRs. Seven different methods were used in current paper (six methods had been used for predicting drug targets before, and one methods proposed by ourselves) and a systematic comparative analysis is conducted in terms of performance of these methods, finally, some general conclusion regarding algorithms and features is obtained, such as, the feature Jaccard coefficient had an important and general effect on the prediction of drug-ADR associations, final formulas of algorithms selected in current study were all converted to linear model in form. Compared with [5], which only used a logistic regression predictive model. In order to facilitate benchmark comparisons between methods in two studies, we tested the performance of the method used in study [5] on data sets used in current paper, and performance evaluated by ten-fold cross validation and prospective evaluation are [AUC, AUPR] = [0.927, 0.616] and [AUC, AUPR] = [0.793, 0.249], respectively. While, in the current paper, for example, the best performance of GWPM evaluated by ten-fold cross validation and prospective evaluation are [AUC, AUPR] = [0.942, 0.657] and [AUC, AUPR] = [0.827, 0.269], respectively. The results showed performance of methods used in current study was competitive with the study [5].
Conclusions
In this paper, three typical algorithms and a new algorithm combining ten features were used to construct models to predict new drug-ADR associations. Different algorithms, features and prediction results were compared and analyzed respectively. Finally, several meaningful conclusions were drawn as follows:
Seven topological features and three intrinsic features of drugs or ADRs were analyzed in this paper. Among these seven topological features  had the most important and general effect on the prediction of drug-ADR associations. In addition, models built using integrated features had better performance than using only topological or intrinsic features, demonstrating that topological and intrinsic features were complementary. However, for rare ADRs (only a few drugs have been currently validated to have these ADRs), models built with integrated features did not correctly predict associations between these ADRs and drugs. Therefore, more effective intrinsic features of drugs and ADRs still require further investigation.
had the most important and general effect on the prediction of drug-ADR associations. In addition, models built using integrated features had better performance than using only topological or intrinsic features, demonstrating that topological and intrinsic features were complementary. However, for rare ADRs (only a few drugs have been currently validated to have these ADRs), models built with integrated features did not correctly predict associations between these ADRs and drugs. Therefore, more effective intrinsic features of drugs and ADRs still require further investigation.
GWPM yielded the best overall performance among all algorithms in this paper as determined from ten-fold cross validation. Additionally, because all algorithms have unified linear formulas, finding an optimal method for constructing the similarity coefficient matrix in the linear formula will be useful to improve accuracy of predicting drug-ADR associations.
Supporting Information
Associations between drug feature covariates.
(DOC)
Associations between ADR feature covariates.
(DOC)
AUC scores of models built with different intrinsic features.
(DOC)
AUPR scores of models built with different intrinsic features.
(DOC)
AUC scores of models built with optimal integrated features.
(DOC)
AUPR scores of models built with optimal integrated features.
(DOC)
Drug features weight coefficients of models built with optimal integrated features.
(DOC)
ADR features coefficients of models built with optimal integrated features.
(DOC)
Supplementary Algorithms. More detailed descriptions regarding algorithms have been provided in this file.
(DOC)
Supplementary Algorithm Analysis. More detailed descriptions of the unified formulas of algorithms have been provided in this file.
(DOC)
MATLAB source code. This file includes MATLAB resource code to implement computational experiments in this paper.
(PDF)
This file lists all drug-ADR association pairs in the training network.
(XLSX)
This file lists all drug-ADR association pairs in the testing network.
(XLSX)
This file lists the chemical structure similarity score of each drug-drug pair.
(TXT)
This file lists the ATC similarity score of each drug-drug pair.
(TXT)
This file lists the MedDRA similarity score of each ADR-ADR pair.
(TXT)
Funding Statement
This work was supported by the National Natural Science Foundation of China (21375095) and the National Natural Science Foundation of China (21305096). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Huang L-C, Wu X, Chen JY (2011) Predicting adverse side effects of drugs. Bmc Genomics 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu M, Wu Y, Chen Y, Sun J, Zhao Z, et al. (2012) Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. Journal Of the American Medical Informatics Association 19: E28–E35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lin J, Kuang Q, Li Y, Zhang Y, Sun J, et al. (2013) Prediction of adverse drug reactions by a network based external link prediction method. Analytical Methods 5: 6120–6127. [Google Scholar]
- 4. Atias N, Sharan R (2011) An Algorithmic Framework for Predicting Side Effects of Drugs. Journal Of Computational Biology 18: 207–218. [DOI] [PubMed] [Google Scholar]
- 5. Cami A, Arnold A, Manzi S, Reis B (2011) Predicting Adverse Drug Events Using Pharmacological Network Models. Science Translational Medicine 3. [DOI] [PubMed] [Google Scholar]
- 6. Yamanishi Y (2008) Supervised bipartite graph inference. 1841–1848. [Google Scholar]
- 7. Yamanishi Y, Araki M, Gutteridge A, Honda W, Kanehisa M (2008) Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 24: I232–I240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bleakley K, Yamanishi Y (2009) Supervised prediction of drug-target interactions using bipartite local models. Bioinformatics 25: 2397–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xia Z, Wu L-Y, Zhou X, Wong STC (2010) Semi-supervised drug-protein interaction prediction from heterogeneous biological spaces. Bmc Systems Biology 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamanishi Y, Kotera M, Kanehisa M, Goto S (2010) Drug-target interaction prediction from chemical, genomic and pharmacological data in an integrated framework. Bioinformatics 26: i246–i254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao S, Li S (2010) Network-Based Relating Pharmacological and Genomic Spaces for Drug Target Identification. Plos One 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Laarhoven T, Nabuurs SB, Marchiori E (2011) Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 27: 3036–3043. [DOI] [PubMed] [Google Scholar]
- 13. Cheng F, Liu C, Jiang J, Lu W, Li W, et al. (2012) Prediction of Drug-Target Interactions and Drug Repositioning via Network-Based Inference. Plos Computational Biology 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X, Liu M-X, Yan G-Y (2012) Drug-target interaction prediction by random walk on the heterogeneous network. Molecular Biosystems 8: 1970–1978. [DOI] [PubMed] [Google Scholar]
- 15. Tabei Y, Pauwels E, Stoven V, Takemoto K, Yamanishi Y (2012) Identification of chemogenomic features from drug-target interaction networks using interpretable classifiers. Bioinformatics 28: I487–I494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Campillos M, Kuhn M, Gavin A-C, Jensen LJ, Bork P (2008) Drug target identification using side-effect similarity. Science 321: 263–266. [DOI] [PubMed] [Google Scholar]
- 17. Brouwers L, Iskar M, Zeller G, van Noort V, Bork P (2011) Network Neighbors of Drug Targets Contribute to Drug Side-Effect Similarity. Plos One 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, et al. (2008) DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic acids research 36: D901–D906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P (2010) A side effect resource to capture phenotypic effects of drugs. Molecular systems biology 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brown EG, Wood L, Wood S (1999) The medical dictionary for regulatory activities (MedDRA). Drug Safety 20: 109–117. [DOI] [PubMed] [Google Scholar]
- 22. Allali O, Magnien C, Latapy M Link prediction in bipartite graphs using internal links and weighted projection; 2011. IEEE 936–941. [Google Scholar]
- 23. Adamic L, Buyukkokten O, Adar E (2003) A social network caught in the web. First Monday 8. [Google Scholar]
- 24. Sketris IS, Metge CJ, Ross JL, MacCara ME, Comeau DG, et al. (2004) The Use of the World Health Organisation Anatomical Therapeutic Chemical/Defined Daily Dose Methodology in Canada*. Drug information journal 38: 15–27. [Google Scholar]
- 25. Wang YC, Deng N, Chen S, Wang Y (2013) Computational Study of Drugs by Integrating Omics Data with Kernel Methods. Molecular Informatics 32: 930–941. [DOI] [PubMed] [Google Scholar]
- 26. Hattori M, Okuno Y, Goto S, Kanehisa M (2003) Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. Journal of the American Chemical Society 125: 11853–11865. [DOI] [PubMed] [Google Scholar]
- 27. Lin D (1998) An information-theoretic definition of similarity. Machine Learning Proceedings of the Fifteenth International Conference (ICML'98) 296–304. [Google Scholar]
- 28. Rifkin R, Yeo G, Poggio T (2003) Regularized least-squares classification. Nato Science Series Sub Series III Computer and Systems Sciences 190: 131–154. [Google Scholar]
- 29. Laub AJ (2005) Matrix analysis for scientists and engineers: Siam. [Google Scholar]
- 30.Raymond R, Kashima H (2010) Fast and Scalable Algorithms for Semi-supervised Link Prediction on Static and Dynamic Graphs. In: Balcazar JL, Bonchi F, Gionis A, Sebag M, editors. Machine Learning And Knowledge Discovery In Databases, Pt Iii. pp. 131–147. [Google Scholar]
- 31. Fawcett T (2006) An introduction to ROC analysis. Pattern recognition letters 27: 861–874. [Google Scholar]
- 32. Raghavan V, Bollmann P, Jung GS (1989) A critical investigation of recall and precision as measures of retrieval system performance. ACM Transactions on Information Systems (TOIS) 7: 205–229. [Google Scholar]
- 33. Cheung B-S, Langevin A, Delmaire H (1997) Coupling genetic algorithm with a grid search method to solve mixed integer nonlinear programming problems. Computers & Mathematics with Applications 34: 13–23. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Associations between drug feature covariates.
(DOC)
Associations between ADR feature covariates.
(DOC)
AUC scores of models built with different intrinsic features.
(DOC)
AUPR scores of models built with different intrinsic features.
(DOC)
AUC scores of models built with optimal integrated features.
(DOC)
AUPR scores of models built with optimal integrated features.
(DOC)
Drug features weight coefficients of models built with optimal integrated features.
(DOC)
ADR features coefficients of models built with optimal integrated features.
(DOC)
Supplementary Algorithms. More detailed descriptions regarding algorithms have been provided in this file.
(DOC)
Supplementary Algorithm Analysis. More detailed descriptions of the unified formulas of algorithms have been provided in this file.
(DOC)
MATLAB source code. This file includes MATLAB resource code to implement computational experiments in this paper.
(PDF)
This file lists all drug-ADR association pairs in the training network.
(XLSX)
This file lists all drug-ADR association pairs in the testing network.
(XLSX)
This file lists the chemical structure similarity score of each drug-drug pair.
(TXT)
This file lists the ATC similarity score of each drug-drug pair.
(TXT)
This file lists the MedDRA similarity score of each ADR-ADR pair.
(TXT)