

Abstract
Cisplatin (CP) is a commonly used anticancer drug, but its notable side effect of nephrotoxicity limits its use in clinic. Epigallocatechin-3-gallate (EGCG), an anti-oxidant, anti-inflammatory, and anti-tumorigenic green tea polyphenol, has been available on the market for its beneficial effects. The aim of this study was to investigate whether EGCG can prevent the nephrotoxic effect of CP and the involved mechanisms. Male C57/BL6 mice were randomly divided into four groups: control group, EGCG group, CP group, and CP+EGCG group. On day 5, mice were sacrificed. Our results showed that EGCG treatment significantly ameliorated the histopathological changes and the increased serum creatinine and blood urea nitrogen (BUN) induced by CP. TUNEL-positive cells significantly reduced in the CP+EGCG group compared with CP group. EGCG also inhibited the expression of the ligand of death receptor Fas (Fas-L), apoptosis regulator BAX (Bax) and tumor-suppressor protein p53, and increased the expression of B-cell lymphoma 2 (Bcl-2). These findings suggest that EGCG can ameliorate CP-induced apoptosis in the kidney by regulating death receptor Fas conducted extrinsic pathway, and the expression of Bax and Bcl-2.
Keywords: Cisplatin (CP), Epigallocatechin-3-gallate (EGCG), renal, apoptosis, mouse
Introduction
Cisplatin or cis-diamine-dichloroplatinum (II) (CDDP) is a platinum-containing anti-cancer drug widely used against multiple solid tumors. Many side effects such as ototoxicity, gastrotoxicity, myelosuppression, and allergic reactions [1,2], especially the nephrotoxicity [3,4], limits its use in cancer therapy. The mechanisms underlying nephrotoxicity induced by CP are not clearly established. Recently many studies have identified that apoptosis plays a pivotal role in renal tubulointerstitial fibrosis [5,6], and the apoptosis of tubular epithelial cells is a major cause [7]. Several cellular signaling pathways include the intrinsic mitochondrial pathway, the extrinsic death-receptor pathway such as TNF-α/TNFR1 and Fas/Fas-L pathway, and endoplasmic reticulum stress related mechanisms have been reported to be involved in regulation of apoptosis in tubular epithelial cells [8-11]. It has been shown that CP-induced nephrotoxicity associated with increased pro-apoptotic protein Bax and decreased anti-apoptotic protein Bcl-2 [11]. Fas is expressed on renal tubular cells (RTCs), and its upregulation during acute and chronic renal failure has been documented by many reports [12,13]. However, whether apoptosis of RTC depends on the Fas/FasL pathway remains controversial [14].
Numerous compounds, such as tomato lycopene complex, grape seed proanthocyanidin extract, extract of Ginkgo biloba, Rosmarinus acid, and cilastatin, can ameliorate cisplatin-induced nephrotoxicity [15-19]. EGCG, the most abundant catechin in tea, has anti-oxidant, anti-inflammatory, and anti-tumorigenic properties. Many studies have showed its effects to blood diseases, cardiovascular diseases, respiratory diseases, eye diseases, and cancers [20-24]. Some studies also demonstrated the renal protect effect EGCG [25-27], leading us to study whether EGCG can ameliorate the acute kidney injury induced by CP. The nephrotoxicity of CP involves Nrf2/HO-1 signaling pathway [28], renal proinflammatory (TNF-α) and oxidant stress signals [29]. The protect mechanism of EGCG on CP-induced nephrotoxicity is still uncertain. So, in this study, we treated mouse model of CP-induced nephropathy with EGCG, and study of whether it has the protect effects to cisplatin nephrotoxicity and the mechanisms.
Materials and methods
Reagents and materials
Cisplatin and Epigallocatechin-3-gallate (EGCG) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies were provided as follows: Fas-L, Bax, Bcl-2, p53 and β-actin antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Antimouse and antirabbit secondary antibodies were obtained from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA, USA). TUNEL staining kit (the In Situ Cell Death Detection kit) was purchased from Roche Diagnostics (Indianapolis, IN, USA).
Animals
Twenty-eight 6-8 week-old adult male C57/BL6 mice (20-25 g) were obtained from the Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Mice were housed separately in metal cages. The cages were placed in a room with controlled temperature (22 ± 0.5°C), humidity (60 ± 10%), and a 12-h light-dark cycle. Food and water were available ad libitum. The animal experiments conformed to the Animal Management Rules of the Chinese Ministry of Health (document No. 55, 2001) and were approved by the Animal Care Committee of Shandong University.
Experimental design
A total of 28 mice were divided randomly in the following five groups: (1) control group (NC; n = 7), only received intraperitoneal (i.p.) injection of vehicle solution (0.9% saline; 10 ml/kg); (2) EGCG group (EGCG; n = 7), received a single i.p. injection of 100 mg/kg EGCG (dissolved in 0.9% saline to reach 20 mg/ml); (3) CP group (CP; n = 7), only received an i.p. injection of 20.0 mg/kg CP (dissolved in 0.9% saline to reach a concentration of 2.0 mg/ml); and (4) CP+EGCG 100 mg/kg group (CP+EGCG 100 mg/kg; n = 7), which successively received i.p. injection of 100 mg/kg EGCG at 30 min before i.p. injection of CP, and i.p. administration of 100 mg/kg EGCG after i.p. injection of CP 48 h.
Five days after i.p. injection of CP, all mice were sacrificed by cervical dislocation, and all of them were weighed and blood was collected from the endocanthion before the sacrifice. Blood samples were centrifuged at 1,500 g at 4°C for 15 min, and sera were collected. Blood urea nitrogen (BUN) and serum creatinine (Scr) levels were measured in a Cobas® 8000 modular analyser (Roche Diagnostics) in Qilu Hospital, Shandong University. Both kidneys were immediately excised, and then cut in half by coronal position after weighed. Half of each excised kidney was stored at -80°C, and the remaining sections were fixed in 4% buffered paraformaldehyde at 4°C and embedded in paraffin.
Histopathologic observation
Histopathological changes in the kidney were examined by periodic acid-Schiff (PAS) staining. Kidneys embedded in paraffin were cut into 4-μm thick sections. Deparaffinized sections were stained with PAS reagent. Tubular damage was assessed by an index of renal tubular necrosis in 10 different fields: 0 = no damage, 1 = less than 25% damage, 2 = 25-50% damage, 3 = 50-75% damage, and 4 = more than 75% damage.
Immunohistochemical study
For immunohistochemical analysis, deparaffinized tissue slices underwent antigen retrieval by microwaved for 10-15 min in 0.01% sodium citrate buffer (pH 6.0). 3% hydrogen peroxide was used to immerse the tissue slices for 10 min in dark to block endogenous peroxidase. The tissue slices were incubated with the primary antibody (anti-Fas-L 1:100, anti-Bax 1:100 and anti-Bcl-2 1:100) at 4°C overnight, while negative controls were incubated with PBS. After washing three times, slices were incubated with secondary antibodies for 60 min at 37°C, and then stained with 3, 3’-diaminobenzidine (DAB) and hematoxylin. Stained slides were analyzed by light microscopy. Brown areas were identified as positive. Semi-quantitative analysis was performed on the colored sections using Image-Pro Plus 5.0.
TUNEL assay
The terminal deoxynucleotidyltransferase-mediated nick end labeling (TUNEL) method was performed to evaluate the in situ apoptosis in kidney tubular cells. The TUNEL staining was conducted following the manufacturer’s instructions. A DAPI filter was used to detect DAPI staining (blue color), and an FITC filter was used to detect TUNEL staining (red color). TUNEL-positive cells were counted in 10 high-power (×400) fields per section in the cortex.
Western blot analysis
Concentration of the protein extraction of the mouse kidney tissue samples was determined according to the procedure described by the Pierce BCA Protein Assay kit (Thermo Fisher Scientific Inc., Rockford, IL, USA). Equal amounts of protein (30 μg) were electrophoresed and subsequently transferred to cellulose acetate membranes. The membranes were blocked in TBS buffer containing nonfat milk for 1 h and then incubated with primary antibodies (anti-Fas-L 1:200, anti-Bax 1:200, antiBcl-2 1:200, anti-p53 1:500 and antiβ-actin 1:2,500) at 4°C overnight. The membranes were then washed and incubated with secondary antibodies for 1 h. Finally the membranes were developed with enhanced chemiluminescence (ECL) reagent (Thermo Fisher Scientific Inc., Rockford, IL, USA) and exposed to an X-ray film. Band intensity was measured using Quantity One software (Bio-Rad, Hercules, CA, USA). Fas-L, Bax, Bcl-2 and p53 relative quantities were expressed as a ratio of luminosity of the respective sample to that of the normal control group.
Statistical analysis
Data are given as means ± standard deviation (S.D.). The intergroup variation between groups was evaluated using one-way analysis variance (ANOVA) followed by Dunnett’s multiple comparison test, and the comparisons between two groups were conducted by unpaired Student’s t-test. P<0.05 was considered statistically significant.
Results
EGCG protects against CP-induced renal injury in mouse
In mice, treatment with CP induced significant increase both in the relative kidney weight and in the levels of serum creatinine and BUN (Table 1, compare CP group with control). Co-treatment of EGCG together with CP suppressed the increased relative kidney weight and creatinine and BUN levels caused by CP treatment alone (Table 1, compare CP+EGCG group with CP group), while treatment with EGCG alone had no effect on these parameters.
Table 1.
The changes in relative kidney weight and the levels of Creatinine and BUN
| Relative kidney weight | Creatinine (μmol/L) | BUN (mmol/L) | |
|---|---|---|---|
| Control | 8.32 ± 0.71 | 30.57 ± 6.95 | 9.99 ± 0.97 |
| EGCG | 8.46 ± 0.85 | 32.00 ± 4.43 | 10.16 ± 1.84 |
| CP | 8.85 ± 0.45a | 178.86 ± 19.58a | 82.76 ± 7.57a |
| CP+EGCG | 8.42 ± 1.08b | 63.71 ± 6.65b | 29.29 ± 3.84b |
Data are presented as means ± SD. Relative kidney weight is expressed as: left kidney weight/body weight * 1000.
p<0.05 versus control group;
p<0.05 versus CP group.
The PAS staining of kidney tissues was conducted to evaluate whether EGCG can ameliorate CP-induced renal tubular damage. Normal tubular morphology was presented in control group (Figure 1A) and EGCG group (Figure 1B). Renal tubular atrophy and dilation, necrosis and desquamation of renal tubular epithelial cells, and intratubular cast formation in the proximal tubules of kidney were observed in CP group (Figure 1C), while the tubular damage was greatly improved by EGCG (Figure 1D). EGCG dramatically reduced the tubular injury scores after CP treatment, and the reduction was statistically significant (P<0.05). These data indicated EGCG suppressed renal injury caused by CP.
Figure 1.
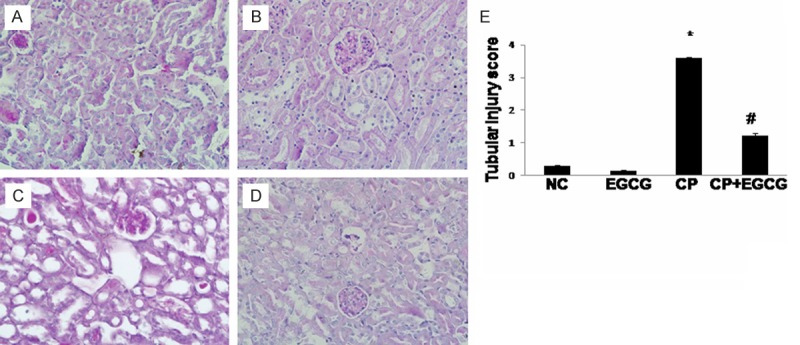
EGCG attenuated the kidney histological abnormalities induced by CP in mice. Mice were treated with vehicle (A), EGCG (B), CP (C), and CP+EGCG (D), separately. In CP group, renal tubular atrophy and dilation, necrosis and desquamation of renal tubular epithelial cells, and intratubular cast formation in the proximal tubules of kidney were obvious. Tubular damage was greatly improved in CP+EGCG group. Tubular injury score (E). Data are presented as means ± SD. *p<0.05 versus control group; #p<0.05 versus CP group. Original magnification, ×400.
EGCG blocks apoptosis of tubular epithelial cells caused by CP
In order to assess whether EGCG can protect against CP-induced renal tubular epithelial cell apoptosis, TUNEL assay was conducted. A large number of TUNEL-positive renal tubular epithelial cells were detected in CP group (Figure 2C), whereas co-treatment with EGCG strongly reduced the percentage of TUNEL-positive cells (Figure 2D). Limited apoptosis was detected in control group (Figure 2A) and the EGCG group (Figure 2B). Thus, EGCG abrogated CP-induced apoptosis.
Figure 2.
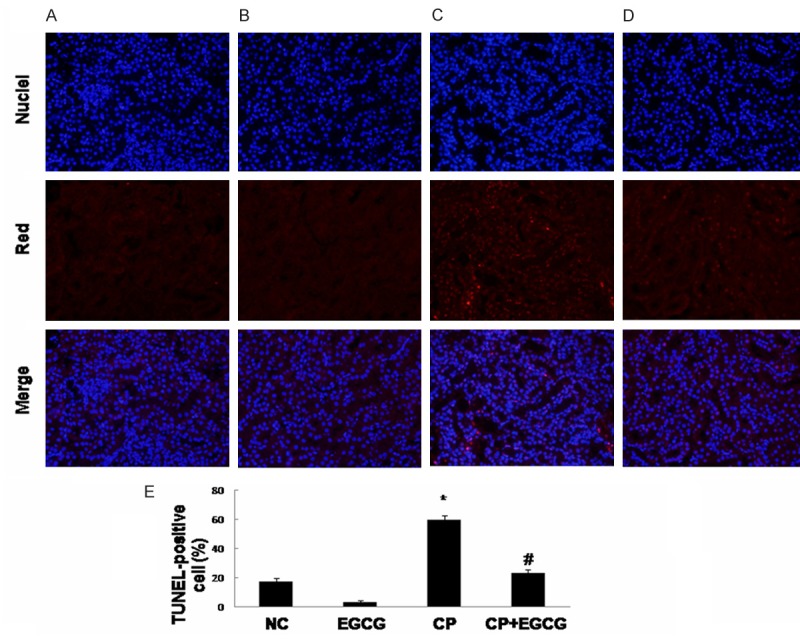
EGCG inhibits the apoptosis of renal tubular epithelial cells induced by CP. Mice were treated with vehicle (A), EGCG (B), CP (C), and CP+EGCG (D), separately. Red staining represents TUNEL‑positive cells. Original magnification, ×400. The percentage of TUNEL-positive cells in different groups (E). Data are presented as means ± SD. *p<0.05 versus control group; #p<0.05 versus CP group.
The mechanism of the protective effect of EGCG to the nephrotoxicity induced by CP in mouse
Death receptor Fas has been implicated in CP-induced renal epithelial cell death. Marker proteins in this pathway such as Fas-L, Bax, and Bcl-2 have been examined by immunohistochemistry and western blotting. As shown in Figures 3, 4 and 5, both two approaches exhibited that Fas-L and Bax were highly expressed in CP group while the expression of Bcl-2 was limited. The antiapoptotic activity of EGCG was evident by elevated Bcl-2, coincided with decreased expression of Fas-L and Bax. In the control and EGCG groups, Fas-L and Bax showed limited expression, and Bcl-2 showed high expression.
Figure 3.
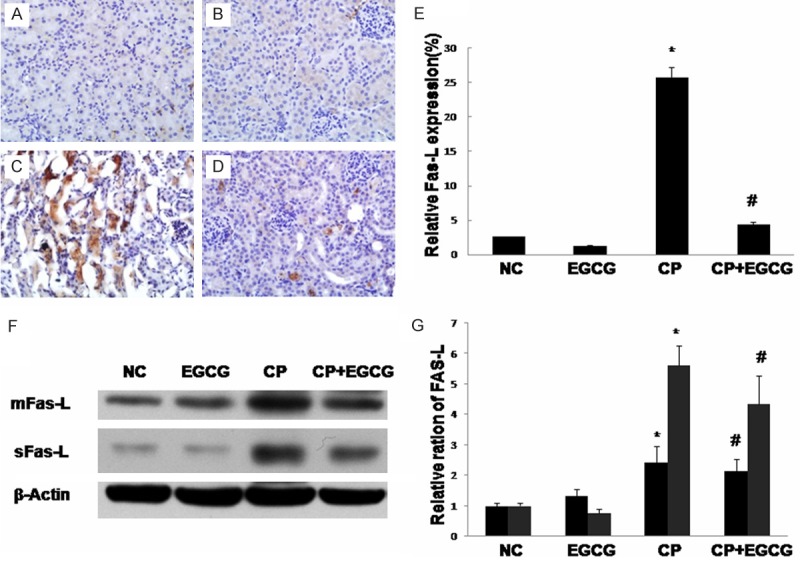
Immunohistochemical of Fas-L in the kidney of mouse. Mice were treated with vehicle (A), EGCG (B), CP (C), and CP+EGCG (D), separately. The brown granules represent positively stained cells. Original magnification, ×400. Measurement of the intensity of Fas-L immunostaining (E). Western blot analysis of Fas-L (F), and quantification of corresponding protein level (G). Data are presented as means ± SD. *p<0.05 versus control group; #p<0.05 versus CP group.
Figure 4.
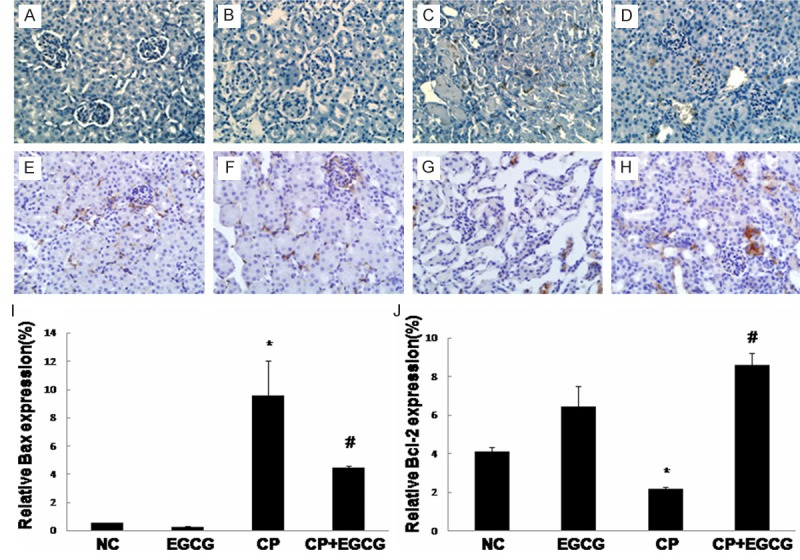
Immunohistochemical detection of Bax and Bcl-2: Mice were treated with vehicle (A, E), EGCG (B, F), CP (C, G), and CP+EGCG (D, H), separately. The brown granules represent positively stained cells. Original magnification, ×400. Measurement of the intensity of Bax (I) and Bcl-2 (J) immunostaining. Data are presented as means ± SD. *p<0.05 versus control group; #p<0.05 versus CP group.
Figure 5.
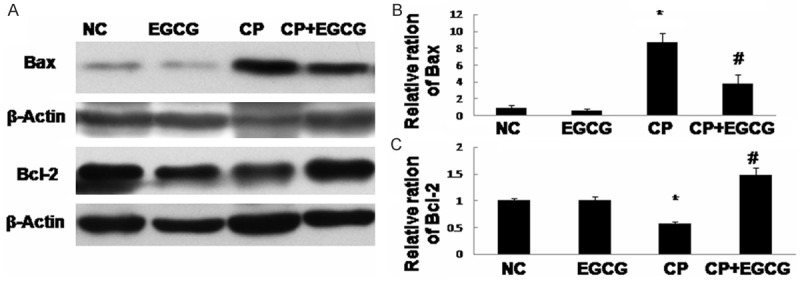
Western blot analysis of Bax and Bcl-2 (A), and quantification of the protein levels (B, C). Mice treated with vehicle, EGCG, CP, and CP+EGCG. Data are expressed as mean ± SD. *p<0.05 versus control group; #p<0.05 versus CP group.
p53 was reported to be involved in Fas/Fas-L-induced apoptosis, and a possible transcriptional regulator of Fas and Bax. Our results showed that the expression of p53 was high in CP group, and inhibited in CP+EGCG group (Figure 6).
Figure 6.
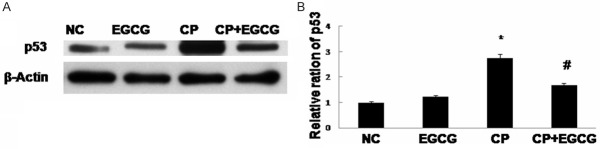
Western blot analysis of p53 (A) and quantification of the protein level (B). Mice treated with vehicle, EGCG, CP, and CP+EGCG. Data are expressed as mean ± SD. *p<0.05 versus control group; #p<0.05 versus CP group.
Discussion
CP is a simple platinum-containing inorganic molecule. It is one of the most remarkable successes in cancer therapy and has been widely used for chemotherapy since the accidental discovery over four decades ago [3,30,31]. Nephrotoxicity, now recognized as the most prevalent side effect, represents a dose and time-related toxicity, occurring in about one-third of patient undergoing CP treatment [3,32]. The most severe and common outcome of nephrotoxicity of CP is acute kidney injury (AKI) [32]. It develops primarily in the proximal tubule [33]. CP-induced AKI involves enhanced oxidative stress, inflammatory reactions, and tubular cell apoptosis. Renal tubular apoptosis has been considered as a key mechanism [34,35]. EGCG is the most abundant catechin in green tea, and it has been shown to reduce and inhibit the growth of various tumors [24]. Based on the antioxidant, anti-inflammatory and antiapoptotic properties of EGCG, the present study was undertaken to examine the protective effects of EGCG against CP-induced nephrotoxicity. Results in this study showed that EGCG effectively ameliorate acute kidney injure in CP-treated mice by suppressing the Fas/Fas-L pathway and regulating expressions of Bcl-2 family members of renal tubular cells. In this study, CP resulted in a severe nephropathy. In CP group, the relative kidney weight, the levels of serum creatinine and BUN, were significantly increased. Apoptosis of renal tubular epithelial cells was also significantly increased. Both the functional defects and apoptosis were markedly ameliorated by EGCG treatment, indicating the renal protective effect of EGCG on CP-induced nephrotoxicity.
Tubulointerstitial inflammation and tubular epithelial cell apoptosis have been demonstrated as the key leading to AKI in CP chemotherapy [14]. Several apoptotic pathways include the extrinsic pathway activated through death receptors, such as TNF receptors or Fas, theintrinsic mitochondrial pathway and the endoplasmic reticulum stress related mechanisms, have been implicated in CP-induced renal tubular epithelial cell death [32]. In the murine kidneys injured by CP administration, Fas expression markedly induced in cultured proximal tubular epithelial cells, while absence of Fas protected them from undergoing apoptosis [11]. Recently studies reported that CP-induced nephropathy is mediated through upregulation of Fas/Fas-L system [12,36]. During the intracellular cascade of caspase activation, activation of caspase-8 can activate the pro-apoptotic protein Bax, and then following the apoptosis in the end.
The intrinsic mitochondrial pathway by which CP activates remains unknown [10]. CP generates reactive oxygen species, which activate the pro-apoptotic Bcl-2 family member Bax, then the activation of Bax induces mitochondrial permeability transition, leading to release of cytochrome c, finally activates caspase-3 and induces the apoptosis [10]. The anti-apoptotic Bcl-2 family members such as Bcl-2, plays a pivotal protective role in preserving mitochondrial structure and function, preventing onset of mitochondrial permeability transition, and finally inhibiting the apoptosis [37,38]. Over-expression of Bcl-2 obviously ameliorated CP-induced apoptosis of renal tubular epithelial cells [39]. The integration of diverse pro- and anti-apoptotic signals occurring at the mitochondria may decide cell fate [40]. Several studies have demonstrated that CP can directly activate Bax, thus down-regulate the expression of Bcl-2 [8,10,41].
Tumor suppressor p53 is activated by CP and pharmacological or genetic inhibition of p53 suppresses CP-induced apoptosis in RTC in vitro and nephrotoxicity in vivo [42-44]. Several studies have emphasized the involvement of p53 in Fas/Fas-L-induced apoptosis [45,46]. In addition, oxidant stress can activate p53 [47], and then p53 can directly activate Bax [48]. p53 may be a transcriptional regulator of Fas and Bax [14,48].
Consistently with previous findings, in this study, the expression of Fas-L, Bax and p53 increased, and the expression of Bcl-2 decreased in the kidneys of CP group. Administration of EGCG before CP treated significantly reduced the overexpression of Fas-L, Bax and p53 and rescued the downregulation of Bcl-2, suggesting the inhibition of tubular apoptosis (Figure 7).
Figure 7.
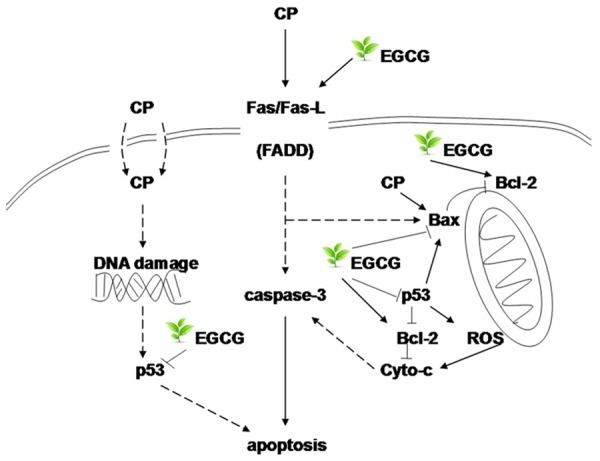
The possible mechanism of the protect effect of EGCG to the nephrotoxicity induced by CP in mouse.
In conclusion, the results of this study showed EGCG can effectively ameliorate CP-induced tubular apoptosis, and the inhibition on CP-induced apoptosis in tubular epithelial cells by EGCG may be through the blockage of the Fas/Fas-L pathway, and regulation of the expression of Bcl-2 family members. The suppression of apoptosis in renal tubular epithelial cells by EGCG could be an effective strategy for the treatment of CP-induced nephropathy. EGCG may have potential value in clinical CP chemotherapy while the therapeutic activity of it still needs to be further confirmed.
Acknowledgements
This research was supported by National Key Technology Support Program (grant no. 2011BAI10B01), the National Natural Science Foundation of China (grant no. 81200529), the Natural Science Foundation of Shandong Province (grant no. ZR2012HQ001 and ZR2013HM100) and the Independent Innovation Foundation of Shandong University (IIFSDU; grant no. 2012TS204 and 2012ST167).
Disclosure of conflict of interest
None.
References
- 1.Hartmann JT, Fels LM, Knop S, Stolt H, Kanz L, Bokemeyer C. A randomized trial comparing the nephrotoxicity of cisplatin/ifosfamide-based combination chemotherapy with or without amifostine in patients with solid tumors. Invest New Drugs. 2000;18:281–289. doi: 10.1023/a:1006490226104. [DOI] [PubMed] [Google Scholar]
- 2.Hartmann JT, Lipp HP. Toxicity of platinum compounds. Expert Opin Pharmacother. 2003;4:889–901. doi: 10.1517/14656566.4.6.889. [DOI] [PubMed] [Google Scholar]
- 3.Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol. 2003;23:460–464. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 4.Sastry J, Kellie SJ. Severe neurotoxicity, ototoxicity and nephrotoxicity following high-dose cisplatin and amifostine. Pediatr Hematol Oncol. 2005;22:441–445. doi: 10.1080/08880010590964381. [DOI] [PubMed] [Google Scholar]
- 5.Choi YJ, Baranowska-Daca E, Nguyen V, Koji T, Ballantyne CM, Sheikh-Hamad D, Suki WN, Truong LD. Mechanism of chronic obstructive uropathy: increased expression of apoptosis-promoting molecules. Kidney Int. 2000;58:1481–1491. doi: 10.1046/j.1523-1755.2000.00310.x. [DOI] [PubMed] [Google Scholar]
- 6.Truong LD, Choi YJ, Tsao CC, Ayala G, Sheikh-Hamad D, Nassar G, Suki WN. Renal cell apoptosis in chronic obstructive uropathy: the roles of caspases. Kidney Int. 2001;60:924–934. doi: 10.1046/j.1523-1755.2001.060003924.x. [DOI] [PubMed] [Google Scholar]
- 7.Hauser P, Oberbauer R. Tubular apoptosis in the pathophysiology of renal disease. Wien Klin Wochenschr. 2002;114:671–677. [PubMed] [Google Scholar]
- 8.Jiang M, Wei Q, Wang J, Du Q, Yu J, Zhang L, Dong Z. Regulation of PUMA-alpha by p53 in cisplatin-induced renal cell apoptosis. Oncogene. 2006;25:4056–4066. doi: 10.1038/sj.onc.1209440. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol. 2005;16:1985–1992. doi: 10.1681/ASN.2004090768. [DOI] [PubMed] [Google Scholar]
- 10.Park MS, De Leon M, Devarajan P. Cisplatin induces apoptosis in LLC-PK1 cells via activation of mitochondrial pathways. J Am Soc Nephrol. 2002;13:858–865. doi: 10.1681/ASN.V134858. [DOI] [PubMed] [Google Scholar]
- 11.Tsuruya K, Ninomiya T, Tokumoto M, Hirakawa M, Masutani K, Taniguchi M, Fukuda K, Kanai H, Kishihara K, Hirakata H, Iida M. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003;63:72–82. doi: 10.1046/j.1523-1755.2003.00709.x. [DOI] [PubMed] [Google Scholar]
- 12.Razzaque MS, Koji T, Kumatori A, Taguchi T. Cisplatin-induced apoptosis in human proximal tubular epithelial cells is associated with the activation of the Fas/Fas ligand system. Histochem Cell Biol. 1999;111:359–365. doi: 10.1007/s004180050368. [DOI] [PubMed] [Google Scholar]
- 13.Schelling JR, Nkemere N, Kopp JB, Cleveland RP. Fas-dependent fratricidal apoptosis is a mechanism of tubular epithelial cell deletion in chronic renal failure. Lab Invest. 1998;78:813–824. [PubMed] [Google Scholar]
- 14.Tsuruya K, Tokumoto M, Ninomiya T, Hirakawa M, Masutani K, Taniguchi M, Fukuda K, Kanai H, Hirakata H, Iida M. Antioxidant ameliorates cisplatin-induced renal tubular cell death through inhibition of death receptor-mediated pathways. Am J Physiol Renal Physiol. 2003;285:F208–218. doi: 10.1152/ajprenal.00311.2002. [DOI] [PubMed] [Google Scholar]
- 15.Camano S, Lazaro A, Moreno-Gordaliza E, Torres AM, de Lucas C, Humanes B, Lazaro JA, Milagros Gomez-Gomez M, Bosca L, Tejedor A. Cilastatin attenuates cisplatin-induced proximal tubular cell damage. J Pharmacol Exp Ther. 2010;334:419–429. doi: 10.1124/jpet.110.165779. [DOI] [PubMed] [Google Scholar]
- 16.Dogukan A, Tuzcu M, Agca CA, Gencoglu H, Sahin N, Onderci M, Ozercan IH, Ilhan N, Kucuk O, Sahin K. A tomato lycopene complex protects the kidney from cisplatin-induced injury via affecting oxidative stress as well as Bax, Bcl-2, and HSPs expression. Nutr Cancer. 2011;63:427–434. doi: 10.1080/01635581.2011.535958. [DOI] [PubMed] [Google Scholar]
- 17.Domitrovic R, Potocnjak I, Crncevic-Orlic Z, Skoda M. Nephroprotective activities of rosmarinic acid against cisplatin-induced kidney injury in mice. Food Chem Toxicol. 2014;66:321–328. doi: 10.1016/j.fct.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Gao Z, Liu G, Hu Z, Li X, Yang X, Jiang B, Li X. Grape seed proanthocyanidin extract protects from cisplatin-induced nephrotoxicity by inhibiting endoplasmic reticulum stress-induced apoptosis. Mol Med Rep. 2014;9:801–807. doi: 10.3892/mmr.2014.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song J, Liu D, Feng L, Zhang Z, Jia X, Xiao W. Protective Effect of Standardized Extract of Ginkgo biloba against Cisplatin-Induced Nephrotoxicity. Evid Based Complement Alternat Med. 2013;2013:846126. doi: 10.1155/2013/846126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khurana S, Hollingsworth A, Piche M, Venkataraman K, Kumar A, Ross GM, Tai TC. Antiapoptotic actions of methyl gallate on neonatal rat cardiac myocytes exposed to H2O2. Oxid Med Cell Longev. 2014;2014:657512. doi: 10.1155/2014/657512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim KC, Lee C. Reversal of Cisplatin resistance by epigallocatechin gallate is mediated by downregulation of axl and tyro 3 expression in human lung cancer cells. Korean J Physiol Pharmacol. 2014;18:61–66. doi: 10.4196/kjpp.2014.18.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi H, Abe N, Zhu B, Murata Y, Nakamura Y. (-)-Epigallocatechin-3-Gallate Ameliorates Photodynamic Therapy Responses in an In Vitro T Lymphocyte Model. Phytother Res. 2014 doi: 10.1002/ptr.5152. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 23.Chen R, Hollborn M, Grosche A, Reichenbach A, Wiedemann P, Bringmann A, Kohen L. Effects of the vegetable polyphenols epigallocatechin-3-gallate, luteolin, apigenin, myricetin, quercetin, and cyanidin in primary cultures of human retinal pigment epithelial cells. Mol Vis. 2014;20:242–258. [PMC free article] [PubMed] [Google Scholar]
- 24.Landis-Piwowar KR, Huo C, Chen D, Milacic V, Shi G, Chan TH, Dou QP. A novel prodrug of the green tea polyphenol (-)-epigallocatechin-3-gallate as a potential anticancer agent. Cancer Res. 2007;67:4303–4310. doi: 10.1158/0008-5472.CAN-06-4699. [DOI] [PubMed] [Google Scholar]
- 25.El-Mowafy AM, Salem HA, Al-Gayyar MM, El-Mesery ME, El-Azab MF. Evaluation of renal protective effects of the green-tea (EGCG) and red grape resveratrol: role of oxidative stress and inflammatory cytokines. Nat Prod Res. 2011;25:850–856. doi: 10.1080/14786419.2010.533669. [DOI] [PubMed] [Google Scholar]
- 26.Nagle DG, Ferreira D, Zhou YD. Epigallocatechin-3-gallate (EGCG): chemical and biomedical perspectives. Phytochemistry. 2006;67:1849–1855. doi: 10.1016/j.phytochem.2006.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng A, Ye T, Rakheja D, Tu Y, Wang T, Du Y, Zhou JK, Vaziri ND, Hu Z, Mohan C, Zhou XJ. The green tea polyphenol (-)-epigallocatechin-3-gallate ameliorates experimental immune-mediated glomerulonephritis. Kidney Int. 2011;80:601–611. doi: 10.1038/ki.2011.121. [DOI] [PubMed] [Google Scholar]
- 28.Sahin K, Tuzcu M, Gencoglu H, Dogukan A, Timurkan M, Sahin N, Aslan A, Kucuk O. Epigallocatechin-3-gallate activates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Life Sci. 2010;87:240–245. doi: 10.1016/j.lfs.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 29.El-Mowafy AM, Al-Gayyar MM, Salem HA, El-Mesery ME, Darweish MM. Novel chemotherapeutic and renal protective effects for the green tea (EGCG): role of oxidative stress and inflammatory-cytokine signaling. Phytomedicine. 2010;17:1067–1075. doi: 10.1016/j.phymed.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 30.Rosenberg B, Vancamp L, Krigas T. Inhibition of cell division in escherichia coli by electrolysis products from a platinum electrode. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 31.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 32.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of Cisplatin nephrotoxicity. Toxins (Basel) 2010;2:2490–2518. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blachley JD, Hill JB. Renal and electrolyte disturbances associated with cisplatin. Ann Intern Med. 1981;95:628–632. doi: 10.7326/0003-4819-95-5-628. [DOI] [PubMed] [Google Scholar]
- 34.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 35.Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci. 2007;334:115–124. doi: 10.1097/MAJ.0b013e31812dfe1e. [DOI] [PubMed] [Google Scholar]
- 36.Linkermann A, Himmerkus N, Rolver L, Keyser KA, Steen P, Brasen JH, Bleich M, Kunzendorf U, Krautwald S. Renal tubular Fas ligand mediates fratricide in cisplatin-induced acute kidney failure. Kidney Int. 2011;79:169–178. doi: 10.1038/ki.2010.317. [DOI] [PubMed] [Google Scholar]
- 37.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 38.Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 39.Jiang M, Wang CY, Huang S, Yang T, Dong Z. Cisplatin-induced apoptosis in p53-deficient renal cells via the intrinsic mitochondrial pathway. Am J Physiol Renal Physiol. 2009;296:F983–993. doi: 10.1152/ajprenal.90579.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brenner C, Kroemer G. Apoptosis. Mitochondria--the death signal integrators. Science. 2000;289:1150–1151. doi: 10.1126/science.289.5482.1150. [DOI] [PubMed] [Google Scholar]
- 41.Wei Q, Dong G, Franklin J, Dong Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007;72:53–62. doi: 10.1038/sj.ki.5002256. [DOI] [PubMed] [Google Scholar]
- 42.Cummings BS, Schnellmann RG. Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways. J Pharmacol Exp Ther. 2002;302:8–17. doi: 10.1124/jpet.302.1.8. [DOI] [PubMed] [Google Scholar]
- 43.Jiang M, Yi X, Hsu S, Wang CY, Dong Z. Role of p53 in cisplatin-induced tubular cell apoptosis: dependence on p53 transcriptional activity. Am J Physiol Renal Physiol. 2004;287:F1140–1147. doi: 10.1152/ajprenal.00262.2004. [DOI] [PubMed] [Google Scholar]
- 44.Wei Q, Dong G, Yang T, Megyesi J, Price PM, Dong Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol. 2007;293:F1282–1291. doi: 10.1152/ajprenal.00230.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bennett M, Macdonald K, Chan SW, Luzio JP, Simari R, Weissberg P. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science. 1998;282:290–293. doi: 10.1126/science.282.5387.290. [DOI] [PubMed] [Google Scholar]
- 46.Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998;188:2033–2045. doi: 10.1084/jem.188.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chandel NS, Vander Heiden MG, Thompson CB, Schumacker PT. Redox regulation of p53 during hypoxia. Oncogene. 2000;19:3840–3848. doi: 10.1038/sj.onc.1203727. [DOI] [PubMed] [Google Scholar]
- 48.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]