

Abstract
Human liver ischemia/reperfusion injury (IRI) is a common and major clinical problem complicating liver surgery and transplantation. The pathogenesis underlying IRI is complex, involving a series of signaling mediators and mechanisms. This study aimed to investigate the effects of Magnesium Isoglycyrrhizinate (MgIG) on the changes of oxidant stress and apoptosis induced by IRI in human hepatic L02 cells. L02 cells with IRI were treated with or without MgIG and mitoKATP (Mitochondrial adenosine triphosphate-dependent potassium) channel modulators. Cell viability was assessed using CCK-8 assay. Cell apoptosis was quantified by flow cytometry. The activity of the antioxidant enzymes superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) were measured. Effects of MgIG on the expression of Bax, Bcl-2, Caspase 3, PARP (poly ADP-ribose polymerase), Akt, and ERK in L02 cells with IRI were examined. Our results showed that MgIG treatment significantly reduced the population of apoptotic cells and the expression of apoptosis-related proteins in hepatic L02 cells with IRI. MgIG also counteract ischemia reperfusion induced oxidative challenge as it effectively reduced malondialdehyde (MDA) and increased the activities of SOD and GSH-Px. L02 cells treated with MgIG showed increased expression of p-Akt and p-ERK, indicating that the protective effect of MgIG might be associated with the activation of Akt and ERK pathways. Moreover, the addition of Diazoxide (DE), a mitoKATP channel opener, enhanced the cytoprotective activity of MgIG, while the mitoKATP blocker 5-hydroxydecanoate (5-HD) reduced the cytoprotective activity of MgIG.
Keywords: L02 cells, ischemia reperfusion injury, liver, magnesium isoglycyrrhizinate
Introduction
Hepatic ischemia/reperfusion injury (IRI) is a common and major complication after liver surgery and transplantation. It impairs liver function, increases postoperative morbidity and mortality, interferes recovery and thus has a major impact on clinical outcomes [1]. Hepatic IRI can be categorized into warm ischemia and cold ischemia. Warm ischemia occurs when hepatic blood supply is temporarily interrupted, and is associated with a variety of clinical situations including trauma, shock transplantation, elective liver surgery, toxic liver injury, sinusoidal obstruction and Budd-Chiari syndrome [2]. On the other hand, the vast majority of cold ischemia occurs during organ preservation prior to transplantation, and is featured by local inflammation mediated by neutrophils, damage to sinusoidal endothelial cells, and disruption of the microcirculation [3].
Hepatic IRI is associated with a series of pathophysiological events, including mitochondrial de-energization, adenosine-5’-triphosphate depletion, Kupffer cell activation, upregulation of pro-inflammatory cytokines and oxidative stress changes [4]. Reactive oxygen species (ROS), including the superoxide, hydroxyl radicals, and hydrogen peroxide, are generated during hepatic IRI as a consequence of altered aerobic metabolism [5], and plays a major role in IRI induced oxidative stress changes [6]. The cellular toxicity of ROS is associated with rapid modifications of cellular constituents, such as the depletion of intracellular glutathione and ATP, a decline in NAD+ level, an increase in free cytosolic Ca2+, and lipid peroxidation [7]. These damages ultimately lead to hepatic cell death either by necrosis or apoptosis [8]. Apoptotic cell death is an active intracellular death program, during which caspases (a family of proteases) are key executors and catalyze the destructive cascade leading to a series of cellular events and changes. The poly ADP-ribose polymerase (PARP), a nuclear enzyme involved in DNA repair, is a well-known substrate for Caspase 3 cleavage [9]. Members in the Bcl-2 protein family are also important regulators of apoptosis [10]. Moreover, studies have shown that prosurvival kinase phosphatidylinositol 3-kinase (PI3K)-Akt and extracellular signal-regulated kinase (ERK) pathways play critical roles in IRI [11]. The PI3K-Akt pathway has also been shown to regulate apoptosis signaling by inhibiting pro-apoptotic Bcl-2 protein BAD [12].
The complex mechanisms involved in IRI, with multiple interacting and interdependent mediators, provide plentiful candidates for therapeutic strategies. The most promising protective treatment against hepatic IRI so far is preconditioning, including ischemic preconditioning, heat shock and pharmacological interventions [7]. Ischemic preconditioning has been demonstrated to limit IRI and cell apoptosis in a number of studies [13-15]. It enhances hepatocyte survival by preventing mitochondrial failure, which involves mitochondrial K-ATP (mitoKATP) channels [16]. Diazoxide (DZ), a specific opener of mitoKATP channel, preserves mitochondrial integrity and suppresses proapoptotic regulators, and has been shown to attenuate IRI [13]. Moreover, many other pharmacological agents, such as methylprednisolone [17], α-tocopherol (vitamin E) [18], and dopamine [19] have also been shown to ameliorate hepatic IRI [20].
Here, we assessed the potential ameliorating effect of Magnesium Isoglycyrrhizinate (MgIG) on hepatic IRI utilizing an in vitro L02 cell model. MgIG, a traditional herbal remedy, is a magnesium salt of 18-α glycyrrhizic acid stereoisomer, and is extracted from the roots of the plant Glycyrrhiza glabra (licorice). It has recently been known for its anti-inflammatory and hepatic protection activity [21,22]. Previous study showed that MgIG provided protection against various organ injuries and diseases, including alcoholic liver disease, lung injury induced by paraquat poisoning, and epithelia ovarian cancers [23-25]. MgIG treatment has also been reported to reduce expression levels of the I/R-induced Tumor necrosis factor alpha (TNFa), Phospholipase A2 (PLA2), and MDA in plasma and liver tissues, and to decrease the I/R-induced Myeloperoxidase (MPO) activity in a rat limb I/R model [26]. However, the effect of MgIG against hepatic IRI, especially its potential antioxidative property and underlying molecular mechanisms remain less studied.
Here, we examined the cytoprotective and anti-apoptotic effects of MgIG on hepatic cells with IRI. This study elucidated that MgIG treatment ameliorated hepatic IRI through enhancing PI3K/Akt activity in human hepatic L02 cells.
Materials and methods
Cell culture and reagents
The human hepatic L02 cell line was obtained from Cell Bank of Peking Union Medical College Hospital, maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma, USA), supplemented with 10% Fetal Bovine Serum (FBS, Sigma, USA), 2 mM glutamine, 100 U/ml penicillin/streptomycin and cultured at 37°C in a humidified atmosphere with 5% CO2. MgIG (50 mg: 10 ml) was purchased from Chia-tai Tianqing Pharmaceutical Co. Ltd, China. 5-hydroxydecanoate (5-HD) and Diazoxide (DE) were purchased from Sigma Chemical Co. SOD, MDA and Glutathione Peroxidase (GSH-Px) Detection Kits were purchased from Nanjing Jiancheng Bioengineering Institute, China. HEPES buffered Tyrode’s lactate and bovine serum albumin (BSA) was purchased from Sigma-Aldrich Chemical Co, USA.
Oxygen deprivation and reperfusion, and MgIG treatment
L02 cells were placed in a cell culture flask till reaching 70% confluence. To create a hypoxic condition, the cells were incubated in a microaerophilic system (Thermo, Cedex, France) with 5% CO2 and 1% oxygen balanced with 94% N2 gas for 4 hours. Then, the cells were cultured in normoxic conditions with 95% oxygen and 5% CO2 at 37°C for 0, 2, 6, 12, 24 h, respectively, to achieve reperfusion. Five groups of differently treated L02 cells were studied. Group I: Normal control (NC) group was incubated with medium only; Group II: L02 cells with ischemia reperfusion injury alone (I/R); Group III: MgIG (10 mg/ml) was added to cultures 24 h prior to ischemia reperfusion condition (MgIG+I/R); Group IV: MgIG was added to cultures as in Group III and 5-HD (a mitoKATP specific ion channel blocker, 100 μmol/L) was given before the IRI treatment (MgIG+5-HD+I/R); Group V: MgIG was added to cultures as in Group III and IV, and DE (a mitoKATP selective channel opener, 100 μmol/L) was administrated before IRI treatment (MgIG+DE+I/R).
Cell viability assay
Cell viability was measured with Cell Counting Kit-8 (CCK-8) according to the manufacture’s protocol (Dojindo, Japan). L02 cells were seeded on 96-well plates (100 μL, 1×104/well) (Falcon, USA). After different treatments, 10 μl of CCK-8 solution was added to each well and cells were incubated for another 2 h at 37°C in a humidified CO2 incubator. The optical absorbance at 450 nm for each sample was measured with a microplate reader (BioRad, USA). Five separate experiments were conducted and each was performed in triplicates. The value was calculated by the formula: Cell viability (%) = [(A450 of treated group-A450 of blank control group)/(A450 of negative control group-A450 of blank control group)] × 100%.
Measurement of apoptotic cell death
To differentiate between apoptotic and healthy cells, we performed double staining with an Annexin V-FITC Apoptosis Detection kit. Differently treated human hepatic L02 cells were collected by removing the DMEM medium, washed twice with ice-cold PBS, gently resuspended in 300 mL binding buffer. 10 mL of Annexin V-FITC was added to cells, followed by gentle vortex and 10 min incubation at 4°C in the dark. 10 mL of propidium iodide (PI) was added to the tube followed by 5 min incubation at 4°C in the dark. After incubation, cells were analyzed by flow cytometry using a BD FACSAria cell sorter (Becton Dickinson, San Jose, CA). The fraction of cell population in different quadrants was analyzed using quadrant statistics. Cells in the lower right quadrant represented early apoptosis and those in the upper right quadrant represented late apoptosis.
Western blot analysis
Human L02 cells in logarithmic growth phase were treated with ischemia reperfusion injury for the above-indicated time. Cells were harvested, rinsed and lysed in an ice-cooled radio-immuno precipitation assay buffer (RIPA, Millipore, USA) to which protease and phosphatase inhibitors had been freshly added. Protein concentration was determined by BCA Protein Assay Kit (Pierce, USA). Samples were boiled for 5 mins at 100°C and insoluble material was removed by centrifugation. About 25 μg-50 μg protein extracts were electrophoresed on 10-15% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) and transferred onto PVDF membranes (0.45 μm, Millipore, BioRad), which were then blocked with 5% BSA in TBST for 1 h, and incubated overnight at 4°C with following primary antibodies: anti-Bax, anti-Bcl-2, anti-p-Akt (pSer473), anti-Akt, anti-p-ERK, anti-ERK, anti-PARP and anti-caspase 3 (1:1000, Cell Signaling Technology, USA). After washing 3 times with TBST, the membranes were incubated for 2 h at room temperature with secondary HRP-conjugated sheep anti-rabbit antibody or HRP-conjugated sheep anti-mouse antibody diluted in TBST with 5% BSA (1:2000, Santa Cruz Biotech, USA). Immunoreactivity was detected by enhanced chemiluminescence (ECL kit, Millipore, USA) and visualized by autoradiography. The level of β-actin was used as a control of the amount of protein loaded into each lane and the optical density of each band was measured using the Bio-Rad Laboratories Quantity One software.
Statistical analysis
The data were presented as mean ± standard deviation. Experimental groups were compared using Kruskal-Wallis variance analysis test. Post hoc analysis using Mann-Whitney U-test was performed to determine the levels of significance, and P < 0.05 was considered to be statistically significant. The statistical analysis was done using the SPSS statistical software (version 12).
Results
Establishment of an in vitro liver ischemia reperfusion model using L02 cells
The CCK-8 results revealed that pretreatment of human hepatic L02 cells with MgIG (10 mg/ml), MgIG (10 mg/ml) plus 5-HD (mitoKATP specific ion channel blocker, 100 μmol/L), or MgIG (10 mg/ml) plus DE (mitoKATP selective channel openers, 100 μmol/L) did not significantly affect cell viability (data not shown). To develop a reproducible in vitro liver ischemia reperfusion model with L02 liver cells, we tested prescribed period (4 h) of hypoxia, followed by a reoxygenation period of different lengths (0, 2, 6, 12 or 24 h, Materials and Methods). Compared with the NC (normal control) group, 4 hours of hypoxia combined with various reoxygenation time (0, 2, 6, 12, or 24 h) significantly decreased the cell viability of I/R group (P < 0.05) (Figure 1). Compared with I/R alone group, MgIG+I/R group showed significantly increased cell viability, with 4 h of hypoxia followed by a 12 h or 24 h reoxygenation period (P < 0.05) (Figure 1). Furthermore, cell viability of MgIG+DE+I/R group was also significantly improved, at 4 h of hypoxia followed by 6, 12, or 24 h reoxygenation period (P < 0.05) (Figure 1); however, the cell viability of MgIG+5-HD+I/R group only significantly increased at the 24 h reoxygenation period compared to I/R group (P < 0.05) (Figure 1).
Figure 1.
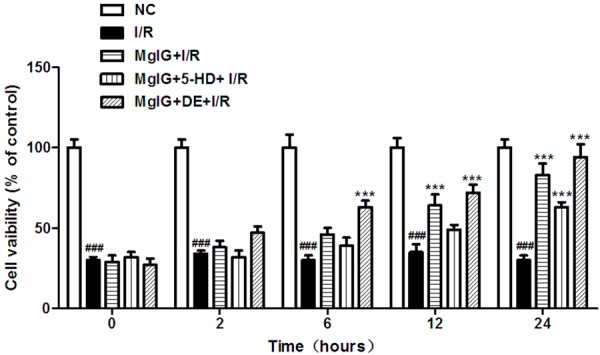
Cell viability was shown in L02 cultures during a combined 4 h-long oxygen deprivation and following 0-2-6-12-24 h of oxygenation. Cell viability was determined by CCK-8 assay as described in Materials and methods. Data represent the mean ± S.D. from five independent experiments (x̅ ± s, n = 5). NC: normal control; I/R: ischemia reperfusion only; MgIG+I/R: MgIG was added to cultures with ischemia reperfusion condition; MgIG+5-HD+I/R: Both MgIG (10 mg/ml) and 5-HD (100 μmol/L) were added to cultures before the IRI treatment; MgIG+DE+I/R: MgIG (10 mg/ml) and DE (100 μmol/L) were administrated to L02 cells before IRI treatment. ###p < 0.001, compared with NC group; ***p < 0.001, compared with I/R group.
MgIG reduces apoptosis in L02 cells with ischemia/reperfusion injury
To evaluate the effect of MgIG on cell apoptosis induced by ischemia/reperfusion in L02 cells, cells pretreated by MgIG with or without mitoKATP channel modulators (5-HD, DE) were incubated under 4 h of hypoxia followed by 24 h reoxygenation. These cells were then stained with Annexin V-FITC/PI and examined by flow cytometry, respectively (Figure 2A, Materials and methods). Compared with normal control, after 4 h hypoxia and 24 h reperfusion treatment, 40.7% of cells were viable, and 54.1% were either at the early apoptotic or late apoptotic stage. However, pretreatment with MgIG 24 h prior to hypoxia significantly reduced the number of early (Annexin V+/PI-) and late apoptosis (Annexin V+/PI+ cells) (Figure 2). In addition, compared with I/R group and MgIG+I/R group, after treatment with both MgIG and DE, cell viability rose up by 90.1% and only 9.7% cells underwent early or late apoptosis, in spite of the ischemia-reperfusion condition. Moreover, we also measured the cell viability of the group with pretreatment of MgIG and 5-HD. During the phases of 4 h hypoxia and 24 h reperfusion, the addition of 5-HD decreased the cytoprotective effects (apoptosis ratio) of MgIG, as compared with MgIG+I/R and MgIG+DE+I/R groups (Figure 2B).
Figure 2.
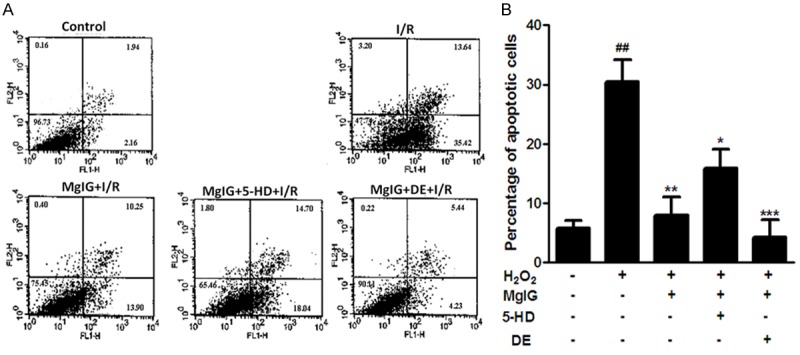
Effect of MgIG on ischemia-reperfusion induced L02 cell apoptosis was determined with Annexin V-FITC/PI binding staining by flow cytometry. A. Flow cytometry results with Annexin V-TITC/PI staining. L02 cells were incubated with 4 h of hypoxia followed by 24 h reoxygenation in the absence (-) or presence (+) of MgIG and mitoKATP channel modulator (5-HD, DE) as described in Materials and methods. After treatment, cells were harvested and apoptosis was examined with flow-cytometry after Annexin-V/PI staining. The horizontal and vertical axes represent labeling with Annexin V-FITC and PI, respectively. Cells are classified as healthy cells (Annexin V-, PI-), early apoptotic cells (Annexin V+, PI-), late apoptotic cells (Annexin V+, PI+), and damaged cells (Annexin V-, PI+). B. The ratio of apoptosis among different experimental groups. Apoptosis ratio was early apoptosis percentage plus late apoptosis percentage. Data represent the mean ± S.D. from five independent experiments (x̅ ± s, n = 5). ##p < 0.01, compared with vehicle controls group; *p < 0.05, compared with H2O2 treated group. **p < 0.01, compared with H2O2 treated group. ***p < 0.001, compared with H2O2 treated group.
MgIG inhibits ischemia/reperfusion induced oxidative stress in hepatic L02 cells model
To investigate the antioxidant ability of MgIG against ROS induced by ischemia/reperfusion in hepatic L02 cells, the activities of SOD and GSH-Px and the level of MDA were measured. SOD is found in all tissues and cells of the aerobic organisms. SOD is a key antioxidants enzyme that efficiently and specifically catalyzes free radical of oxygen to H2O2 and O2 , while GSH-Px also protects cells from oxidative damage. Compared with normal control, the activities of SOD and GSH-Px were significantly decreased in I/R group (Figure 3A, 3B), while MDA levels were obviously higher in I/R group than in the control group (Figure 3C). Nevertheless, the activities of SOD and GSH-Px were significantly up-regulated in MgIG+I/R and MgIG+DE+I/R groups, compared to I/R group, while the MDA level was significantly down-regulated. Moreover, we also noticed that this antioxidative stress effect of MgIG against IR injury is partially offset by the addition of 5-HD (Figure 3A-C).
Figure 3.
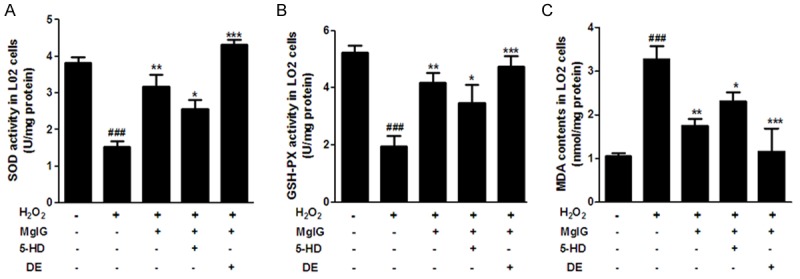
Inhibition of ROS by MgIG in ischemia reperfusion induced L02 cell injury. Effects of MgIG treatment on SOD activity (A), GSH-PX activity (B) and MDA level (C) in L02 Cells. L02 cells were incubated with 4 h of hypoxia followed by 24 h reoxygenation in the absence (-) or presence (+) of MgIG, 5-HD and DE, as described in Materials and methods. Data represent the mean ± S.D. from five independent experiments (x̅ ± s, n = 5), performed in triplicates ###p < 0.001, compared with vehicle controls group; *p < 0.05, compared with H2O2 treated group. **p < 0.01, compared with H2O2 treated group. ***p < 0.001, compared with H2O2 treated group.
MgIG alters expression of Bax, Bcl-2, Caspase 3 and PARP in L02 cells with ischemia reperfusion injury
In order to explore the influence of MgIG on apoptosis in hepatic L02 with IRI, the expression of two apoptosis-related proteins, Bcl-2 and Bax were examined. Bcl-2 is an anti-apoptotic gene and promotes cell survival [27], while Bax is proapoptotic, and is an indicator of apoptotic cells [28]. I/R injury significantly reduced Bcl-2 expression (Figure 4A) and increased Bax expression (Figure 4B), compared with normal control cells, respectively. However, pretreatment of IRI L02 cells with MgIG, or the combination of MgIG and DE, result in elevated Bcl-2 expression and attenuated Bax expression as compared to I/R group, respectively (Figure 4A, 4B). And compared to MgIG+I/R group, the cells treated with both MgIG and 5-HD inhibit the up-regulation of Bcl-2 and down-regulation of Bax in IRI L02 cells to some extent (Figure 4A, 4B). Similarly, compared with normal control group, I/R treatment promoted an additional and significant increase in the cleavage rate of Caspase 3, a gene activated in apoptotic cells [29], and its substrate PARP (Figure 4C). The enhanced Caspase 3 expression and activity in I/R alone group were significantly reduced by the pretreatment of MgIG or MgIG+DE, to the levels comparable to that of the NC group (Figure 4C). Notably, the addition of 5-HD, the mitoKATP channel blocker, reversed the effects of MgIG on the expression of Caspase 3 (Figure 4C).
Figure 4.
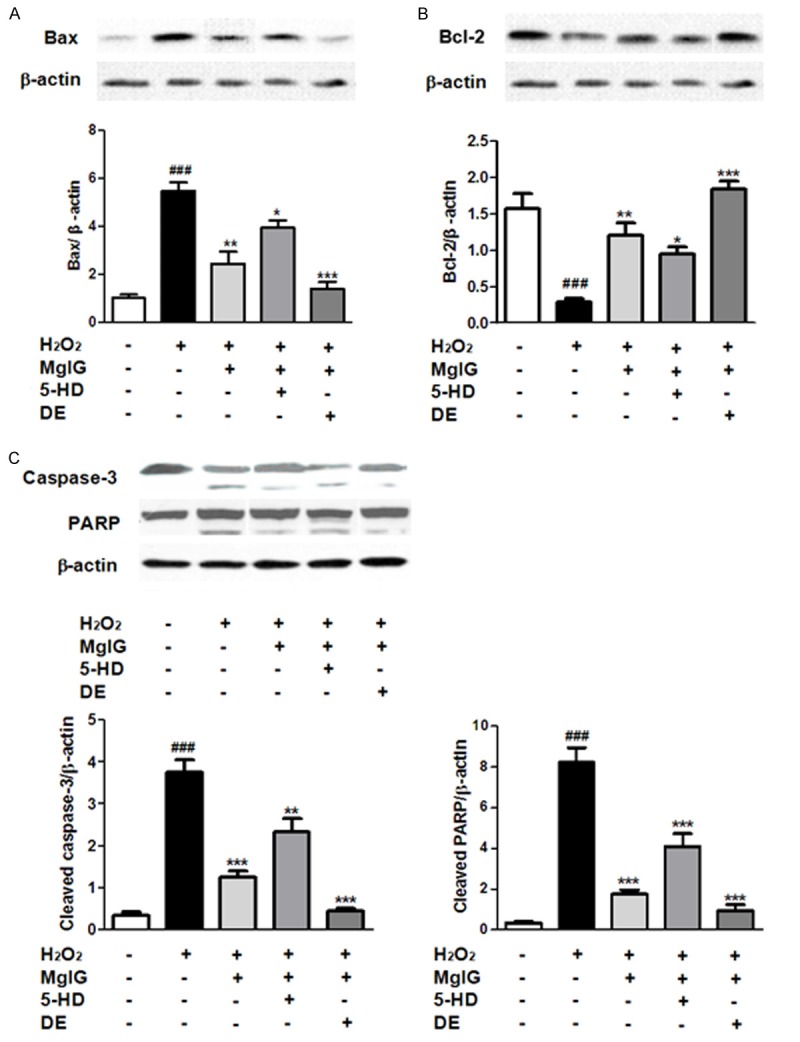
The expression of apoptosis related proteins Bax, Bcl-2, Caspase 3 and PARP were analyzed by Western blot. Effects of MgIG treatment on Bax (A); Bcl-2 (B); Caspase 3 and PARP (C) expression. Upper: Western Blot analysis for proteins. Lower: Densitometric analysis using Quantity one software. Data were normalized by the level of β-actin expression. Data represent the mean ± S.D. from five independent experiments (x̅ ± s, n = 5), performed in triplicates. ###p < 0.001, compared with vehicle controls group; *p < 0.05, compared with H2O2 treated group. **p < 0.01, compared with H2O2 treated group. ***p < 0.001, compared with H2O2 treated group.
MgIG enhances Akt and ERK pathways in L02 cells with ischemia reperfusion injury
Immunoblot analysis showed that ischemia reperfusion treatment resulted in a significant reduction in the protein levels of phosphorylated Akt (Ser473, p-Akt) and ERK (p-ERK), but not regular Akt and ERK, as compared to the control group (Figure 5). Additionally, our data demonstrated that treatment with MgIG, or the combination of MgIG and DE, result in a significant increase in Akt and ERK phosphorylation level, compared with I/R alone group (Figure 5). And MgIG combined with 5-HD relatively reduced the upregulated expression of p-Akt (Ser473) and p-ERK induced by MgIG pretreated. Thus, Akt and ERK pathway activity may play an essential role in the prosurvival effect of MgIG on L02 cells injury induced by IR exposure.
Figure 5.
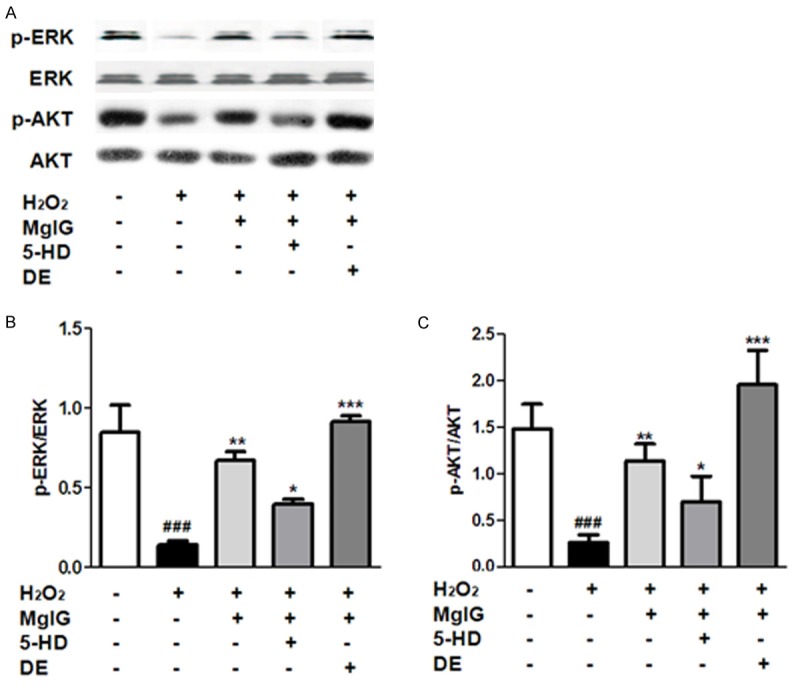
The expression of p-Akt, Akt, p-ERK, ERK was analyzed by Western Blot. L02 cells were incubated with 4 h of hypoxia followed by 24 h reoxygenation in the absence (-) or presence (+) of MgIG and mitoKATP channel modulator (5-HD, DE) as described in Materials and methods. A. Western Blot analysis for p-Akt, Akt, p-ERK, ERK proteins expression. Densitometric analysis using Quantity one software in left was at bottom. Data were normalized by the level of β-actin expression. B. p-ERK/ERK; C. p-Akt/Akt. Data represent the mean ± S.D. from five independent experiments (x̅ ± s, n = 5), performed in triplicate. ###p < 0.001, compared with vehicle controls group; *p < 0.05, compared with H2O2 treated group. **p < 0.01, compared with H2O2 treated group. ***p < 0.001, compared with H2O2 treated group.
Discussion
In this study, we established an in vitro model using hepatic L02 cells to study liver IRI. Our results demonstrated that in this assay a substantial degree of cell injury occurred during the progression of the hepatic L02 cells reoxygenation period. And the addition of MgIG alleviated cell death. This protective effect could be enhanced by the combinatorial treatment of MgIG and DE, while be compromised by the concurrent treatment of MgIG with 5-HD. This study also showed that MgIG treatment alone significantly inhibit the accumulation of oxidation products and reduced apoptosis in L02 cells with IRI. Moreover, co-treatment cell with both MgIG and DE further downregulated the apoptosis rate and oxidation stress in IRI L02 cells, and thus resulted in enhanced cytoprotective effects compared to MgIG treatment alone. We also exposed L02 cells to the ischemia/reperfusion condition with the combinatorial pretreatment of MgIG and 5-HD, a common mitoKATP channel blocker. The extent of oxidant stress and apoptosis attenuated by MgIG treatment alone was partly reversed when 5-HD was added. In addition, both Akt and ERK pathways were restored, after MgIG treatment, possibly as a compensatory pro-survival mechanism to suppress cell death rates. Thus, our results suggest that MgIG have the synergic effect of strengthening the protective effect with the activation of mitoKATP channel to some extent, and this beneficial effect may also be associated with the activation of Akt and ERK signaling pathways.
Our study is the first report that MgIG, a pure magnesium salt of 18-α glycyrrhizic acid stereoisomer, inhibits IRI in L02 cells. In the cellular level, MgIG potently protects L02 cells against ischemia reperfusion injury by decreasing the ROS accumulation and cell apoptosis. We also demonstrated that the hepatoprotective effect of MgIG is associated with its antioxidant properties. Previous studies showed that MgIG administration significantly ameliorated CCl4-induced biochemical alterations and lipid peroxidation in the liver of mice or rats [30,31]. The protective effects of MgIG might be resulted from the elimination of oxygen free radicals before the damage to macromolecular and metabolism of the cells. In this study, we also explored putative effects of MgIG on detoxifying enzymes, such as SOD and GSH-Px, which are known to be down-regulated in severe hepatic IRI [32]. After L02 cells were exposed to ischemia reperfusion condition, the formation of MDA was significantly elevated, while SOD and GSH-Px activity decreased. However, all these parameters exhibited amelioration when the exposure was combined with pretreatment with MgIG, alone or combined with DE.
MgIG treatment not only enhances the defense capacity of L02 cells against the oxidization stress induced by IRI, but also up-regulates anti-apoptosis protein Bcl-2 and decreases the expression of proapoptotic proteins, such as Bax, Caspase 3 and PARP. Bcl-2 family of proteins, including Bcl-2 (anti-apoptotic) and Bax (proapoptotic), mediate apoptosis by opening or closing mitochondrial permeability transition pore. Down-regulation of the ratio of Bcl-2 to Bax has been reported to be correlated with the activation of Caspase cascade, which further cleaves or activates downstream regulators, such as Caspase 3 and PARP, resulting in the subsequent apoptosis events [33].
The main function of mitochondria is ATP synthesis, which plays a major role in mediating cellular responses to ischemia reperfusion stress [34]. Previous studies showed that calcium influx into mitochondria and opening of permeability transition pores lead to apoptotic cell death in various organs including liver [35,36]. Additionally, the pharmacological targeting of mitochondrial ATP-sensitive K+ (mitoKATP) channels, with administration of drugs such as DE, protects myocytes from subsequent prolonged ischemia [37]. The major new finding from our study is that the MgIG pretreatment with combined with the mitoKATP channel opener DE shows an enhanced cytoprotective effect against hepatic IRI in vitro. This protective effect was reversed by co-administration of MgIG with 5-HD, an antagonist of mitoKATP channels. While the mechanism of exerted cellular protection by DE is not completely clear yet, one possible explanation is that the opening of mitoKATP channel minimizes calcium influx, mitochondrial swelling, and subsequent injury by depolarizing the membranes of mitochondrion [38,39].
The Akt and ERK signaling pathway plays a central role in intracellular processes, including cell survival and proliferation [40]. We observed that MgIG pretreatment significantly increased the levels of phosphorylated Akt and ERK in L02 cells with IRI, suggesting that the Akt and ERK signaling pathway was activated due to effects of MgIG. Decreased Akt phosphorylation when treated with 5-HD suggests that the mitoKATP channel is cross talking with Akt and ERK pathway. Our study suggests that MgIG, and the combined pretreatment of MgIG and DE may activate Akt/PI3K and ERK pathways, directly or indirectly. However, activation of ERK or Akt by MgIG may not be the only reason that leads to opening of mitoKATP, inhibition of apoptosis and cytoprotection when L02 cells with IRI was treated with MgIG. Hepatic IRI is caused by multifactorial processes, and numerous signaling cascades are involved. Further studies will be needed to elucidate the upstream and downstream regulators of these pathways in response to MgIG treatment.
In summary, our study showed that MgIG ameliorated the in vitro hepatic IRI by decreasing levels of oxidative stress and cell apoptosis. Our results may shed light on the utilization of MgIG pretreatment as a promising approach to combat liver IRI in vivo.
Acknowledgements
This work was supported by a grant from The Chinese Foundation for Hepatitis Prevention and Control (CFHPC), NO. 20132107.
Disclosure of conflict of interest
None.
References
- 1.Teoh NC. Hepatic ischemia reperfusion injury: Contemporary perspectives on pathogenic mechanisms and basis for hepatoprotection-the good, bad and deadly. J Gastroenterol Hepatol. 2011;26(Suppl 1):180–187. doi: 10.1111/j.1440-1746.2010.06584.x. [DOI] [PubMed] [Google Scholar]
- 2.Fernández V, Tapia G, Videla LA. Recent advances in liver preconditioning: Thyroid hormone, n-3 long-chain polyunsaturated fatty acids and iron. World J Hepatol. 2012;4:119–128. doi: 10.4254/wjh.v4.i4.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaouali MA, Abdennebi H, Padrissa-Altés S, Mahfoudh-Boussaid A, Roselló-Catafau J. Pharmacological strategies against cold ischemia reperfusion injury. Expert Opin Pharmacother. 2010;11:537–555. doi: 10.1517/14656560903547836. [DOI] [PubMed] [Google Scholar]
- 4.Papadopoulos D, Siempis T, Theodorakou E, Tsoulfas G. Hepatic ischemia and reperfusion injury and trauma: current concepts. Arch Trauma Res. 2013;2:63–70. doi: 10.5812/atr.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaeschke H, Mitchell JR. Mitochondria and xanthine oxidase both generate reactive oxygen species in isolated perfused rat liver after hypoxic injury. Biochem Biophys Res Commun. 1989;160:140–147. doi: 10.1016/0006-291x(89)91632-x. [DOI] [PubMed] [Google Scholar]
- 6.Glantzounis GK, Salacinski HJ, Yang W, Davidson BR, Seifalian AM. The contemporary role of antioxidant therapy in attenuating liver ischemia-reperfusion injury: a review. Liver Transpl. 2005;11:1031–1047. doi: 10.1002/lt.20504. [DOI] [PubMed] [Google Scholar]
- 7.Song X, Zhang N, Xu H, Cao L, Zhang H. Combined preconditioning and postconditioning provides synergistic protection against liver ischemic reperfusion injury. Int J Biol Sci. 2012;8:707–718. doi: 10.7150/ijbs.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 9.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly (ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 10.Burlacu A. Regulation of apoptosis by Bcl-2 family proteins. J Cell Mol Med. 2003;7:249–257. doi: 10.1111/j.1582-4934.2003.tb00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: processes in inflammatory networks--a review. Liver Transpl. 2010;16:1016–1032. doi: 10.1002/lt.22117. [DOI] [PubMed] [Google Scholar]
- 12.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 13.Akao M, O’Rourke B, Kusuoka H, Teshima Y, Jones SP, Marbán E. Differential actions of cardioprotective agents on the mitochondrial death pathway. Circ Res. 2003;92:195–202. doi: 10.1161/01.res.0000051862.16691.f9. [DOI] [PubMed] [Google Scholar]
- 14.Akao M, Ohler A, O’Rourke B, Marbán E. Mitochondrial ATP-sensitive potassium channels inhibit apoptosis induced by oxidative stress in cardiac cells. Circ Res. 2001;88:1267–1275. doi: 10.1161/hh1201.092094. [DOI] [PubMed] [Google Scholar]
- 15.Ardehali H, O’Rourke B. Mitochondrial K (ATP) channels in cell survival and death. J Mol Cell Cardiol. 2005;39:7–16. doi: 10.1016/j.yjmcc.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gross GJ, Auchampach JA. Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs. Circ Res. 1992;70:223–233. doi: 10.1161/01.res.70.2.223. [DOI] [PubMed] [Google Scholar]
- 17.Saidi RF, Chang J, Verb S, Brooks S, Nalbantoglu I, Adsay V, Jacobs MJ. The effect of methylprednisolone on warm ischaemia-reperfusion injury in the liver. Am J Surg. 2007;193:345–347. doi: 10.1016/j.amjsurg.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Soltys K, Dikdan G, Koneru B. Oxidative stress in fatty livers of obese Zucker rats: rapid amelioration and improved tolerance to warm ischaemia with tocopherol. Hepatology. 2001;34:13–18. doi: 10.1053/jhep.2001.25452. [DOI] [PubMed] [Google Scholar]
- 19.Kullmann R, Breull WR, Reinsberg J, Wassermann K, Konopatzki A. Dopamine produces vasodilatation in specific regions and layers of the rabbit gastrointestinal tract. Life Sci. 1983;32:2115–2122. doi: 10.1016/0024-3205(83)90100-5. [DOI] [PubMed] [Google Scholar]
- 20.Abu-Amara M, Gurusamy K, Hori S, Glantzounis G, Fuller B, Davidson BR. Systematic review of randomized controlled trials of pharmacological interventions to reduce ischaemia-reperfusion injury in elective liver resection with vascular occlusion. HPB (Oxford) 2010;12:4–14. doi: 10.1111/j.1477-2574.2009.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bao QD, Yang LL, Wang L, Cui DL. Protective effects of magnesium isoglycyrrhizinate against carbon tetrachloride-induced acute liver injury in mice. World Chinese J Digestology. 2008;16:1004–1007. [Google Scholar]
- 22.Dong LP, Yu F, Liu J, Mu XM. Protective effect of magnesium isoglycyrrhizinate on acute hepatic injury in mice. China Pharmacy. 2006;17:902–904. [Google Scholar]
- 23.Zhang ZL, Li ZJ, Liu SK, Zhou YL. [Effect of notoginseng radix on expression quantity of TGF-beta1/Smads and CTGF mRNA in rats with alcoholic liver disease] . Zhongguo Zhong Yao Za Zhi. 2013;38:2859–2862. [PubMed] [Google Scholar]
- 24.Xiao ZW, Zhang W, Ma L, Qiu ZW. Therapeutic effect of magnesium isoglycyrrhizinate in rats on lung injury induced by paraquat poisoning. Eur Rev Med Pharmacol Sci. 2014;18:311–320. [PubMed] [Google Scholar]
- 25.Chen KJ, Chen WY, Chen X. Increased elimination of paclitaxel by magnesium isoglycyrrhizinate in epithelial ovarian cancer patients treated with paclitaxel plus cisplatin: a pilot clinical study. Euro J Drug Metabo Pharmacokinet. 2014;39:25–31. doi: 10.1007/s13318-013-0136-y. [DOI] [PubMed] [Google Scholar]
- 26.Zhang JC, Zheng GF, Wu MX, Wu JW, Ouyang LY, Liu XQ. [Effect of magnesium isoglycyrrhizinate on PLA2 during liver tissue injury following limb ischemia/reperfusion in rats] . Zhonghua Gan Zang Bing Za Zhi. 2012;20:537–541. doi: 10.3760/cma.j.issn.1007-3418.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 27.Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47:19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- 28.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 29.Salvesen GS. Caspases: opening the boxes and interpreting the arrows. Cell Death Differ. 2002;9:3–5. doi: 10.1038/sj.cdd.4400963. [DOI] [PubMed] [Google Scholar]
- 30.Bao QD, Yang LL, Wang L, Cui DL. Protective effects of magnesium isoglycyrrhizinate against carbon tetrachloride-induced acute liver injury in mice. World Chinese J Digestology. 2008;16:1004–1007. [Google Scholar]
- 31.Wang P, Wu XM. Therapeutic effects of magnesium isoglycyrrhizinate on chronic liver injury induced by CCl4 in rats. Chin J New Drugs Clin Rem. 2004;23:833–836. [Google Scholar]
- 32.Casillas-Ramírez A, Mosbah IB, Ramalho F, Roselló-Catafau J, Peralta C. Past and future approaches to ischemia-reperfusion lesion associated with liver transplantation. Life Sci. 2006;79:1881–1894. doi: 10.1016/j.lfs.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 33.Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Bio. 2013;5:a008722. doi: 10.1101/cshperspect.a008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4:177–197. doi: 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeng Z, Huang HF, He F, Wu LX, Lin J, Chen MQ. Diazoxide attenuates ischemia/reperfusion injury via upregulation of heme oxygenase-1 after liver transplantation in rats. World J Gastroenterol. 2012;18:1765–1772. doi: 10.3748/wjg.v18.i15.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffiths JE. Mitochondria-potential role in cell life and death. Cardiovasc Res. 2000;46:24–27. doi: 10.1016/s0008-6363(00)00020-1. [DOI] [PubMed] [Google Scholar]
- 37.Ockaili R, Emani VR, Okubo S, Brown M, Krottapalli K, Kukreja RC. Opening of mitochondrial KATP channel induces early and delayed cardioprotective effect: role of nitric oxide. Am J Physiol Heart Circ Physiol. 1999;277:H2425–H2434. doi: 10.1152/ajpheart.1999.277.6.H2425. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Wang S, Harvat T, Kinzer K, Zhang L, Feng F, Qi M, Oberholzer J. Diazoxide, a KATP Channel Opener, Prevents Ischemia? Reperfusion Injury in Rodent Pancreatic Islets. Cell Transplant. 2013 doi: 10.3727/096368913X673441. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Csonka C, Kupai K, Bencsik P, Görbe A, Pálóczi J, Zvara A, Puskás LG, Csont T, Ferdinandy P. Cholesterol-enriched diet inhibits cardioprotection by ATP-sensitive K+ channel activators cromakalim and diazoxide. Am J Physiol Heart Circ Physiol. 2014;306:H405–413. doi: 10.1152/ajpheart.00257.2013. [DOI] [PubMed] [Google Scholar]
- 40.Liu RG, Song N, Li JM, Cui X, Chen YQ. [Akt involved in diazoxide preconditioning against rat hippocampal neuronal apoptosis induced by anoxia-reoxygenation injury] . Zhonghua Yi Xue Za Zhi. 2010;90:492–495. [PubMed] [Google Scholar]