

Abstract
DNA damage response and repair are carried out by certain proteins following damage by environmental clastogens, such as ionizing radiation and reactive oxygen species. It has been reported that many carcinomas that are characterized by resistance to chemotherapy and poor outcomes show dysfunction of these proteins. Chromobox homologue 8 (CBX8), a member of the polycomb group of proteins, has been identified as a factor that protects tumor cells from the detrimental effects of ionizing radiation (IR) or hydrogen peroxide (H2O2). In this study, we found that CBX8 was up-regulated in esophageal carcinoma tissues compared with adjacent non-cancerous tissues (P<0.01) and correlated with TNM stage in esophageal squamous cell carcinoma patients. Depletion of CBX8 decreased cell proliferation both in vitro and in vivo and increased the phosphorylation levels of p21, Wee1, and CHK1, which result in cyclin-dependent kinase inhibition and cell-cycle delay. CBX8 depletion also led to accumulation of spontaneous DNA damage and raised the sensitivity of tumor cells to IR or H2O2. We also found that the total level of CBX8 in the cells was increased after treating tumor cells with clastogens. In addition, our data showed that decreased CBX8 expression was accompanied by the reduction of EZH2 and EED, which have been reported to participate in DNA damage repair. Collectively, CBX8 might emerge as an oncogene for promoting the proliferation of tumor cells and raising the resistance of neoplasms to chemotherapy.
Keywords: CBX8, DNA repair, esophageal carcinoma, G2M cell cycle, tumorigenesis
Introduction
The polycomb group (PcG) of proteins is essential for regulating cell proliferation and can contribute to the development of cancer [1-4]. PcG proteins participate in the formation of two major protein complexes, known as the polycomb repressive complexes 1 and 2 (PRC1 and PRC2), whose function is to maintain transcriptional repression. In humans, PRC2 is comprised of three PcG proteins (EZH2, EED, and SUZ12), and RbAp48 [5-8]. EZH2 possesses histone methyltransferase activity and trimethylates lysine 27 on histone H3 (H3K27me2/3) [9]. Trimethylated H3K27 is believed to facilitate recruitment of the PRC1 complex, which is essential for maintaining the repression of target genes during differentiation and development [10].
Chromobox homolog 8 (CBX8), a member of PRC1, belongs to the CBX protein family (which includes CBX2, 4, 6, 7, and 8), which is homologous to the Drosophila polycomb (Pc) protein [11]. CBX8 has been reported to regulate cell mitosis in human and mouse fibroblasts through direct binding to the INK4A-ARF locus, and the ectopic expression of CBX8 leads to cellular immortalization [12,13]. The efficient and accurate repair of chromosomal lesions is critical in preventing cell death and carcinogenesis when cells are exposed to detrimental environmental factors. It has been reported that the members of the polycomb complex (CBX8 and EZH2 are enriched on sites of DNA damage. Silencing EZH2 or EED by several independent shRNAs renders U2OS cells sensitive to ionizing radiation (IR) [14].
The current treatments for cancer, which typically include radiotherapy, chemotherapy and surgery, are often not enough to cure patients of their cancers [15]. How cancer cells become resistant to treatments is an important research focus [16]. Thus many cancers may also become resistant to the DNA damage that results from chemotherapy drugs [17]. For example, ABL could be regulated by AXL to promote cisplatin resistance in esophageal cancer [18]. Tics (also called cancer stem cells) are partly responsible for resistance to DNA-damaging treatment. Research has shown that enhancing the DNA repair capability of tumor cells results in tumorigenesis [19]. For example, the over-expression of apoptosis-resistant protein promotes tumor formation and development by increasing cancer cell DNA-repair capability [20]. Enhanced survival mechanisms of malignant cells against DNA damaging agents can sustain resistance against chemotherapy.
In this study, we found that CBX8 knockdown increased the sensitivity of esophageal tumor cells to DNA clastogen and decreased the expression levels of P21, EZH2, and EED. Moreover, CBX8 could prevent the accumulation of spontaneous DNA damage. Consequently, loss of CBX8 causes defects in DNA repair and checkpoint activity, rendering cells hypersensitive to IR or H2O2. Thus, the PcG protein CBX8 is a novel DNA damage response factor that mediates DNA damage repair and tumorigenesis.
Materials and methods
Cell culture
EC109 and EC9706 cells were obtained from the Tumor Center of the Chinese Academy of Medical Science. HeLa, CCD-1079Sk and HaCaT were purchased from the cell bank of the Chinese Academy of Science. All cell lines were maintained at 37°C under a 5% CO2 atmosphere in Dulbecco’s Modified Eagle Medium (DMEM) high glucose (Invitrogen, USA) with 10% fetal bovine serum (FBS).
Depletion of CBX8 in cells
We transfected 50 nM CBX8-siRNA oligonucleotides (Sigma, USA), with the following sequences: siCBX8-1 5’-CTCGCTTGCTCGCAGCCTT-3’, siCBX8-2 5’-GAGAGTGAGCGTGAGCTTG-3’, into cells using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions. The transfected cells were harvested 72 h after the initiation of transfection. For stable CBX8 depletion, the cells were infected with lentivirus (LV) that expressed CBX8-shRNA (Genchem, China) or its negative control at 30-50% confluence. The monoclonal population of the stably infected cells was selected by a limiting dilution assay using the green fluorescent protein marker.
Cell proliferation assay
The treated cells (1 × 103 cells per well) were seeded in 96-well plates. The MTT assay was performed on days 1, 3, 5, and 6. Sample absorbance was measured with a spectrophotometer reader at 490 nm. Each assay was performed in triplicate.
Colony formation assay
The treated cells were seeded in 12-well plates (200 cells per well), incubated for 7-10 days for colony growth. Then, these cells were fixed in 4% formaldehyde for 20 min and stained with 0.1% crystal violet for 15 min. Colonies containing 50 or more cells were counted.
Cell cycle analysis
Cells were harvested by trypsinization, fixed in 70% ethanol, and resuspended in 500 μL PBS buffer, 10 μL propidium iodide (Sigma, USA), 2.5 μL RNase1, and 0.5 μL 20% NP40. Samples were incubated for 15 min before analysis. DNA content was analyzed by flow cytometry (BD Biosciences, USA).
Real-time PCR
Total RNA from tissue samples and cell lines was extracted using TRIzol reagent (Invitrogen, USA), following the manufacturer’s protocol. Real-time RT-PCR was performed using SYBR premix Ex Taq (TaKaRa, Japan) and the Applied Biosystems 7900 Real-Time PCR System (Applied Biosystems, USA), including the supplied analytical software. The specific primer sequences were as follows: CBX8-sense 5’-CTGCTGCTTTTCTGCTCC’-3’ and CBX8-antisense 5’-CATCCCTTCCATTTCACG-3’, 18S rRNA-sense 5’-CCTGGATACCGCAGCTAGGA-3’ and (18S rRNA-antisense 5’-CGCCGCGTTATGCTTACGGGG-3’.
Western blot
For western blot, cells were harvested and lysed in RIPA lysis buffer (Beyotime, China). A total of 30 μg protein/sample was processed for SDS-PAGE. Primary antibodies used were: CBX8 (Santa Cruz Biotechnology, USA); Wee1, p21, p-PLK1 (Thr210), and CHK1 (all from Cell Signaling Technology, USA); β-actin (Sigma, USA); and γ-H2AX (Abcam, USA). The membrane was scanned on an Odyssey infrared imaging system (LI-COR, USA) at a wavelength of 700 or 800 nm.
Immunohistochemistry
For immunohistochemistry, the slides were deparaffinized, rehydrated, and immersed in 3% hydrogen peroxide solution for 10 min. Following treatment with 10% goat serum at 37°C for 30 min, the slides were incubated with anti-CBX8 (1:100 dilution, Santa Cruz, USA) at 4°C for 12 h. After washing with PBS, the slides were incubated with Signalstain Boost IHC Detection Reagent (Cell Signaling Technology, USA) for 1 h at room temperature. The slides were developed using DAB solution (Vector, SK-4100, USA), and counterstaining was performed with hematoxylin.
Comet assay
DNA damage was evaluated in EC109 cells using an Oxiselect Comet Assay Kit (Cell Biolab, USA) according to the manufacturer’s instructions. A total of 100 cells were analyzed per sample.
Generation of DNA damage
DNA damage was induced by IR, which was delivered by an X-ray generator (Gammacell-40γ) or H2O2 (Sangon Biotech, China).
Xenograft mouse model
In vivo studies of the inhibitory function of CBX8-shRNA-lentivirus (LV) were conducted in nude mice. Ten nude mice aged 4 weeks were purchased from BIkai Laboratory Animal Corp. Ltd (Shanghai, China). According to the recommendations guidelines of the Animal Care and Use Committee of The Tenth People’s Hospital of Shanghai, the studies were performed strictly with the permit number: 2011-RES1. The protocol was approved by Science and Technology Commission of Shanghai Municipality (ID: SYXK 2007-0006). Then the immune-deficient mice were subcutaneously injected with 5 × 106 EC109 cells expressing CBX8-shRNA-LV or the control (ctrl)-LV. Tumor size was measured using a caliper, and tumor volume was calculated as 0.5 × L × W2, with L indicating length and W indicating width. The mice were sacrificed under sodium pentobarbital anesthesia 30 days after injection. Additionally all efforts were made to minimize suffering.
Results
CBX8 was abnormally expressed in esophageal carcinoma tissues
Although CBX8 has been reported to regulate and co-localize on the INK4a tumor suppressor locus [11,13] and to be essential for MLL-AF9-induced leukemogenesis [21], the relationship between CBX8 and solid tumor has rarely been researched. In order to explore the impact of CBX8 on tumorigenesis, we detected CBX8 expression from 59 pairs of samples, each consisting of a sample of esophageal carcinoma tissue and a corresponding sample of adjacent normal tissue. The results of real-time reverse transcriptase-polymerase chain reaction (RT-PCR) showed that the average CBX8 mRNA levels were 3-fold higher in esophageal carcinoma tissues than in adjacent normal tissues (P<0.01) (Figure 1A). Expression was also assessed by western blot and immunohistochemistry, and the results further confirmed that the protein levels of CBX8 in esophageal carcinoma tissues were higher than in adjacent normal tissues (Figure 1B and 1C). In addition, we analyzed the expression of CBX8 in human skin fibroblasts CCD-1079Sk, human keratinocyte cell line HaCaT, and esophageal squamous cell carcinoma (ESCC) cell lines EC109 and EC9706. As shown in Figure 1D, no endogenous CBX8 expression was observed in normal human cell lines CCD-1079Sk and HaCaT, but high CBX8 expression was found in EC109 and EC9706. These data suggest that abnormally high expression of CBX8 might have an important role in esophageal tumorigenesis.
Figure 1.

Expression of CBX8 in esophageal tissues and cell lines. A. CBX8 mRNA levels were measured in 59 pairs of esophageal carcinoma and corresponding adjacent normal tissues by real-time RT-PCR, where 18S was used as an internal reference. The line in each box is the median -ΔCt value, while the upper and lower edges of each box show the 75th and 25th percentile, respectively. P value was calculated by paired t-test (P<0.01). B. The difference in CBX8 expression was tested by western blot analysis. β-Actin was the loading control. N represents adjacent normal tissues, T represents carcinoma tissues. C. Representative immunohistochemical detection of the CBX8 protein expression in paired esophageal tissues, N represents adjacent normal tissues, T represents carcinoma tissues. Left: hematoxylin-eosin staining (H&E); Right: immunohistochemistry. D. CBX8 expression patterns in normal cell lines and ESCC cell lines.
Expression of CBX8 is correlated with TNM stage in ESCC patients
To reveal the functions of CBX8 in tumorigenesis, we analyzed the relationship between abnormal CBX8 expression and the clinicopathologic status of ESCC patients. As shown in Table 1, no significant association was found between CBX8 expression and other clinical characteristics such as age, gender, gross pathological type, tumor position, tumor size, but relative CBX8 expression significantly increased with TNM stage (P=0.027), suggesting that CBX8 expression is correlated with TNM stage in ESCC patients.
Table 1.
Association between CBX8 expression and clinicopathologic information of ESCC patients
| Characteristic | Total (n=59) | CBX8 expression | P | ||
|---|---|---|---|---|---|
|
| |||||
| Low | High | Normal | |||
| Age (years) | 0.91 | ||||
| <60 | 25 | 1 | 11 | 13 | |
| ≥60 | 34 | 1 | 13 | 20 | |
| Gender | 0.667 | ||||
| Female | 31 | 1 | 15 | 15 | |
| Male | 27 | 1 | 8 | 18 | |
| Position of tumor | 0.06 | ||||
| Upper | 3 | 1 | 1 | 1 | |
| Middle | 44 | 1 | 18 | 25 | |
| Lower | 11 | 0 | 5 | 6 | |
| Gross Pathology | 0.099 | ||||
| Fungating | 3 | 0 | 1 | 2 | |
| Medullary | 26 | 1 | 9 | 16 | |
| Others | 30 | 1 | 14 | 15 | |
| N Classification | 0.175 | ||||
| N0 | 28 | 2 | 15 | 21 | |
| N1 | 21 | 0 | 9 | 12 | |
| TNM Stage | 0.027 | ||||
| I | 8 | 1 | 2 | 5 | |
| II | 39 | 0 | 20 | 19 | |
| III | 12 | 1 | 2 | 9 | |
| Tomor Size (cm3) | 0.681 | ||||
| <10 | 23 | 1 | 11 | 11 | |
| ≥10, ≤20 | 23 | 0 | 6 | 17 | |
| >20 | 13 | 1 | 7 | 5 | |
CBX8 expression was detected by real-time RT-PCR in paired tumor tissues and normal adjacent tissues from 59 ESCC patients. 18S was taken for normalization of the data. The patients were divided into high and low CBX8 expression by the 50th percentiles of 2-ΔΔCt. The association between relative CBX8 expression and clinicopathologic information was analyzed. P values were obtained using the χ2 test.
CBX8 knockdown prevents the tumor cell proliferation and slows down cell cycle progression
To knockdown endogenous CBX8 in ESCC cells, two independent siRNAs against CBX8 were transfected into EC109 cells, and the efficiency was verified as in Figure 2A. The results of the MTT assay showed that the proliferation of EC109 and EC9706 cells was significantly inhibited by transfection of siRNAs that target CBX8 (Figure 2B). Furthermore, we found that CBX8 knockdown suppressed the colony-forming efficiency of EC109 and EC9706 cells (Figure 2C). To test whether depletion of CBX8 could suppress tumorigenesis in vivo, EC109 cells infected with CBX8-ShRNA lentivirus (CBX8-shRNA-LV) or control lentivirus (Ctrl-LV) were subcutaneously inoculated into nude mice. As shown in Figure 2D, the xenograft tumors formed in the absence of CBX8 were of lower mean volume and weight compared with those formed in the control. All these data demonstrate that CBX8 depletion can decrease cell proliferation both in vitro and in vivo.
Figure 2.
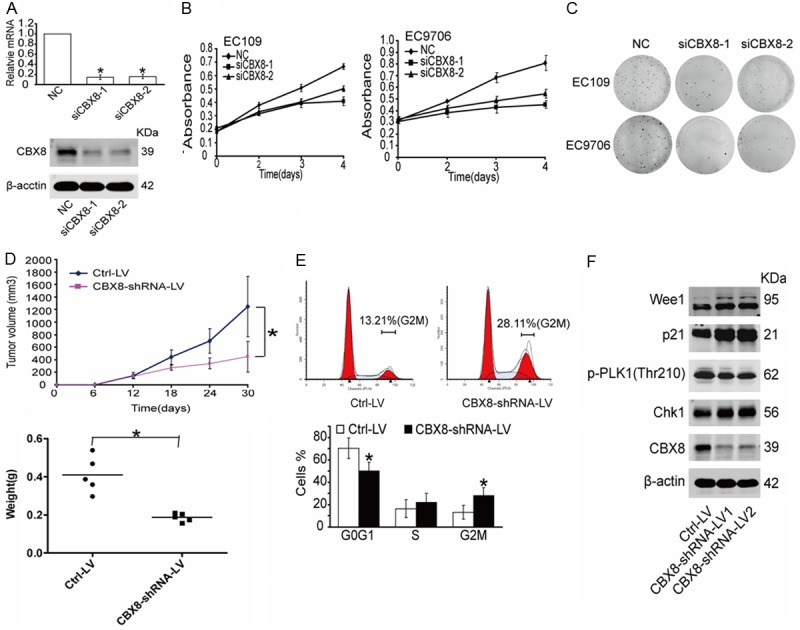
CBX8 is required for the proliferation of ESCC cells and cell cycle progression. (A) The interference efficiency of two CBX8-siRNAs was tested by both RT-PCR and western blot. (B, C) The relative cell proliferation was measured by MTT (B) and colony formation assays (C) in EC109 and EC9706 cells. (D) Xenograft mouse model. Tumor volume and weight were measured for 30 days after subcutaneous injection. n=5 per group. (E) FACS analysis showed that CBX8-depleted cells accumulated in the G2M phase in EC109. (F) G2M checkpoint-related proteins were detected in EC109 cells. (NC: Negative Control).
To further identify whether CBX8 depletion affects cell cycle progression, EC109 cells infected with CBX8-shRNA-LV or Ctrl-LV were harvested 24 h after synchronization in serum-free medium, and the cell cycle distribution of these cells was analyzed by flow cytometry. Our results showed that the population of cells in the G2M phase in the CBX8-depleted cells was twice that in the control cells (Figure 2E). In addition, we also detected the expression of key cell cycle factors and found that expression of Wee1, p21, and Chk1 were up-regulated, while PLK1 thr210 phosphorylation was down-regulated after CBX8 depletion; all these changes could lead to accumulation of cells in the G2M phase. These results were consistent with the flow cytometry analysis. Collectively, these data show that CBX8-depletion prevents tumor cell proliferation as a result of cell cycle arrest (Figure 2F).
Inhibition of CBX8 expression sensitizes cells to DNA damage
PcG proteins had been reported to be recruited to damaged chromatin after cells exposed to IR, suggesting that PcG proteins may play a direct role in the cellular response to DNA damage [22]. In addition, we have proved that depletion of CBX8, a member of the PcG group of proteins, could induce G2M arrest, and alter protein or phosphorylation levels of cell-cycle regulators. Therefore, we proposed that CBX8 depletion may influence DNA damage repair, leading to accumulation of spontaneous DNA damage, and subsequently induced cell cycle arrest and apoptosis. To assess this, neutral comet assays were performed to determine the effect of CBX8 knockdown on accumulation of DNA damage. The results showed that CBX8-depleted cells showed more damage than control cells in the absence of H2O2 and IR, and these cells were more sensitive to treatment with H2O2 and IR (Figure 3A and 3B). Additionally, the expression of phosphorylated H2AX (γ-H2AX), which is a well-established marker for DNA damage [23,24], was significantly up-regulated by inhibition of CBX8 in EC109 and EC9706 cells (Figure 3C and 3D). These data imply that CBX8 depletion results in accumulation of spontaneous DNA damage and decreases sensitivity to H2O2 and IR in ESCC cells.
Figure 3.
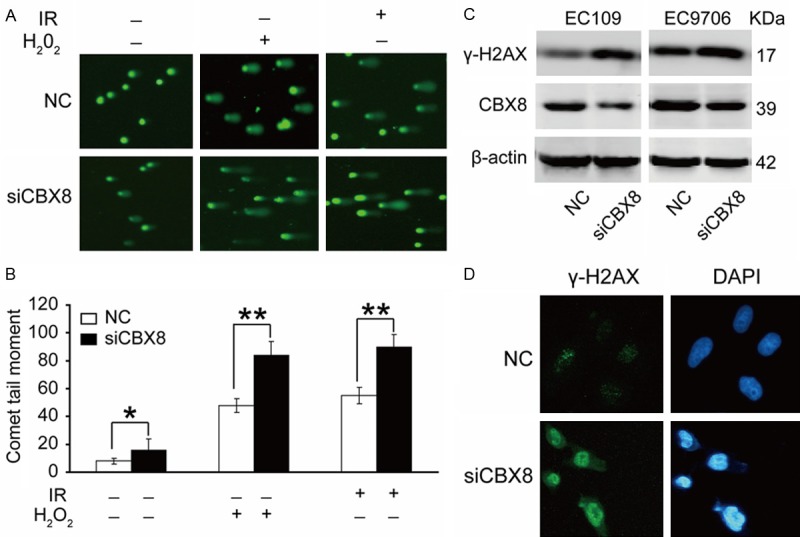
CBX8 is related to DNA damage. (A) CBX8-depletion spontaneously induces DNA damage. EC109 cells were transfected with the indicated siRNA for 48 h, exposed to 10 Gy of IR or 0.1 mM H2O2 for 20 min, and subjected to neutral comet analysis. (B) Quantification of tail moments using cells from (A). Tail moments for each condition were calculated on a minimum of 300 cells for each data point. Three independent experiments have been done for each result. *P<0.05, without IR or H2O2 treatment; **P<0.01, IR treatment or H2O2 treatment. (C) A marker for DNA DSBs, γ-H2AX, was tested by western blot both in EC109 and EC9706 cells. (D) EC109 cells transfected with NC or siCBX8 were fixed and immunostained with anti-γ-H2AX antibody.
EZH2 and EED, members of the PRC2 complex [5-8], have been reported to be involved in DNA damage repair in U2OS cells [14]. Trimethylation of H3K27 by EZH2 facilitates the recruitment of the PRC1 complex [10]. To investigate whether DNA damage induced by CBX8 depletion is related to EZH2 and EED, we detected the expression of EZH2 and EED in CBX8-depleted cells. As shown in Figure S1, inhibition of CBX8 decreased the protein levels of EZH2 and EED in both EC109 and EC9706 cells. These data suggest that knockdown of CBX8 may induce accumulation of spontaneous DNA damage by decreasing the expression of EZH2 and EED, resulting in impairment of repair capacity (Figure S1).
CBX8 promotes DNA damage repair
Increased expression is a characteristic hallmark shared by many important regulators involved in DNA damage response [25,26]. Therefore, to investigate whether CBX8 is involved in DNA damage repair, we analyzed the endogenous expression of CBX8 in EC109 and HeLa cells treated with H2O2 or IR. As shown in Figure 4A and 4B, CBX8 expression increased rapidly, reaching a maximum 2 or 3 h after treatment of EC109 and HeLa cells with H2O2 or IR. The expression of γ-H2AX increased with the dose of H2O2 or IR. We determined the repair capability of EC-109 cells in the absence of CBX8 by neutral comet assays and western blot. Compared with the NC group, in which DNA damage returned to basal levels at 6 h, the level of H2O2-induced damage remained higher at 6 h in CBX8-depleted cells and recovered at 8 h (Figure 4C). This result was confirmed by western blot (Figure 4D). These results demonstrated that CBX8 may play a direct role in the cellular response to DNA damage repair.
Figure 4.
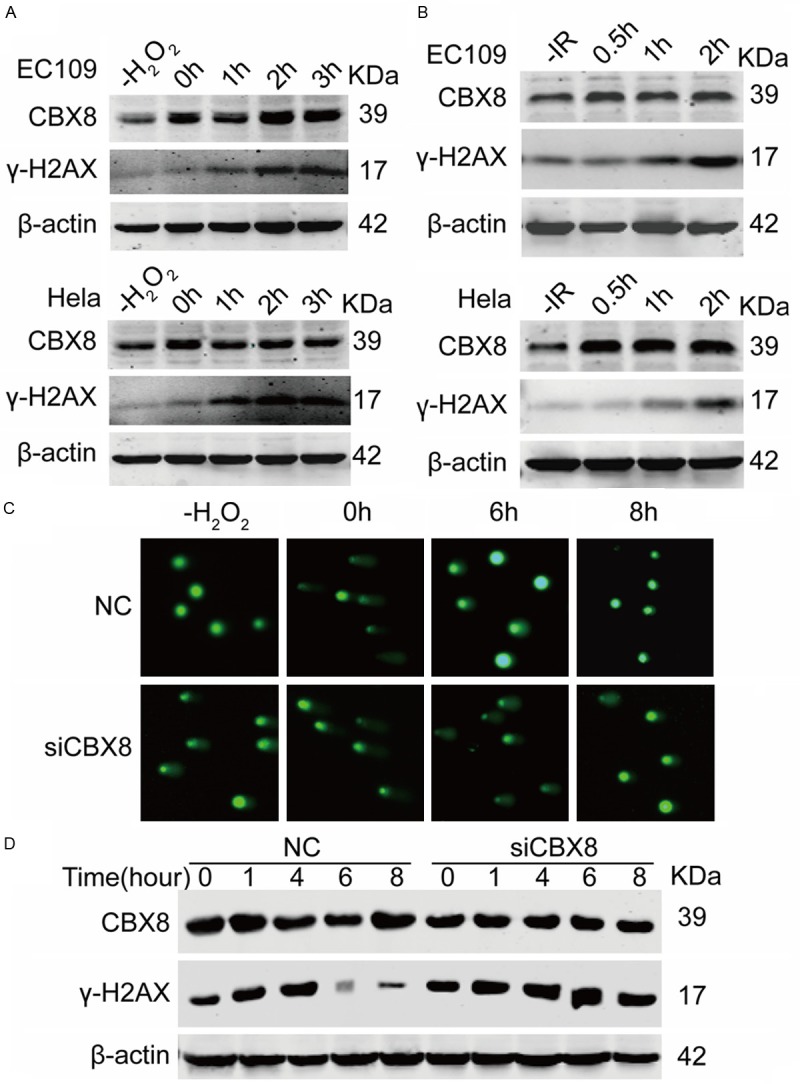
Depletion of CBX8 decreases resistance to H2O2 or IR. A, B. Both EC109 and HeLa cells were treated with H2O2 or IR. For H2O2 treatment, cells were exposed to 0.1 mM H2O2 for 20 min and then maintained in culture for indicated time points at 37°C, 5% CO2. For IR exposure, cells were exposed to 10 Gy IR and maintained in culture for indicated time points at 37°C, 5% CO2. CBX8 and γ-H2AX were determined by Western blot. C. EC109 cells were transfected with siCBX8 for 48 h, then exposed to 0.1 mM H2O2 for 20 min, and subjected to neutral comet assay at the indicated time points. D. EC109 cells were transfected with siCBX8; exposed to 0.1 mM H2O2 for 20 min; and maintained in culture for indicated time points. CBX8 and γ-H2AX levels were monitored by western blot analysis; β-actin was used as an internal reference. Results indicate that CBX8 depletion increases sensitivity of EC109 cells to H2O2.
Discussion
Our study demonstrated, by detecting the expression of CBX8 after cells were treated with IR or H2O2 that CBX8 participates in DNA damage repair. Knockdown of CBX8 in several tumor cells led to a dramatic increase in γ-H2AX expression, suggesting that CBX8 depletion induced massive DNA damage [27]. CBX8 also upregulates the expression of EZH2 and EED. The proteins EZH2 and EED were reported to accumulate at the sites of DNA lesions and to participate in DNA damage repair [28]. We speculate that CBX8 is involved in DNA damage response and repair through the regulation of EZH2 and EED. FACS analysis showed that the G2M checkpoint was activated in CBX8-depleted cells. Therefore, the slowdown of cell proliferation after CBX8 depletion might result from the G2M phase arrest. The relationship between CBX8 and tumorigenesis was investigated by detecting the differences in CBX8 expression between esophageal carcinoma tissues and corresponding normal tissues and then analyzing the correlations between CBX8 expression and clinicopathological variables, which demonstrated that CBX8 expression was correlated with the generation and development of esophageal carcinoma.
It is now evident that CBX8 is involved in DNA damage repair, but the mechanism of repair remains unclear. It is possible that the cells initiate a process known as “transcription-coupled repair”, when confronting DNA damage factors [29-31]. As a transcriptional repressor, CBX8 will be recruited to DNA damage sites to suppress the transcriptional process at the lesions and to reduce the activities of RNA polymerase, through which the generation of broken mRNA could be avoided. Meanwhile, other DNA repair factors, such as EZH2, EED, and TIP60 will also be recruited by CBX8 [21,32,33].
It has been reported that esophageal carcinoma is characterized by resistance to chemotherapy and radiotherapy, so the high expression of CBX8 in carcinoma tissues seems likely to play an important role in tumorigenesis. Moreover, over-expression of CBX8 can inhibit apoptosis and promote cell proliferation. Thus, CBX8 is a novel oncogene that reinforces the resistance of tumor cells to DNA-damaging drugs, mediating the correlation between the occurrence of DNA damage and tumorigenesis.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81272292).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.van Kemenade FJ, Raaphorst FM, Blokzijl T, Fieret E, Hamer KM, Satijn DP, Otte AP, Meijer CJ. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 2001;97:3896–3901. doi: 10.1182/blood.v97.12.3896. [DOI] [PubMed] [Google Scholar]
- 2.Bea S, Tort F, Pinyol M, Puig X, Hernandez L, Hernandez S, Fernandez PL, van Lohuizen M, Colomer D, Campo E. BMI-1 gene amplification and overexpression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res. 2001;61:2409–2412. [PubMed] [Google Scholar]
- 3.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 6.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 7.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 9.Akhtar A, Lanzuolo C, Lo Sardo F, Diamantini A, Orlando V. PcG complexes set the stage for epigenetic inheritance of gene silencing in early S Phase before replication. PLoS Genet. 2011;7:e1002370. doi: 10.1371/journal.pgen.1002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Blagosklonny MV, Maertens GN, El Messaoudi-Aubert S, Racek T, Stock JK, Nicholls J, Rodriguez-Niedenführ M, Gil J, Peters G. Several distinct polycomb complexes regulate and co-localize on the INK4a tumor suppressor locus. PLoS One. 2009;4:e6380. doi: 10.1371/journal.pone.0006380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Monch K, Minucci S, Porse BT, Marine JC, Hansen KH, Helin K. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dietrich N, Bracken AP, Trinh E, Schjerling CK, Koseki H, Rappsilber J, Helin K, Hansen KH. Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. EMBO J. 2007;26:1637–48. doi: 10.1038/sj.emboj.7601632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley MR, Fishel ML. DNA repair proteins as molecular targets for cancer therapeutics. Anticancer Agents Med Chem. 2008;8:417–25. doi: 10.2174/187152008784220294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurosawa A, Adachi N. Functions and regulation of Artemis: a goddess in the maintenance of genome integrity. J Radiat Res. 2010;51:503–509. doi: 10.1269/jrr.10017. [DOI] [PubMed] [Google Scholar]
- 17.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong J, Peng D, Chen Z, Sehdev V, Belkhiri A. ABL regulation by AXL promotes cisplatin resistance in esophageal cancer. Cancer Res. 2013;73:331–340. doi: 10.1158/0008-5472.CAN-12-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burhans WC, Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007;35:7545–7556. doi: 10.1093/nar/gkm1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kojic M, Holloman WK. Brh2 domain function distinguished by differential cellular responses to DNA damage and replication stress. Mol Microbiol. 2012;83:351–361. doi: 10.1111/j.1365-2958.2011.07935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan J, Jones M, Koseki H, Nakayama M, Muntean AG, Maillard I, Hess Jay L. CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer Cell. 2011;20:563–575. doi: 10.1016/j.ccr.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shao Z, Raible F, Mollaaghababa R, Guyon J, Wu C, Bender W, Kingston R. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell. 1999;98:37–46. doi: 10.1016/S0092-8674(00)80604-2. [DOI] [PubMed] [Google Scholar]
- 23.Altaf M, Saksouk N, Côté J. Histone modifications in response to DNA damage. Mutat Res. 2007;618:81–90. doi: 10.1016/j.mrfmmm.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Cha H, Lowe JM, Li H, Lee JS, Belova GI, Bulavin DV, Fornace AJ Jr. Wip1 directly dephosphorylates-H2AX and attenuates the DNA damage response. Cancer Res. 2010;70:4112–4122. doi: 10.1158/0008-5472.CAN-09-4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol. 2010;190:741–749. doi: 10.1083/jcb.201001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimura T, Torres MJ, Martin MM, Rao VA, Pommier Y, Katsura M, Miyagawa K, Aladjem MI. Bloom’s syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J Mol Biol. 2008;375:1152–1164. doi: 10.1016/j.jmb.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gutierrez L, Oktaba K, Scheuermann JC, Gambetta MC, Ly-Hartig N, Muller J. The role of the histone H2A ubiquitinase Sce in Polycomb repression. Development. 2011;139:117–127. doi: 10.1242/dev.074450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Z, Lee ST, Qiao Y, Li Z, Lee PL, Lee YJ, Jiang X, Tan J, Aau M, Lim CZH, Yu Q. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ. 2011;18:1771–1779. doi: 10.1038/cdd.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andreassen PR, Ho GP, D’Andrea AD. DNA damage responses and their many interactions with the replication fork. Carcinogenesis. 2005;27:883–892. doi: 10.1093/carcin/bgi319. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Jørgensen M, Kolde R, Zhao X, Parker B, Valen E, Wen J, Sandelin A. Prediction of RNA Polymerase II recruitment, elongation and stalling from histone modification data. BMC Genomics. 2011;12:544. doi: 10.1186/1471-2164-12-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lagerwerf S, Vrouwe MG, Overmeer RM, Fousteri MI, Mullenders LH. DNA damage response and transcription. DNA Repair (Amst) 2011;10:743–750. doi: 10.1016/j.dnarep.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 32.Zeidler M, Varambally S, Cao Q, Chinnaiyan AM, Ferguson DO, Merajver SD, Kleer CG. The Polycomb group protein EZH2 Impairs DNA repair in breast epithelial cells. Neoplasia. 2005;7:1011–1019. doi: 10.1593/neo.05472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006;16:433–442. doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.