

Abstract
Spindle cell rhabdomyosarcoma (RMS) is an uncommon histiologic variant of RMS that has spindle cell morphology. This tumor occurs almost exclusively in childhood and more rarely in adults. Only a few adult cases, including two retroperitoneal cases in male patients, have been documented previously. We describe a rare case of spindle cell RMS of the retroperitoneum in a 37-year-old woman developed during pregnancy and incidentally discovered after vaginal delivery. Computed tomography showed a huge tumor mass, measured 20 × 20 × 15 cm in size, arising in retroperitoneal space. Histologically, the tumor consisted of spindle cells arranged in a fascicular or herringbone growth pattern, morphologically mimicking adult fibrosarcoma, intermingled with scattered rhabdomyoblasts. Mitotic activity ranged from 20 to 28 mitoses per 10 high-power fields and tumor necrosis was evident. Immunohistochemically, tumor cells were stained diffusely positive for muscle specific actin, desmin, and vimentin, scattered positive for myogenin, MyoD1 and myoglobin, with a Ki-67 (MIB-1) proliferative labeling index of 46.11%. This tumor also stains positively for CD99, strong cytoplasmic WT1, and nuclear p53. Other markers such as S100 protein, smooth muscle specific actin, CD34, cytokeratin, and epithelial membrane antigen were all negative in the tumor cells. On the basis of the findings, a spindle cell RMS was diagnosed. The neoplasm was incompletely excised because of encasement of major vessels and invasion to adjacent structures, and additional chemotherapy was given.
Keywords: Rhabdomyosarcoma, spindle cell, adult, retroperitoneum, immunohistochemistry
Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue tumor manifesting features of skeletal muscle differentiation, usually occurring in childhood or adolescence [1,2]. Histologically, RMS is classified as embryonal, alveolar, and pleomorphic subtypes that differ in the degree of differentiation, clinical aggressiveness, and prognosis [2]. Spindle cell and sclerosing, pseudovascular RMS, a rare histological variant of RMS [3-6], has been previously included as a subtype of embryonal RMS [1], and now represents a distinct subtype of RMS [7].
Spindle cell RMS was first recognized in the pediatric population in 1992 as a rare variant that occurred mainly in males, arose preferentially in the paratesticular area and the head and neck region, and carried a better prognosis when compared with other RMS variants [8]. Adult cases were first described in 1998 [9], subsequently, larger adult series showed it to be a disease predominantly affecting males in a wide age range [5,6]. Spindle cell RMS accounts for less than 10% of adult RMSs, most commonly occurs on the head and neck, but are found at a wide range of sites, including the diaphragm [9], paratesticular area [10] and uterus [11]. Other rarely affected anatomical locations include the viscera and retroperitoneum, and only two male cases of retroperitoneal cases have been previously described [5,12]. These lesions in adults appear to have distinct clinicopathologic features and a more aggressive clinical course when compared with cases occurring in the pediatric population [5].
We report here the first female case of spindle cell RMS that closely mimics adult fibrosarcoma arising in the retroperitoneal space in a 37-year-old woman during pregnancy. The unique features of this case including the presentation of a spindle cell RMS in a pregnant woman in an unusual location have not been reported previously.
Case report
A 37-year-old woman, gravida 1, para 1, abortus 0, had ill-defined left flank pain and noticed abnormal abdominal enlargement during the latter half of her first pregnancy. The patient attributed those to pregnancy and did not seek medical attention. She was otherwise healthy. The patient was admitted in labor at 40+6 weeks. A healthy female weighing 3,850 g, with Apgar scores of nine at 1 and 5 min, was born by spontaneous vaginal delivery. Immediately after delivery, however, a distinct mass in the left lower quadrant was shown without abdominal tenderness. Routine investigations of the mother’s and baby’s blood and urine revealed normal results. Her past and a family history were otherwise unremarkable. Abdominal computed tomography revealed a mass arising from the left retroperitoneal region measuring 20 × 20 × 15 cm with homogeneous density and without any areas of necrosis or calcification, suspected to be an abdominal tumor, while other abdominal organs were essentially normal (Figure 1A, 1B).
Figure 1.
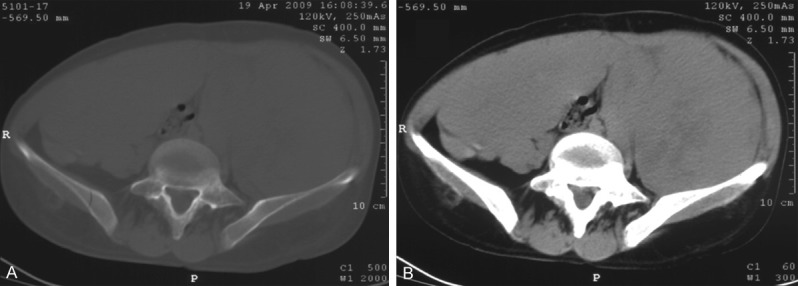
Abdominal computed tomography. A. Non-enhanced axial CT image demonstrates a large, homogeneous, soft tissue mass about 20 × 15 cm in diameter, occupying the whole abdominal and pelvic cavity and compressing the intraabdominal organs to the caudal side. B. Contrast-enhanced CT shows a tumor with early enhancement in mainly dorsal side of the tumor.
A laparotomy was performed under general anesthesia two weeks after delivery. Exploration revealed a poorly encapsulated, firm, globular mass lay retroperitoneally in the left paravertebral gutter, displacing the left uterus and left ovary. The abdomen and pelvis demonstrated no evidence of tumor involvement in any specific organ as the site of origin of the tumor and the peritoneal cavity was found to be free of tumor, suggestive of a retroperitoneal rather than gynecologic origin. Intraoperative frozen-section revealed densely cellular spindle cell neoplasm suspected malignant. The tumor was too large to completely excise because of encasement of major vessels and invasion to adjacent structures.
Materials and methods
Tumor tissues were fixed in 10% neutral buffered formalin, routinely processed, and embedded in paraffin wax. Tissue sections of 4-5 μm thickness were prepared and stained with hematoxylin and eosin (H&E stain), and additional sections were used for immunohistochemical staining after deparaffinization, rehydration, and antigen retrieval treatments. The following primary antibodies and dilutions were used: actin (HHF35, 1:50), α-smooth muscle actin (SMA) (1A4, 1:50), Bcl-2 (8C8, Neat), Bcl-6 (P1F6+PG-B6P, 1:80), CD34 (QBEND10, Neat), CD45 (PD7/26+2B11, 1:100), CD68 (PG-M1, 1:50), CD99 (12E7, 1:50), CD117 (Polyclonal, 1:200), CD163 (10D6, 1:50), clusterin (41D, 1:100), cytokeratin (AE1/AE3, 1:50), desmin (D33, 1:50), epithelial membrane antigen (EMA) (E29, 1:50), EGFR (EGFR.113, Neat), ER (SP1, 1:70), fascin (55K-2, 1:100), Her-2 (Polyclonal, 1:200), HMB45 (HMB-45, 1:50), HSP70 (W-27, 1:100), Ki-67 (MIB-1, neat), LCK (35BH11, 1:50), lysozyme (Polyclonal, 1:50), MyoD1 (5.8A, 1:100), myoglobulin (MGN01, 1:50), myogenin (F5D, 1:100), p53 (DO-7, 1:50), PR (SP2, 1:200), WT-1 (6F-H2, 1:50), S-100 protein (polyclonal, 1:200), and vimentin (V9, 1:200). All the primary antibodies used in this study were mouse monoclonal antibodies and the DakoCytomation products (Dako, Carpentaria, CA, USA) unless otherwise stated. The specific bindings of the antibodies on the sections were stained using the Dako EnVision Detection System, Peroxidase/DAB (Dako, Carpentaria, California, USA), according to the manufacturer’s instructions. Appropriate positive and negative controls were run in parallel.
Results
Grossly, the tumor was a huge, firm and fibrous, solid mass that measured 20 × 20 × 15 cm and weighed 1,000 g. The cut surface was glistening, faintly bulging, and lobulated with a whorled cut surface resembling leiomyoma, with cystic and necrotic areas. There was no discrete capsule.
Histologically, the neoplasm was composed mainly of hypercellular spindle cells, arranged in fascicular and herringbone patterns, closely resembling leiomyosarcoma or fibrosarcoma (Figure 2A). The spindle cells showed oval, elongated and tapered nuclei, vesicular chromatin, inconspicuous to small nucleoli, and indistinct eosinophilic cytoplasm (Figure 2B). Occasionally, the neoplastic cells also showed rather more rounded or epithelioid morphology (Figure 2B). In addition, scattered enlarged plump spindle- or polygonal-shaped rhabdomyoblasts with abundant eosinophilic cytoplasm and a large nucleus with a prominent nucleolus admixed within dense whorls and fascicles of spindle cells throughout the tumor (Figure 2B, 2C). Mitoses, including atypical forms ranged from 20 to 28 per 10 high-power fields (Figure 2B, 2C). Geographic necrosis was present (Figure 2D).
Figure 2.
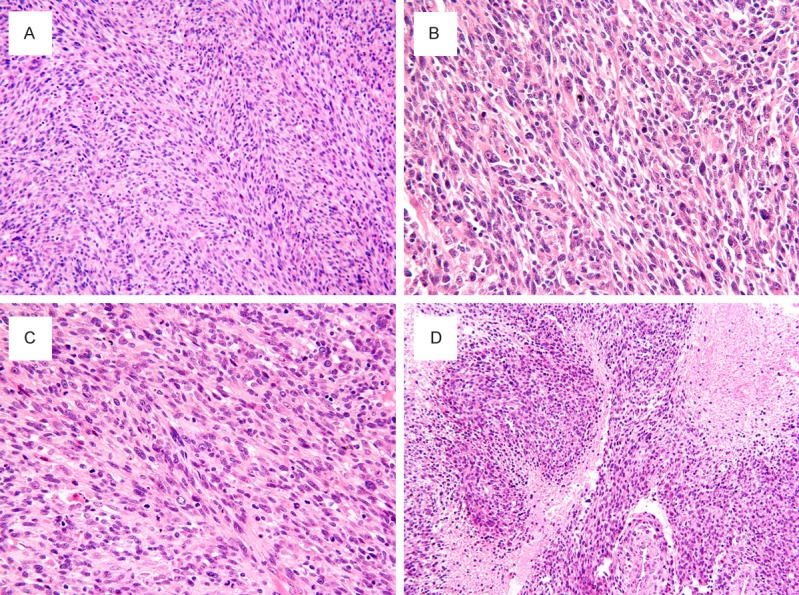
Spindle cell RMS. (A) The tumor showed high cellularity consisted predominately of spindle cells in long fascicles, arranged in a herringbone pattern, resembling adult-type fibrosarcoma. The spindle cells had elongated and hyperchromatic nuclei, prominent nucleoli and scant cytoplasm, often arranged in a fascicular or herringbone growth pattern. (B) Scattered rhabdomyoblasts containing abundant eosinophilic cytoplasm and eccentrically placed nuclei are admixed with spindled cells. (D) Tumor cells demonstrated a higher mitotic activity and mitotic figures. (E) Spindle cells interspersed with larger geographic areas of necrosis. H & E, original magnification, (A, D), × 200; (B, C), × 400.
By immunohistochemistry, the tumor cells were stained uniformly positively for muscle-specific actin (Figure 3A), desmin (Figure 3B), myoglobin (Figure 3C) and vimentin (Figure 3D). Tumor cells also showed strongly positive for myogenin (Figure 3E), and MyoD1 (Figure 3F) in 21.9% and 23.64% of tumor cell nuclei, respectively. In addition, a strong and diffuse cytoplasmic staining for WT1 (Figure 4A), and CD99 (Figure 4B) was noted. A strong nuclear accumulation of p53 (Figure 4C) has been detected in more than 82.2% of the tumor cells. The proliferative fraction, evaluated by Ki-67 (MIB-1), was 46.11% of the tumor cells (Figure 4D). Other markers such as S100 protein, smooth muscle actin, cytokeratin, EMA, HMB45 and CD34, histiocyte marker CD45, CD68, CD163, and lysozyme were all negative.
Figure 3.

Immunohistochemistry of spindle cell RMS. Tumor cells are stained positively for muscle specific actin (A), desmin (B), myoglobin (C), and vimentin (D). Scattered tumor cells show a nuclear positivity for myogenin (E) and MyoD1 (F). DAKO Envision peroxidase detection system, (A), × 200; (B-F), × 400.
Figure 4.

Immunohistochemistry of spindle cell RMS. A. WT1 immunostaining reveals the fascicular features more clearly. The neoplastic cells show strong cytoplasmic stain for WT1. B. The tumor cells are diffusely positive for CD99 in a cytoplasmic and membranous fashion. C. More than 82.2% of the tumor cells show nuclear accumulation of p53. D. MIB-1 positivity is found in 46.11% of tumor cells. DAKO Envision peroxidase detection system, × 400.
Based on histopathologic and immunohistochemical findings, a diagnosis of spindle cell RMS was made. Subsequent imaging studies of the lungs and brain showed no evidence of metastatic disease. Adjuvant chemotherapy therapy was administered.
Discussion
Spindle cell RMS of the retroperitoneum is an extremely rare disease in adults, and only two male cases have been previously described in the literature [5,12]. This report describes an unusual case of retroperitoneal RMS in a pregnant woman whose disease was obscured by pregnancy, and discovered only after vaginal delivery. The overall clinical, radiological, histologic and immunohistochemical patterns of this case are most consistent with spindle cell variant RMS. To our knowledge, this is the first case report of retroperitoneal spindle cell RMS in a pregnant woman.
The clinical presentation of this tumor includes abdominal pain, unexplained weight loss, and mass, which are usually not specific. This may be mistakenly attributed to the physical changes that occur during pregnancy if the symptoms are obscured or masked by pregnancy. Histological examination showed a highly cellular tumor composed mainly of fascicles of spindle cells with elongated, vesicular nuclei, inconspicuous nucleoli and palely eosinophilic cytoplasm, arranged in a herringbone pattern, reminiscent to what is seen in rare fibrosarcoma in adults, leiomyosarcoma or malignant peripheral nerve sheath tumor. Occasionally, scattered tumor cells showed a rather polygonal, rounded, or strap-shaped shape with abundant brightly eosinophilic cytoplasm resembling rhabdomyoblasts. The spindle cell morphology in the tumor presented here initially provided some diagnostic challenges, thus an immunohistochemical staining using a panel of markers for screening was essential. Traditionally, intermediate filament desmin, the contractile protein actin, or the oxygen transport molecule, myoglobin immunohistochemistry has been used to detect myoid differentiation. However, these traditional markers require considerable differentiation along the myogenic pathway before cellular expression occurs, and only 30% of embryonal RMS and 70% of alveolar RMS cases are immunohistochemical staining for myoglobin [13]. In contrast, myogenin and MyoD1, the myogenic transcriptional regulatory proteins expressed early in skeletal muscle differentiation, are considered sensitive and specific markers for RMS and are more specific than desmin and muscle-specific actin and more sensitive than myoglobin [14]. These markers stain the overwhelming majority (> 95%) of RMSs [14-16] including the primitive undifferentiated forms, regardless of morphologic evidence of skeletal muscle differentiation [15], whereas almost no in other paediatric tumors demonstrate positive immunostaining [14,17-19]. Although essentially all RMS samples show some degree of nuclear immunostaining with MyoD and myogenin, the expression patterns differ between embryonal RMS and alveolar RMS. RMS of the alveolar subtype, alveolar or solid variants, demonstrate widespread and strong myogenin expression in a high proportion of cells (extensive nuclear myogenin staining in more than 75% of tumor cells in most cases of alveolar RMS) compared with those of the embryonal subtype (less uniform, and in many cases less than 25% of tumor cells) [14,18,20]. A recurrent mutation and transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma [21-23]. Thus, MyoD1 and myogenin immunohistochemistry is useful in the diagnosis of RMS, including its subtypes [15], and also able to distinguish alveolar RMS from the more common embryonal subtype [20,24]. In our case, in contrast to the classic embryonal and alveolar forms [20], the spindle tumor cells are uniformly and strongly positive for all muscle cell markers (actin, myoglobin, desmin, vimentin), and also positive for myogenin and MyoD1 in 21.9% and 23.64% spindled tumor cells in addition to obvious rhabdomyoblasts, confirming its myogenic origin and keeping with immunophenotypic features of an embryonal RMS [3,5]. Thus, the diagnosis of spindle cell RMS was preferred.
Spindle cell RMS poses special problems in differential diagnosis when arising in adults and it should be distinguished from other spindle cell malignancy [9], including sarcomatoid carcinoma, leiomyosarcoma, malignant peripheral nerve sheath tumor with heterologous rhabdomyoblastic differentiation, and fibrosarcoma. Again, immunohistochemical staining for skeletal muscle markers can be helpful in differentiating spindle cell RMS from other spindle cell tumors. In our case, these spindle cells were immunonegative with markers for epithelial, mesothelial, endothelial, neurogenic, leiomyogenic, lipogenic, melanocytic or other hematopoietic derivation, thus, excluding other spindle cell mimics of these origins.
Previous studies have shown that a strong cytoplasmic rather than the specific nuclear staining for WT1, and CD99 is observed in RMSs, including in adult-type spindle cell RMS in occasional reports [6,25]. WT1, the Wilms tumor suppressor gene, located on chromosome 11p13, encodes a putative transcription factor implicated in tumorigenesis and in normal urogenital development. WT1 nuclear staining in the blastemal area is seen in 70% to 100% of cases [25,26]. Although the blastemal component is reactive for vimentin and desmin (partially in up to 90%), the absence of staining for myogenin and MyoD1 discriminates blastomatous Wilms tumor from RMS [27]. CD99, the product of the MIC2 gene, is a cell surface glycoprotein that is expressed in more than 95% of Ewing sarcoma/primitive neuroectodermal tumors (EWS/PNETs) with a diffuse membranous staining pattern [28,29]. It is interesting to note that our case stained strongly positive for WT1 and CD99 emphasizing that WT1 represents a further sensitive, but not specific marker for RMS [25], and that the cytoplasmic expression of CD99 has to be considered in the differential diagnosis of CD99 positive neoplasms. The expression of p53 protein in RMS has been recognized in 19-75% of cases [30-35]. It has been reported that p53 overexpression has no association with the patients’ prognoses or with any of the other clinicopathologic parameters, including age, sex or histopathologic subtype [30,33], but related to tumor aggressiveness in RMS because tumors with p53 overexpression have a high proliferative activity [30,34]. In line with these reports, the tumor in the present case showed a higher nuclear accumulation of p53 and a marked cell proliferation as evidenced by extremely high mitosis counts and high MIB-1 labeling indices, indicative of a high rate of cell proliferation.
Spindle cell RMS are treated aggressively with similar protocols as for other RMS and owing to their rarity, very little research has been done on the use of a different approach for this variant. The standard protocols involve combined therapy including surgery, chemotherapy, and adjuvant radiation. Spindle cell RMS behaves more aggressively, with worse overall survival in adults in which up to 40% of patients had uncontrolled local disease, 25% developed metastases, and 17% died of disease [5], in striking contrast to children and adolescents in which it has an excellent prognosis [8,36]. Because of this marked difference between the clinical findings in children and adults with spindle cell RMS, it is possible that these two neoplasms in fact represent different disease processes. As in other types of sarcomas in adults, patient age, tumor size, extent of disease, and margin status after resection represent the most important predictors of outcome in adult patients with RMS [37]. Unfortunately, our patient has a large tumor invading vital structures that result in incomplete surgery. The patient in the present study developed multiple metastases and died of disease within a short time period, although she receives treatment with surgical resection and chemotherapy.
In summary, spindle cell RMS of retroperitoneal origin in adults is uncommon, and its occurrence in a pregnant woman has not been reported previously. The presentation of spindle cell RMS arising in adult patients during pregnancy was obscured by pregnancy, and delayed the diagnosis in this case, thus affecting the efficiency of disease management. In addition to its distinct clinical presentation, this case highlights the histological features of a rare form of RMS, and emphasizes the importance of awareness of its existence and the utility of skeletal muscle markers in distinguishing spindle cell RMS from its mimics.
Acknowledgements
This research was supported by grants No. 31201037 (Dr. L. Yu) and No. 30972589 (Dr. S.J. Yang) from the National Natural Science Foundation of China.
Disclosure of conflict of interest
None.
References
- 1.Fletcher CDM, Unni KK, Mertens F. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2002. World Health Organization Classification of Tumors. [Google Scholar]
- 2.Parham DM, Ellison DA. Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med. 2006;130:1454–1465. doi: 10.5858/2006-130-1454-RIAACA. [DOI] [PubMed] [Google Scholar]
- 3.Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002;26:1175–1183. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Mentzel T, Katenkamp D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 2000;436:305–311. doi: 10.1007/s004280050451. [DOI] [PubMed] [Google Scholar]
- 5.Nascimento AF, Fletcher CD. Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol. 2005;29:1106–1113. [PubMed] [Google Scholar]
- 6.Mentzel T, Kuhnen C. Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch. 2006;449:554–560. doi: 10.1007/s00428-006-0284-4. [DOI] [PubMed] [Google Scholar]
- 7.Nascimento AF, Barr FG. Spindle cell/sclerosing rhabdomyosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO Classification of Tumours of Soft Tissue and Bone. 4th edn. Lyon: IARC Press; 2013. pp. 134–135. [Google Scholar]
- 8.Cavazzana AO, Schmidt D, Ninfo V, Harms D, Tollot M, Carli M, Treuner J, Betto R, Salviati G. Spindle cell rhabdomyosarcoma: A prognostically favorable variant of rhabdomyosarcoma. Am J Surg Pathol. 1992;16:229–235. doi: 10.1097/00000478-199203000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Rubin BP, Hasserjian RP, Singer S, Janecka I, Fletcher JA, Fletcher CD. Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. Am J Surg Pathol. 1998;22:459–464. doi: 10.1097/00000478-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 10.Kizer WS, Dykes TE, Brent EL, Chatham JR, Schwartz BF. Paratesticular spindle cell rhabdomyosarcoma in an adult. J Urol. 2001;166:606–607. [PubMed] [Google Scholar]
- 11.McCluggage WG, Lioe TF, McClelland HR, Lamki H. Rhabdomyosarcoma of the uterus: report of two cases, including one of the spindle cell variant. Int J Gynecol Cancer. 2002;12:128–132. doi: 10.1046/j.1525-1438.2002.01069.x. [DOI] [PubMed] [Google Scholar]
- 12.Fernando Val-Bernal J, Fernandez N, Gomez-Roman JJ. Spindle cell rhabdomyosarcoma in adults. A case report and literature review. Pathol Res Pract. 2000;196:67–72. doi: 10.1016/s0344-0338(00)80024-2. [DOI] [PubMed] [Google Scholar]
- 13.Kahn HJ, Yeger H, Kassim O, Jorgensen AO, MacLennan DH, Baumal R, Smith CR, Phillips MJ. Immunohistochemical and electron microscopic assessment of childhood rhabdomyosarcoma. Increased frequency of diagnosis over routine histologic methods. Cancer. 1983;51:1897–1903. doi: 10.1002/1097-0142(19830515)51:10<1897::aid-cncr2820511023>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 14.Kumar S, Perlman E, Harris CA, Raffeld M, Tsokos M. Myogenin is a specific marker for rhabdomyosarcoma: an immunohistochemical study in paraffin-embedded tissues. Mod Pathol. 2000;13:988–993. doi: 10.1038/modpathol.3880179. [DOI] [PubMed] [Google Scholar]
- 15.Sebire NJ, Malone M. Myogenin and MyoD1 expression in paediatric rhabdomyosarcomas. J Clin Pathol. 2003;56:412–416. doi: 10.1136/jcp.56.6.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang NP, Marx J, McNutt MA, Rutledge JC, Gown AM. Expression of myogenic regulatory proteins (myogenin and MyoD1) in small blue round cell tumors of childhood. Am J Pathol. 1995;147:1799–1810. [PMC free article] [PubMed] [Google Scholar]
- 17.Folpe AL. MyoD1 and myogenin expression in human neoplasia: a review and update. Adv Anat Pathol. 2002;9:198–203. doi: 10.1097/00125480-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Cessna MH, Zhou H, Perkins SL, Tripp SR, Layfield L, Daines C, Coffin CM. Are myogenin and myoD1 expression specific for rhabdomyosarcoma? A study of 150 cases, with emphasis on spindle cell mimics. Am J Surg Pathol. 2001;25:1150–1157. doi: 10.1097/00000478-200109000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Cui S, Hano H, Harada T, Takai S, Masui F, Ushigome S. Evaluation of new monoclonal anti-MyoD1 and anti-myogenin antibodies for the diagnosis of rhabdomyosarcoma. Pathol Int. 1999;49:62–68. doi: 10.1046/j.1440-1827.1999.00825.x. [DOI] [PubMed] [Google Scholar]
- 20.Dias P, Chen B, Dilday B, Palmer H, Hosoi H, Singh S, Wu C, Li X, Thompson J, Parham D, Qualman S, Houghton P. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol. 2000;156:399–408. doi: 10.1016/S0002-9440(10)64743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szuhai K, de Jong D, Leung WY, Fletcher CD, Hogendoorn PC. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol. 2014;232:300–307. doi: 10.1002/path.4307. [DOI] [PubMed] [Google Scholar]
- 22.Agaram NP, Chen CL, Zhang L, LaQuaglia MP, Wexler L, Antonescu CR. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: Evidence for a common pathogenesis. Genes Chromosomes Cancer. 2014;53:779–87. doi: 10.1002/gcc.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohsaka S, Shukla N, Ameur N, Ito T, Ng CK, Wang L, Lim D, Marchetti A, Viale A, Pirun M, Socci ND, Qin LX, Sciot R, Bridge J, Singer S, Meyers P, Wexler LH, Barr FG, Dogan S, Fletcher JA, Reis-Filho JS, Ladanyi M. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet. 2014;46:595–600. doi: 10.1038/ng.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgenstern DA, Rees H, Sebire NJ, Shipley J, Anderson J. Rhabdomyosarcoma subtyping by immunohistochemical assessment of myogenin: tissue array study and review of the literature. Pathol Oncol Res. 2008;14:233–238. doi: 10.1007/s12253-008-9012-5. [DOI] [PubMed] [Google Scholar]
- 25.Carpentieri DF, Nichols K, Chou PM, Matthews M, Pawel B, Huff D. The expression of WT1 in the differentiation of rhabdomyosarcoma from other pediatric small round blue cell tumors. Mod Pathol. 2002;15:1080–1086. doi: 10.1097/01.MP.0000028646.03760.6B. [DOI] [PubMed] [Google Scholar]
- 26.Jimenez RE, Folpe AL, Lapham RL, Ro JY, O’Shea PA, Weiss SW, Amin MB. Primary Ewing’s sarcoma/primitive neuroectodermal tumor of the kidney: a clinicopathologic and immunohistochemical analysis of 11 cases. Am J Surg Pathol. 2002;26:320–327. doi: 10.1097/00000478-200203000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Bahrami A, Truong LD, Ro JY. Undifferentiated tumor: true identity by immunohistochemistry. Arch Pathol Lab Med. 2008;132:326–348. doi: 10.5858/2008-132-326-UTTIBI. [DOI] [PubMed] [Google Scholar]
- 28.Folpe AL, Goldblum JR, Rubin BP, Shehata BM, Liu W, Dei Tos AP, Weiss SW. Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol. 2005;29:1025–1033. [PubMed] [Google Scholar]
- 29.Devoe K, Weidner N. Immunohistochemistry of small round-cell tumors. Semin Diagn Pathol. 2000;17:216–224. [PubMed] [Google Scholar]
- 30.Takahashi Y, Oda Y, Kawaguchi K, Tamiya S, Yamamoto H, Suita S, Tsuneyoshi M. Altered expression and molecular abnormalities of cell-cycle-regulatory proteins in rhabdomyosarcoma. Mod Pathol. 2004;17:660–669. doi: 10.1038/modpathol.3800101. [DOI] [PubMed] [Google Scholar]
- 31.Yoo J, Park SY, Kang SJ, Shim SI, Kim BK. Altered expression of G1 regulatory proteins in human soft tissue sarcomas. Arch Pathol Lab Med. 2002;126:567–573. doi: 10.5858/2002-126-0567-AEOGRP. [DOI] [PubMed] [Google Scholar]
- 32.Wurl P, Taubert H, Bache M, Kroll J, Meye A, Berger D, Siermann A, Holzhausen HJ, Hinze R, Schmidt H, Rath FW. Frequent occurrence of p53 mutations in rhabdomyosarcoma and leiomyosarcoma, but not in fibrosarcoma and malignant neural tumors. Int J Cancer. 1996;69:317–323. doi: 10.1002/(SICI)1097-0215(19960822)69:4<317::AID-IJC14>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 33.Ayan I, Dogan O, Kebudi R, Bavbek B, Alatli C, Dervisoglu S, Disci R, Demiryont M. Immunohistochemical detection of p53 protein in rhabdomyosarcoma: association with clinicopathological features and outcome. J Pediatr Hematol Oncol. 1997;19:48–53. doi: 10.1097/00043426-199701000-00007. [DOI] [PubMed] [Google Scholar]
- 34.Leuschner I, Langhans I, Schmitz R, Harms D, Mattke A, Treuner J. p53 and mdm-2 expression in Rhabdomyosarcoma of childhood and adolescence: clinicopathologic study by the Kiel Pediatric Tumor Registry and the German Cooperative Soft Tissue Sarcoma Study. Pediatr Dev Pathol. 2003;6:128–136. doi: 10.1007/s10024-001-0097-z. [DOI] [PubMed] [Google Scholar]
- 35.Yoo J, Lee HK, Kang CS, Park WS, Lee JY, Shim SI. p53 gene mutations and p53 protein expression in human soft tissue sarcomas. Arch Pathol Lab Med. 1997;121:395–399. [PubMed] [Google Scholar]
- 36.Leuschner I, Newton WA Jr, Schmidt D, Sachs N, Asmar L, Hamoudi A, Harms D, Maurer HM. Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol. 1993;17:221–230. doi: 10.1097/00000478-199303000-00002. [DOI] [PubMed] [Google Scholar]
- 37.Hawkins WG, Hoos A, Antonescu CR, Urist MJ, Leung DH, Gold JS, Woodruff JM, Lewis JJ, Brennan MF. Clinicopathologic analysis of patients with adult rhabdomyosarcoma. Cancer. 2001;91:794–803. [PubMed] [Google Scholar]