

Abstract
Purpose.
To analyze the genetic test results of probands referred to eyeGENE with a diagnosis of hereditary maculopathy.
Methods.
Patients with Best macular dystrophy (BMD), Doyne honeycomb retinal dystrophy (DHRD), Sorsby fundus dystrophy (SFD), or late-onset retinal degeneration (LORD) were screened for mutations in BEST1, EFEMP1, TIMP3, and CTRP5, respectively. Patients with pattern dystrophy (PD) were screened for mutations in PRPH2, BEST1, ELOVL4, CTRP5, and ABCA4; patients with cone-rod dystrophy (CRD) were screened for mutations in CRX, ABCA4, PRPH2, ELOVL4, and the c.2513G>A p.Arg838His variant in GUCY2D. Mutation analysis was performed by dideoxy sequencing. Impact of novel variants was evaluated using the computational tool PolyPhen.
Results.
Among the 213 unrelated patients, 38 had BMD, 26 DHRD, 74 PD, 8 SFD, 6 LORD, and 54 CRD; six had both PD and BMD, and one had no specific clinical diagnosis. BEST1 variants were identified in 25 BMD patients, five with novel variants of unknown significance (VUS). Among the five patients with VUS, one was diagnosed with both BMD and PD. A novel EFEMP1 variant was identified in one DHRD patient. TIMP3 novel variants were found in two SFD patients, PRPH2 variants in 14 PD patients, ABCA4 variants in four PD patients, and p.Arg838His GUCY2D mutation in six patients diagnosed with dominant CRD; one patient additionally had a CRX VUS. ABCA4 mutations were identified in 15 patients with recessive CRD.
Conclusions.
Of the 213 samples, 55 patients (26%) had known causative mutations, and 13 (6%) patients had a VUS that was possibly pathogenic. Overall, selective screening for mutations in BEST1, PRPH2, and ABCA4 would likely yield the highest success rate in identifying the genetic basis for macular dystrophy phenotypes. Because of the overlap in phenotypes between BMD and PD, it would be beneficial to screen genes associated with both diseases.
Keywords: genetic testing, eyeGENE, macular dystrophy
This paper discusses the known causative mutations and variants of unknown significance detected in a set of patients diagnosed with one of six specific macular dystrophies.
Introduction
The National Ophthalmic Disease Genotyping and Phenotyping Network, eyeGENE, is a multicomponent genetics initiative created by the National Eye Institute to facilitate research in inherited eye disease. The program provides access to deidentified DNA, phenotype, and genotype information of its participants.1 This study describes eyeGENE data on six specific conditions involving central vision loss: Best macular dystrophy, Doyne honeycomb retinal dystrophy, Sorsby fundus dystrophy, late-onset retinal degeneration, pattern dystrophy, and dominant and recessive cone-rod dystrophy. All of these conditions are autosomal dominant inherited diseases except recessive cone-rod dystrophy (CRD).
Best macular dystrophy (BMD) is characterized by a yellow yolk-like lesion in the macula with loss of central vision. Onset of disease is generally in childhood or early teenage years. Mutations in the bestrophin-1 (BEST1 or VMD2) gene are known to cause BMD2,3 (Table 1). Although BMD is an autosomal dominant disease, recessive mutations in BEST1 are associated with recessive bestrophinopathy with symptoms similar to those of dominant BMD.4
Table 1.
Retinal Disease Genes Screened for Mutations and the Phenotypes Associated With These Genes
|
Genes Screened |
Location |
Gene Accession Number |
Chromosome |
Associated Diagnosis |
| ABCA4 | 1p22.1 | NM_000350.2 | 1 | Pattern dystrophy, cone-rod dystrophy |
| Recessive retinitis pigmentosa | ||||
| BEST1 | 11q12.3 | NM_004183.3 | 11 | Best macular dystrophy |
| CRX | 19q13.32 | 19 | Dominant cone-rod dystrophy | |
| CTRP5 | 11q23.3 | NM_001278431.1 | 11 | Late-onset retinal degeneration |
| EFEMP1 | 2p16.1 | NM_001039348.2 | 2 | Doyne honeycomb retinal dystrophy |
| ELOVL4 | 6q14.1 | NM_022726.3 | 6 | Autosomal dominant Stargardt's-like macular dystrophy |
| GUCY2D | 17p13.1 | NM_000180.3 | 17 | Dominant cone-rod dystrophy |
| PRPH2 | 6p21.1 | NM_000322.4 | 6 | Pattern dystrophy, cone-rod dystrophy |
| Macular degeneration | ||||
| TIMP3 | 22q12.3 | NM_000362.4 | 22 | Sorsby fundus dystrophy |
Doyne honeycomb retinal dystrophy (DHRD) or malattia leventinese is clinically diagnosed by the presence of drusen in the (sub)retinal epithelium (RPE) of the posterior region of the eye, especially in areas associated with the macula and the optic disc. Over time, a honeycomb pattern of drusen deposition can be seen in the fundus of some patients. Mutations in the epidermal growth factor containing fibulin-like extracellular matrix protein 1 gene (EFEMP1) are associated with DHRD5 (Table 1).
Sorsby fundus dystrophy (SFD) is a late-onset disease with central vision loss but also includes atrophy of the peripheral choroid, drusen-like deposits, and choroidal neovascularization in most cases.6 Mutations in the tissue inhibitor of metallopeptidase 3 gene (TIMP3) have been implicated in the development of SFD7 (Table 1). Due to the late age of onset, overlap in clinical symptoms, and lack of information on additional family members, SFD is often misdiagnosed as the more common condition, age-related macular degeneration (AMD).
Late-onset retinal degeneration (LORD) is a disease with early onset of abnormal lens zonules, abnormalities in dark adaptation, late-onset drusen-like deposits, central vision loss, and neovascularization of the retina.8 A single missense mutation, p.Ser163Arg, in the C1q tumor necrosis factor-related protein 5 gene (CqQTNF5/CTRP5) has been reported in patients with LORD9 (Table 1). The clinical symptoms of LORD overlap with the symptoms of SFD and AMD.
Pattern dystrophy (PD) patients often exhibit yellow, orange, or gray deposits in the macula that disrupt central vision. Occasionally, the pattern will appear in the shape of a butterfly.10 Mutations in the peripherin 2 (PRPH2) and adenosine triphosphate (ATP) binding cassette subfamily A, member 4 (ABCA4) genes have been reported in patients with the PD phenotype10 (Table 1).
Cone-rod dystrophies are characterized by peripheral vision loss along with central vision loss, and this phenotype is inherited in both dominant and recessive fashion. Dominant CRD is caused by mutations in the guanylate cyclase 2D (GUCY2D) and the cone-rod homeobox (CRX) genes11–13 (Table 1). Mutations changing the codon arginine at amino acid position 838 to histidine, proline, or cysteine in GUCY2D have been identified in unrelated patients with CRD. The p.Arg828His mutation is the most common among all GUCY2D mutations.13 Recessive CRD is usually associated with missense mutations in ABCA4.14
We summarize the genetic variations identified in 213 patients from the eyeGENE database with a retinal dystrophy phenotype involving central vision loss.
Methods
Since genetic and phenotypic heterogeneity are common in retinal degenerations with central vision loss, analysis of multiple genes may be needed to determine the underlying cause of these diseases. Molecular analysis by eyeGENE involves systematic screening of relevant genes beginning with the most likely to be associated with the patient's clinical phenotype as available through collaborating CLIA (Clinical Laboratory Improvement Amendment) laboratories. All research followed the tenets of the Declaration of Helsinki and had Institutional Review Board approval.
Two hundred thirteen patients listed in the eyeGENE database with a retinal dystrophy phenotype of central vision loss were selected for this study. Mutation screening was carried out by arrayed primer extension (APEX) technology and/or dideoxy sequencing by eyeGENE collaborating laboratories including ours (Downs CA, et al. IOVS 2004;45:E-Abstract 2474). The genes selected for analysis were based on the primary and secondary diagnosis provided by the physician. When mutations were not detected in genes associated with the primary diagnosis, other genes associated with similar phenotypes were tested. Patients with a primary diagnosis of Stargardt's were not included in the study.
Late-Onset Retinal Dystrophy
Six probands with a primary diagnosis of LORD were screened for mutations in exons 1 to 3 of the CTRP5 gene.
Sorsby Fundus Dystrophy
Eight probands with a primary diagnosis for SFD were screened for mutations in exons 1 to 5 of the TIMP3 gene.
Doyne Honeycomb Retinal Dystrophy
Twenty-six probands with a primary diagnosis for DHRD were screened for mutations in the EFEMP1 gene (exons 1–11 in 25 patients; exons 3–12 in one patient).
Best Macular Dystrophy
Forty-four probands with a clinical diagnosis of BMD were screened for mutations in the BEST1 gene (exons 2–11 in 40 families; exons 1–11 in 5 families).
Pattern Dystrophy
Eighty probands with a clinical diagnosis for PD were screened for mutations in the PRPH2 gene.
Twenty-seven probands also had clinical signs consistent with Stargardt's disease (STGD1), BMD, SFD, DHRD, or other retinal dystrophies. They were additionally screened for mutations in the ABCA4, ELOVL4, CTRP5, BEST1, and/or PRPH2 genes.
Cone-Rod Dystrophy
Fifty-four probands with a primary diagnosis of CRD were tested based on the pattern of inheritance determined by family history and/or differential diagnoses. Thirteen CRD probands with presumably autosomal dominant inheritance were screened for the p.Arg838His mutation in GUCY2D, and mutations in the CRX, ELOVL4, PRPH2, and/or ABCA4 genes. Fifteen CRD families with autosomal recessive inheritance were tested for mutations in the ABCA4 gene using dideoxy sequencing of the coding region, using the ABCR genotyping microarray (the ABCR400 chip) constructed by APEX technology, or by solid state sequencing.15,16 Twenty-six patients that were isolated cases and/or lacked a family history for CRD were screened for mutations in ABCA4. Ten of the 26 CRD isolated cases also had a differential diagnosis of STGD1 based on early age of onset, presence of central scotoma, and fundus appearance including foveal atrophy, perimacular flecks or a beaten bronze appearance, and increased fundus autoflourescence. These patients underwent additional mutation screening for the CRX, PRPH2, and ELOVL4 genes.
When candidate disease-causing variants were identified, several criteria were used to predict their pathogenicity: examination of whether the variant had been previously reported, the bioinformatics score determined by PolyPhen (http://genetics.bwh.harvard.edu/pph2/ [in the public domain]), the association of the genotype with the phenotype, and whether the presence of the variant was consistent with observed pattern of inheritance. When rare (i.e., allele frequency < 0.01) or novel variants of unknown significance (VUS) were present, the variants were analyzed by PolyPhen to predict their potential impact. Novel variants identified as “potentially damaging” support the clinical diagnosis but were validated by segregation analysis to confirm the likelihood that the variants were disease causing. The Berkeley Drosophila Genome Project Splice Site Prediction analysis tool was used to determine whether rare or novel silent variants occurring within the exon were splice altering (http://www.fruitfly.org/seq_tools/splice.html [in the public domain]). Variants identified as “possibly damaging” require additional studies to confirm their involvement in causing pathology. When no mutations were found in the genes associated with the clinical phenotype, then the molecular basis of disease in this patient is unknown, and the molecular diagnosis neither excludes nor supports the clinical diagnosis.
Results
Among the patients registered in the eyeGENE database, 213 unrelated patients were diagnosed with a retinal dystrophy phenotype involving central vision loss. Out of the 213, 6 were diagnosed with LORD, 8 with SFD, 26 with DHRD, 38 with BMD, 74 with PD, 54 with CRD, 6 with both PD and BMD, and 1 with no specific clinical diagnosis.
Genetic Analysis
Late-Onset Retinal Degeneration.
No causative mutations were found in the exonic regions of CTRP5, demonstrating that coding mutations in CTRP5 were not the cause of disease in these six patients, and therefore they did not have a positive confirmation of diagnosis (Table 2).
Table 2.
Mutations and Unknown Variants Detected in Patients With Central Vision Loss
|
Patient |
Gene |
Exon |
DNA Change |
Protein Change |
Genotype |
Result |
PolyPhen Description |
PolyPhen Score |
Molecular Diagnosis |
| Late-onset retinal degeneration | |||||||||
| NA | CTRP5 | NA | NA | NA | NA | NA | NA | ||
| Sorsby fundus dystrophy | |||||||||
| Patient 1 | TIMP3 | 1 | c.113C>G | p.Ser38Cys | Het | vAR/us | Probably damaging |
1 | Positive |
| Patient 2 | TIMP3 | 1 | c.113C>G | p.Ser38Cys | Het | vAR/us | Probably damaging |
1 | Positive |
| Patient 3 | TIMP3 | 5 | c.610A>T | p.Ser204Cys | Het | Mut | Positive | ||
| Doyne honeycomb dystrophy | |||||||||
| Patient 1 | EFEMP1 | 9 | c.1033C>T | p.Arg345Trp | Het | Mut | Positive | ||
| Patient 2 | EFEMP1 | 9 | c.1033C>T | p.Arg345Trp | Het | Mut | Positive | ||
| Patient 3 | EFEMP1 | IVS10 | c.IVS10-14C>T | None | Het | vAR/us | NA | NA | Unconfirmed |
| Best macular dystrophy | |||||||||
| Patient 1 | BEST1 | 2 | c.28G>A | p.Ala10Thr | Het | Mut | Positive | ||
| Patient 2 | BEST1 | 2 | c.47C>T | p.Ser16Phe | Het | Mut | Positive | ||
| Patient 3 | BEST1 | 2 | c.72G>T | p.Trp24Cys | Het | Mut | Positive | ||
| Patient 4 | BEST1 | 3 | c.240C>A | p.Phe80Leu | Het | Mut | Positive | ||
| Patient 5 | BEST1 | 3 | c.240C>A | p.Phe80Leu | Het | Mut | Positive | ||
| Patient 6 | BEST1 | 4 | c.248G>C | p.Gly83Ala | Het | vAR/us | Probably damaging |
1 | Positive |
| Patient 7 | BEST1 | 4 | c.277T>C | p.Trp93Arg | Het | vAR/us | Probably damaging |
1 | Positive |
| Patient 8 | BEST1 | 4 | c.279G>C | p.Trp93Cys | Het | Mut | Positive | ||
| Patient 9 | BEST1 | 6 | c.652C>T | p.Arg218Cys | Het | Mut | Positive | ||
| Patient 10 | BEST1 | 6 | c.652C>T | p.Arg218Cys | Het | Mut | Positive | ||
| Patient 11 | BEST1 | 6 | c.680A>G | p.Tyr227Cys | Het | Mut | Positive | ||
| Patient 12 | BEST1 | 6 | c.741G>A | p.Arg218His | Het | Mut | Positive | ||
| Patient 13 | BEST1 | 6 | c.741G>A | p.Arg218His | Het | Mut | Positive | ||
| Patient 14 | BEST1 | 7 | c.727G>A | p.Ala243Thr | Het | Mut | Positive | ||
| Patient 15 | BEST1 | 7 | c.727G>A | p.Ala243Thr | Het | Mut | Positive | ||
| Patient 16 | BEST1 | 7 | c.728C>T | p.Ala243Val | Het | Mut | Positive | ||
| Patient 17 | BEST1 | 7 | c.728C>T | p.Ala243Val | Het | Mut | Positive | ||
| Patient 18 | BEST1 | 8 | c.880C>T | p.Leu294Phe | Het | vAR/us | Probably damaging |
1 | Positive |
| Patient 19 | BEST1 | 8 | c.887A>G | p.Asn296Ser | Het | Mut | Positive | ||
| Patient 20 | BEST1 | 8 | c.903T>G | p.Asp301Glu | Het | Mut | Positive | ||
| Patient 21 | BEST1 | 8 | c.903T>G | p.Asp301Glu | Het | Mut | Positive | ||
| Patient 22 | BEST1 | 8 | c.910G>A | p.Asp304Asn | Het | Mut | Positive | ||
| Patient 23 | BEST1 | 8 | c.925T>C | p.Trp309Arg | Het | vAR/us | Probably damaging |
1 | Positive |
| Patient 24 | BEST1 | 8 | c.929T>C | p.Ile310Thr | Het | Mut | Positive | ||
| Patient 25, case 3 | BEST1 | 4 | c.250T>G | p.Phe84Val | Het | vAR/us | Probably damaging |
1 | Positive |
| Pattern dystrophy | |||||||||
| Patient 1 | ABCA4 | 6 | c.634C>T | p.Arg212Cys | Het | Mut | Positive | ||
| ABCA4 | 30 | c.4469G>A | p.Cys1490Tyr | Het | Mut | ||||
| Patient 2 | ABCA4 | 17 | c.2588G>C | p.Gly863Ala | Het | Mut | Unconfirmed | ||
| Patient 3 | ABCA4 | IVS26 | c.3862+3A>G | Abnormal splicing | Het | vAR/us | Unconfirmed | ||
| Patient 4 | PRPH2 | 1 | c.271T>A | p.Tyr91Asn | Het | vAR/us | Probably damaging |
0.909 | Positive |
| PRPH2 | 1 | c.310-313del(AT) | p.Ile104Val | Het | Mut | ||||
| Patient 5, case 6 | PRPH2 | 1 | c.422A>G | p.Tyr141Cys | Het | Mut | Positive | ||
| Patient 6 | PRPH2 | 1 | c.422A>G | p.Tyr141Cys | Het | Mut | Positive | ||
| Patient 7 | PRPH2 | 1 | c.515G>A | p.Arg172Gln | Het | Mut | Positive | ||
| Patient 8 | PRPH2 | 2 | c.583C>T | p.Arg195Stop | Het | Mut | Positive | ||
| Patient 9 | PRPH2 | 2 | c.629C>G | p.Pro210Arg | Het | Mut | Positive | ||
| Patient 10 | PRPH2 | 2 | c.635G>C | p.Ser212Thr | Het | Mut | Positive | ||
| Patient 11 | PRPH2 | 2 | c.683C>T | p.Thr228Ile | Het | Mut | Positive | ||
| Patient 12 | PRPH2 | 2 | c.708C>G | p.Tyr236Stop | Het | Mut | Positive | ||
| Patient 13, case 4 | PRPH2 | IVS2 | c.828+3A>T | Splice | Het | Mut | Positive | ||
| Patient 14 | PRPH2 | IVS2 | c.828+3A>T | Splice | Het | Mut | Positive | ||
| Patient 15 | PRPH2 | IVS2 | c.828+3A>T | Splice | Het | Mut | Positive | ||
| Patient 16 | PRPH2 | IVS2 | c.828+3A>T | Splice | Het | Mut | Positive | ||
| Patient 17, case 2 | ABCA4 | IVS38 | c.5461-10T>C | None | Het | Mut | Unconfirmed | ||
| Patient 18 | PRPH2 | 2 | c.584G>A | p.Arg195Gln | Het | vAR/us | Probably damaging |
1 | Positive |
| Cone-rod dystrophy | |||||||||
| Patient 1, dominant | GUCY2D | 13 | c.2512C>T | p.Arg838Cys | Het | Mut | Positive | ||
| Patient 2, dominant | GUCY2D | 13 | c.2513G>A | p.Arg838His | Het | Mut | Positive | ||
| Patient 3, dominant | GUCY2D | 13 | c.2513G>A | p.Arg838His | Het | Mut | Positive | ||
| Patient 4, dominant | GUCY2D | 13 | c.2513G>A | p.Arg838His | Het | Mut | Positive | ||
| Patient 5, dominant | GUCY2D | 13 | c.2513G>A | p.Arg838His | Het | Mut | Positive | ||
| CRX | 3 | c.607T>C | p.Ser213Pro | Het | vAR/us | Probably damaging |
0.999 | ||
| Patient 6, recessive | ABCA4 | 2 | c.156T>G | p.His52Gln | Het | vAR/us | Probably damaging |
0.998 | Positive |
| ABCA4 | 3 | c.161G>A | p.Cys54Tyr | Het | Mut | ||||
| ABCA4 | 28 | c.4169T>C | p.Leu1390Pro | Het | Mut | ||||
| Patient 7, recessive | ABCA4 | 16 | c.2385C>T | p.Ser795Arg | Het | vAR/us | Probably damaging |
0.99 | Positive |
| ABCA4 | IVS40 | c.5714+5G>A | Splice | Het | Mut | ||||
| Patient 8, recessive | ABCA4 | 42 | c.5882G>A | p.Gly1961Glu | Het | Mut | Positive | ||
| ABCA4 | 45 | c.6221G>T | p.Gly2074Val | Het | vAR/us | Probably damaging |
1 | ||
| Patient 9, recessive | ABCA4 | IVS42 | c.5898+1G<A | Splice | Het | Mut | Positive | ||
| ABCA4 | IVS42 | c.5899-2delA | Splice | Het | Mut | ||||
| Patient 10, recessive | ABCA4 | 5 | c.559C>T | p.Arg187Cys | Het | Mut | Positive | ||
| ABCA4 | 40 | c.5645T>C | p.Met1882Thr | Het | Mut | ||||
| Patient 11, recessive | ABCA4 | 6 | c.768G>T | p.Val256Val (abnlspl) | Het | Mut | Positive | ||
| ABCA4 | 31 | c.4577C>T | p.Thr1526Met | Het | Mut | ||||
| Patient 12, recessive | ABCA4 | 12 | c.1622T>C | p.Leu541Pro | Het | Mut | Positive | ||
| ABCA4 | 21 | c.3113C>T | p.Ala1038Val | Het | Mut | ||||
| ABCA4 | 12 | c.1622T>C | p.Leu541Pro | Hom | Mut | ||||
| ABCA4 | 21 | c.3113C>T | p.Ala1038Val | Hom | Mut | ||||
| ABCA4 | 22 | c.3322C>T | p.Arg1108Cys | Het | Mut | ||||
| Patient 13, recessive | ABCA4 | 12 | c.1622T>C | p.Leu541Pro | Hom | Mut | Positive | ||
| ABCA4 | 21 | c.3113C>T | p.Ala1038Val | Hom | Mut | ||||
| Patient 14, recessive | ABCA4 | 13 | c.1927G>A | p.Val643Met | Het | Mut | Positive | ||
| ABCA4 | 24 | c.3602T>G | p.Leu1201Arg | Het | Mut | ||||
| ABCA4 | 36 | c.5186T>C | p.Leu1729Pro | Het | Mut | ||||
| Patient 15, recessive | ABCA4 | 23 | c.3364G>A | p.Glu1122Lys | Het | Mut | Positive | ||
| ABCA4 | 48 | c.6529G>A | p.Asp2177Asn | Het | Mut | ||||
| Patient 16, recessive | ABCA4 | 35 | c.4918C>T | p.Arg1640Trp | Het | Mut | Positive | ||
| ABCA4 | 28 | c.4222T>C | p.Trp1408Arg | Het | Mut | ||||
| Patient 17, recessive | ABCA4 | 11 | c.1532G>A | p.Arg511His | Het | Mut | Unconfirmed | ||
| Patient 18, recessive | ABCA4 | 27 | c.3899G>A | p.Arg1300Gln | Het | vAR/us | Benign | 0.143 | Unconfirmed |
| Patient 19, recessive | ABCA4 | 13 | c.1933G>A | p.Asp645Asn | Het | Mut | Unconfirmed | ||
| Patient 20, recessive | ABCA4 | 35 | c.4918C>T | p.Arg1640Trp | Het | Mut | Unconfirmed | ||
| Patient 21, recessive | ABCA4 | IVS7 | c.859-9T>C | Unknown | Hom | vAR/us | NA | NA | Unconfirmed |
| Patient 22 | ABCA4 | 42 | c.5882G>A | p.Gly1961Glu | Hom | Mut | Positive | ||
| Patient 23, recessive | ABCA4 | 43 | c.5917delG | Deletion | Hom | Mut | Positive | ||
| Patient 24, recessive | ABCA4 | 32 | c.4661A>G | p.Glu1554Gly | Het | vAR/us | Benign | 0.326 | Unconfirmed |
| ABCA4 | 30 | c.4383G>A | p.Trp1461Stop | Het | Mut | ||||
| Patient 25, recessive | ABCA4 | IVS38 | c.5461-10T>C | None | Het | Mut | Positive | ||
| ABCA4 | 22 | c.3259G>A | p.Glu1087Lys | Het | Mut | ||||
| Patient 26, recessive | ABCA4 | IVS38 | c.5461-10T>C | None | Het | Mut | Positive | ||
| ABCA4 | 42 | c.5882G>A | p.Gly1961Glu | Het | Mut | ||||
| Patient 27, dominant | GUCY2D | 13 | c.2513G>A | p.Arg838His | Het | Mut | Positive | ||
| Patient 28, recessive, case 5 | PRPH2 | 1 | c.514C>T | p.Arg172Trp | Het | Mut | Positive | ||
| No specific clinical diagnosis | |||||||||
| Patient 1, case 1 | ABCA4 | 35 | c.4919G>A | p.Arg1640Gln | Het | Mut | Positive | ||
| ABCA4 | 42 | c.5882G>A | p.Gly1961Glu | Het | Mut | ||||
| ABCA4 | IVS42 | c.5898-11G>A | NA | Het | vAR/us | NA | NA | ||
| ABCA4 | IVS48 | c.6729+21C>T | NA | Het | vAR/us | NA | NA | ||
Het, heterozygous; Mut, mutation; vAR, variant; VUS, variant of unknown significance.
Sorsby Fundus Dystrophy.
Of the eight SFD samples, one patient carried a known heterozygous mutation in the TIMP3 gene and two had the same novel variant (p.Ser38Cys) with the PolyPhen score of 11 (Table 2). Thus, 38% of the SFD patients had a positive molecular diagnosis.
Doyne Honeycomb Retinal Dystrophy.
Of the 26 patients with DHRD, two patients were heterozygous for known causative mutations in EFEMP1 leading to a positive molecular diagnosis (Table 2). One of the 26 patients had a novel VUS in EFEMP1: a heterozygous variant in the 5′ flanking intronic region of exon 10 (IVS10-14C>T). A disease-causing mutation was identified in only 8% of DHRD patients when testing was limited to only the exonic regions of EFEMP1.
Best Macular Dystrophy.
Of the 44 patients diagnosed with BMD, causative mutations in BEST1 were found in 25 patients (Table 1). Twenty patients had known heterozygous mutations; five patients had novel heterozygous variants (p. Leu294Phe, p.Phe84Val, p.Gly83Ala, p.Trp93Arg, p.Trp309Arg) in BEST1; each novel variant had a PolyPhen score of 1, predicting that all variants were “probably damaging” mutations. Thus, 57% of the BMD patients had a molecular diagnosis consistent with their clinical diagnosis.
Pattern Dystrophy.
Pattern dystrophy is an autosomal dominant inherited disease. Mutations in PRPH2 (peripherin 2 gene) are known to cause PD. Isolated cases with a PD phenotype were also screened for mutations in ABCA4 (ATP binding cassette subfamily A, member 4 gene) when no mutations were detected in PRPH2. Of the 80 PD patients, 15 showed disease-causing variants in either the PRPH2 or ABCA4 gene (Table 2). Thirteen patients were heterozygous for known mutations in PRPH2; one additional patient had a novel p.Arg195Gln PRPH2 variant that was predicted to be “probably damaging,” leading to a positive molecular diagnosis. One of the 13 patients was a compound heterozygote for a VUS and a known causative mutation in PRPH2 (p.Tyr91Asn and p.Ile104Val, respectively) (Table 2). The VUS had a PolyPhen score of 0.909, predicting it to be “probably damaging.” Two patients had a single heterozygous mutation in ABCA4 leading to an unconfirmed molecular diagnosis, while another patient had two known compound heterozygous ABCA4 mutations leading to a positive molecular diagnosis (Table 2). One patient had a VUS in ABCA4 (c.3862+3A>G). This analysis resulted in a positive molecular diagnosis for 19% of the 80 probands with a diagnosis of PD.
Of the 80 patients with PD, six were additionally screened for mutations in ELOVL4, one for mutations in CTRP5, and six for mutations in BEST1. None of the patients with a diagnosis of PD had disease-associated variants in ELOVL4 or CTRP5. One of the patients was found to carry a novel variant in BEST1, c.250T>G (p.Phe84Val) (Table 2).
Cone-Rod Dystrophy.
Of the 54 CRD patients, 13 showed an autosomal dominant pattern of inheritance. Six patients carried the p.Arg838His or p.Arg838Cys GUCY2D mutation (Table 2). One of the six samples also carried a novel CRX variant, p.Ser213Pro (PolyPhen score of 0.998, “probably damaging”) (Table 2). These six samples had a positive molecular diagnosis.
Of the 41 CRD patient samples with autosomal recessive inheritance, 21 patient samples carried previously known mutations or novel VUS in ABCA4. However, only 15 patients had causative homozygous or compound heterozygous variants in ABCA4 resulting in a positive molecular diagnosis for recessive CRD. Of these 15, five patients did not have additional family members to test for segregation. The causative mutations included three novel variants: p.His52Gln (PolyPhen score 0.998), p.Ser795Arg (PolyPhen score 0.990), and p.Gly2074Val (PolyPhen score 1). The six cases with unconfirmed molecular diagnoses had variants predicted to be benign, had only a single heterozygous mutation, or had no mutations. These variants included p.Arg1300Gln and p.Glu1554Gly. The impact of c.859-9T>C detected in one of the patients is unknown, and thus the molecular analysis neither confirms nor excludes the clinical diagnosis in these patients (Table 2). In summary, 41% of the combined dominant and recessive CRD patient samples had a positive molecular diagnosis that confirmed their clinical diagnosis.
Phenotype and Genotype of Six Selected Patients With Late-Onset Retinal Pathology and Drusen
Case 1 (Multiple Diagnoses, No. 1).
A 44-year-old female with a visual acuity of 20/25 OD and 20/60 OS became symptomatic at the age of 35. Submacular yellow deposits of approximately 120 μm, some aggregated with a pigmented center, were observed. Retinal atrophy of the foveal region was more prominent in the right eye than the left eye (Figs. 1A, 1B). On fluorescein angiography, the deposits stained with fluorescein, and there was no suggestion of a dark choroid (Figs. 1C, 1D). The patient was enrolled with the potential diagnoses of DHCD, STGD1, or SFD.
Figure 1.

Clinical phenotype of case 1. A 44-year-old female with visual acuities of 20/25 OD and 20/60 OS became symptomatic at the age of 35. Submacular yellow deposits are circumscribed and measure approximately 120 μm; some have aggregated and form a pigmented center. Outer retinal atrophy of the foveal region is more prominent in the right eye than the left eye. On fluorescein angiography, these deposits stained with fluorescein, and there was no suggestion of a dark choroid.
Analysis of EFEMP1 did not identify causative mutations. Subsequent analysis of ABCA4 revealed two previously reported heterozygous mutations, c.4919G>A (p.Arg1640Gln) and c.5882G>A (p.Gly1961Gln), and six additional heterozygous VUS. Four of these novel changes were synonymous variants and were predicted to not cause splice alterations. Two additional novel variants were located in the intronic region (c.5898-11G>A and c.6729+21C>T) (Table 2).
Case 2 (PD, No. 17).
A 39-year-old female with a visual acuity of 20/25 OD and 20/200 OS, with central scotomas in both eyes, became symptomatic at age 35. Her optical coherence tomography (OCT) showed predominant outer retinal atrophy at the fovea with sub-RPE drusenoid deposits (Figs. 2A, 2B). The retinal atrophy was worse in the left eye than the right eye (Figs. 2C, 2D). The yellow circumscribed sub-RPE deposits aggregated as a ring in the parafoveal region and stained on fluorescein angiography, and there was no suggestion of a dark choroid (Figs. 2E, 2F). The 24/2 Humphrey visual field test showed central visual field deficits in both eyes.
Figure 2.

Clinical phenotype of case 2. A 39-year-old female with visual acuities of 20/25 OD and 20/200 OS and central scotomas in both eyes became symptomatic at the age of 35. Her OCT showed predominant outer retinal atrophy at the fovea with sub-RPE drusenoid-like deposits. The retinal atrophy is worse in the left eye than the right eye. The yellow circumscribed sub-RPE deposits aggregate as a ring in the parafoveal region. These stained on fluorescein angiography, and there was no suggestion of a dark choroid.
This patient was enrolled as having STGD1, PD, SFD or DHRD. Analysis of ABCA4, ELOVL4, PRPH2, and TIMP3 did not reveal the presence of known mutations. A single heterozygous known mutation, c.5461-10T>C, was detected in the ABCA4 gene (Table 2).
Case 3 (BMD, No. 25).
An 81-year-old male with a visual acuity of 20/200 OD, counting finger OS, and central scotoma in both eyes became symptomatic at age 47. He had normal full-field electroretinography (ERG) findings with retinal atrophy in the fovea of both eyes. This patient was enrolled as having BMD.
This patient was screened for mutations in PRPH2 and BEST1. Causative changes were not detected in PRPH2. Analysis of the BEST1 revealed two homozygous novel silent variants, c.1557C>T (p.Ser519Ser) and c.1608T>C (p.Thr536Thr) in exon 10, and one novel heterozygous missense variant, c.250T>G (p.Phe84Val) in exon 4, with the PolyPhen score of 1. Neither of the silent variants was predicted to alter splicing (Table 2).
Case 4 (PD, No. 13).
A female with a visual acuity of 20/20 OD, 20/30 OS presented with visual symptoms at age 39. She had yellow sub-RPE deposits in both maculas. She was enrolled with a diagnosis of BMD or PD. Mutation analysis did not detect causative mutations in BEST1, while a previously reported splice site mutation c.828+3A>T was detected in PRPH2 in the heterozygous state (Table 2).
Case 5 (CRD, No. 28).
A 63-year-old woman with a visual acuity of 20/40 OU and central scotoma in both eyes became symptomatic at age 46. Her full-field ERG showed reduced photopic and scotopic responses, and she exhibited signs of macular degeneration on examination.
This patient was enrolled with a diagnosis of dominant CRD. Initially the coding sequence of the CRX gene and targeted testing of codon Arg838 of GUCY2D were performed. When both these tests came up negative, PRPH2 was screened, and a heterozygous mutation, c.514C>T (p.Arg172Trp) that segregated with disease in the family was found (Table 2; Fig. 3).
Figure 3.
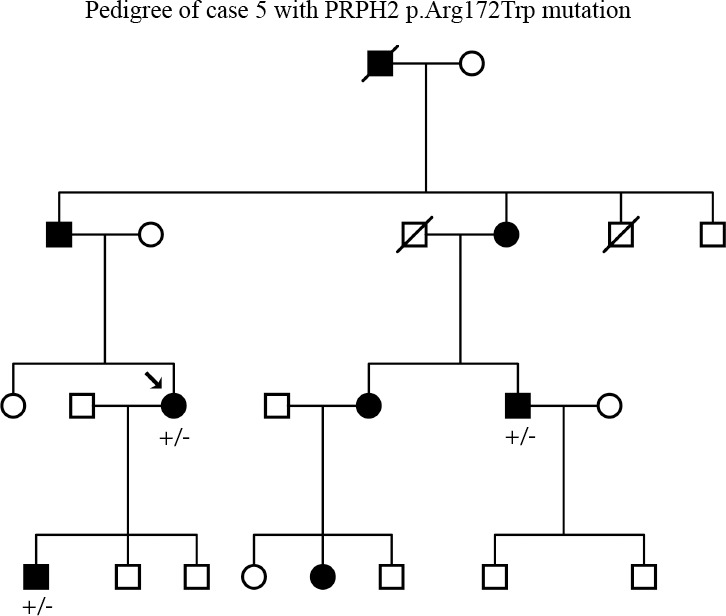
Pedigree of case 5. Segregation of the PRPH2 mutation at c.514C>T (p.Arg172Trp) with disease.
Case 6 (PD, No. 5).
An 82-year-old man with visual acuities of 20/20 OD and 20/25 OS became symptomatic at age 40. He had signs of macular degeneration on examination. The patient and two additional affected family members (a sister and a niece) were enrolled in eyeGENE with a diagnosis of PD. Analysis of CRX and ELOVL4 did not detect causative mutations, but PRPH2 testing revealed a heterozygous mutation, c.422A>G (p.Tyr141Cys), in the PRPH2 gene. The PRPH2 mutation was also observed in both affected relatives (Table 2; Fig. 4).
Figure 4.
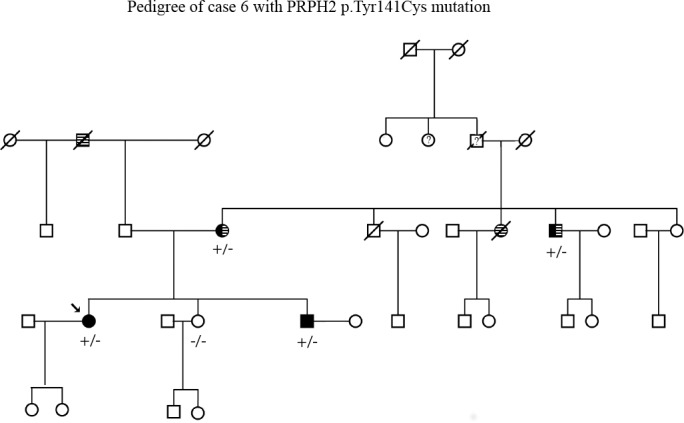
Pedigree of case 6. Segregation of the PRPH2 mutation at c.422A>G (p.Tyr141Cys) with disease. Striped background designates family members afflicted with hearing loss, and solid black designates family members affected by retinal degeneration. Question mark designates status unknown.
Discussion
Of the 213 samples, 55 patients (26%) had known causative mutations and 13 (6%) patients had VUS that were possibly pathogenic (Table 3). Best macular dystrophy had the highest success rate (57% of patients) for molecular diagnosis, likely contributed to by the relatively limited variation in phenotype: abnormal electro-oculography (EOG) and vitelliform lesions. The lowest rate of success was found in LORD patients, with none of the six patients having a positive molecular diagnosis. Late-onset retinal degeneration, an extremely rare disease with a phenotype that overlaps with many other retinal dystrophies including the common AMD, is often misdiagnosed.17 So far, only one mutation in CTRP5/C1QTNF5 has been reported in families of European origin.9 Lack of CTRP5 gene mutations in patients diagnosed with LORD may indicate involvement of other genes or the presence of mutations in the unscreened regions of the genes (introns or the promoter region).
Table 3.
Mutations or Unknown Variants Detected in Patients With Central Vision Loss
|
Gene |
Exon |
DNA Change |
Protein Change |
Genotype |
Result |
PolyPhen Description |
PolyPhen Score |
Frequency* |
Variant ID |
| Late-onset retinal degeneration | |||||||||
| CTRP5 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Sorsby fundus dystrophy | |||||||||
| TIMP3 | 1 | c.113C>G | p.Ser38Cys | Het | vAR/us | Probably damaging |
1 | 2 | |
| TIMP3 | 5 | c.610A>T | p.Ser204Cys | Het | Mut | 1 | CM941325/rs137853298 | ||
| Doyne honeycomb dystrophy | |||||||||
| EFEMP1 | 9 | c.1033C>T | p.Arg345Trp | Het | Mut | 2 | CM990504 | ||
| EFEMP1 | IVS10 | c.IVS10-14C>T | None | Het | vAR/us | NA | NA | 1 | |
| Best macular dystrophy | |||||||||
| BEST1 | 2 | c.28G>A | p.Ala10Thr | Het | Mut | 1 | CM982017 | ||
| BEST1 | 2 | c.47C>T | p.Ser16Phe | Het | Mut | 1 | CM010520 | ||
| BEST1 | 2 | c.72G>T | p.Trp24Cys | Het | Mut | 1 | CM982018 | ||
| BEST1 | 3 | c.240C>A | p.Phe80Leu | Het | Mut | 2 | CM004423 | ||
| BEST1 | 4 | c.248G>C | p.Gly83Ala | Het | vAR/us | Probably damaging |
1 | 1 | |
| BEST1 | 4 | c.277T>C | p.Trp93Arg | Het | vAR/us | Probably damaging |
1 | 1 | |
| BEST1 | 4 | c.279G>C | p.Trp93Cys | Het | Mut | 1 | rs28940273/CM982021 | ||
| BEST1 | 6 | c.652C>T | p.Arg218Cys | Het | Mut | 2 | CM982023 | ||
| BEST1 | 6 | c.680A>G | p.Tyr227Cys | Het | Mut | 1 | CM982024 | ||
| BEST1 | 6 | c.741G>A | p.Arg218His | Het | Mut | 2 | CM003486 | ||
| BEST1 | 7 | c.727G>A | p.Ala243Thr | Het | Mut | 2 | CM004434 | ||
| BEST1 | 7 | c.728C>T | p.Ala243Val | Het | Mut | 2 | rs28940570/CM00841 | ||
| BEST1 | 8 | c.880C>T | p.Leu294Phe | Het | vAR/us | Probably damaging |
1 | 1 | |
| BEST1 | 8 | c.887A>G | p.Asn296Ser | Het | Mut | 1 | CM010524 | ||
| BEST1 | 8 | c.903T>G | p.Asp301Glu | Het | Mut | 2 | CM991243 | ||
| BEST1 | 8 | c.910G>A | p.Asp304Asn | Het | Mut | 1 | CM024219 | ||
| BEST1 | 8 | c.925T>C | p.Trp309Arg | Het | vAR/us | Probably damaging |
1 | 1 | |
| BEST1 | 8 | c.929T>C | p.Ile310Thr | Het | Mut | 1 | CM000843 | ||
| BEST1 | 4 | c.250T>G | p.Phe84Val | Het | vAR/us | Probably damaging |
1 | 1 | |
| Pattern dystrophy | |||||||||
| ABCA4 | 6 | c.634C>T | p.Arg212Cys | Het | Mut | 1 | rs61750200 | ||
| ABCA4 | 17 | c.2588G>C | p.Gly863Ala | Het | Mut | 1 | CM970003/rs76157638 | ||
| ABCA4 | IVS26 | c.3862+3A>G | Abnormal splicing | Het | vAR/us | 1 | NA | ||
| ABCA4 | 30 | c.4469G>A | p.Cys1490Tyr | Het | Mut | 1 | CM990056/rs61751402 | ||
| ABCA4 | IVS38 | c.5461-10T>C | None | Het | Mut | 1 | CS057513 | ||
| PRPH2 | 1 | c.271T>A | p.Tyr91Asn | Het | vAR/us | Probably damaging |
.909 | 1 | |
| PRPH2 | 1 | c.310-313del(AT) | p.Ile104Val | Het | Mut | 1 | NA/Deletion | ||
| PRPH2 | 1 | c.422A>G | p.Tyr141Cys | Het | Mut | 2 | CM010125/rs61755781 | ||
| PRPH2 | 1 | c.515G>A | p.Arg172Gln | Het | Mut | 1 | CM930637/rs61755792 | ||
| PRPH2 | 2 | c.583C>T | p.Arg195Stop | Het | Mut | 1 | CM032999 | ||
| PRPH2 | 2 | c.629C>G | p.Pro210Arg | Het | Mut | 1 | CM941210 | ||
| PRPH2 | 2 | c.635G>C | p.Ser212Thr | Het | Mut | 1 | CM971289/rs61755801 | ||
| PRPH2 | 2 | c.683C>T | p.Thr228Ile | Het | Mut | 1 | TMP_ESP_6_ 42672248 | ||
| PRPH2 | 2 | c.708C>G | p.Tyr236Stop | Het | Mut | 1 | rs61755813 | ||
| PRPH2 | IVS2 | c.828+3A>T | Splice | Het | Mut | 4 | CS010139 | ||
| PRPH2 | 2 | c.584G>A | p.Arg195Gln | Het | vAR/us | Probably damaging |
1 | 1 | |
| Cone-rod dystrophy | |||||||||
| ABCA4 | 2 | c.156T>G | p.His52Gln | Het | vAR/us | Probably damaging |
0.998 | 1 | |
| ABCA4 | 3 | c.161G>A | p.Cys54Tyr | Het | Mut | 1 | CM990012/rs150774447 | ||
| ABCA4 | 28 | c.4169T>C | p.Leu1390Pro | Het | Mut | 1 | CM014810/rs61752430 | ||
| ABCA4 | 16 | c.2385C>T | p.Ser795Arg | Het | vAR/us | Probably damaging |
0.99 | 1 | |
| ABCA4 | IVS40 | c.5714+5G>A | Splice | Het | Mut | 1 | CS982057 | ||
| ABCA4 | 27 | c.3899G>A | p.Arg1300Gln | Het | vAR/us | Benign | 0.143 | 1 | |
| ABCA4 | 32 | c.4661A>G | p.Glu1554Gly | Het | vAR/us | Benign | 0.326 | 1 | |
| ABCA4 | 30 | c.4383G>A | p.Trp1461Stop | Het | Mut | 1 | Stop/NA | ||
| ABCA4 | IVS38 | c.5461-10T>C | None | Het | Mut | NA | NA | 2 | CS057513 |
| ABCA4 | 22 | c.3259G>A | p.Glu1087Lys | Het | Mut | 1 | CM970008/rs61751398 | ||
| ABCA4 | 42 | c.5882G>A | p.Gly1961Glu | Het | Mut | 2 | CM970016/rs1800553 | ||
| ABCA4 | 45 | c.6221G>T | p.Gly2074Val | Het | vAR/us | Probably damaging |
1 | 1 | |
| ABCA4 | IVS42 | c.5898+1G<A | Splice | Het | Mut | 1 | CS011524 | ||
| ABCA4 | IVS42 | c.5899-2delA | Splice | Het | Mut | 1 | rs3112831 | ||
| CRX | 3 | c.607T>C | p.Ser213Pro | Het | vAR/us | Probably damaging |
0.999 | 1 | |
| ABCA4 | 5 | c.559C>T | p.Arg187Cys | Het | Mut | 1 | COSM913472 | ||
| ABCA4 | 40 | c.5645T>C | p.Met1882Thr | Het | Mut | 1 | rs4147830 | ||
| ABCA4 | 6 | c.768G>T | p.Val256Val (abnlspl) | Het | Mut | 1 | CM990057/rs61750152 | ||
| ABCA4 | 31 | c.4577C>T | p.Thr1526Met | Het | Mut | 1 | rs62645944 | ||
| ABCA4 | 11 | c.1532G>A | p.Arg511His | Het | Mut | 1 | rs140482171 | ||
| ABCA4 | 12 | c.1622T>C | p.Leu541Pro | Het | Mut | 1 | CM990022/rs61751392 | ||
| ABCA4 | 21 | c.3113C>T | p.Ala1038Val | Het | Mut | 1 | CM970006/rs61751374 | ||
| ABCA4 | 12 | c.1622T>C | p.Leu541Pro | Hom | Mut | 2 | CM990022/rs61751392 | ||
| ABCA4 | 21 | c.3113C>T | p.Ala1038Val | Hom | Mut | 2 | CM970006/rs61751374 | ||
| ABCA4 | 22 | c.3322C>T | p.Arg1108Cys | Het | Mut | 1 | CM990039/rs61750120 | ||
| ABCA4 | 13 | c.1927G>A | p.Val643Met | Het | Mut | 1 | CM014293/rs61749417/ rs143548435 | ||
| ABCA4 | 24 | c.3602T>G | p.Leu1201Arg | Het | Mut | 1 | CM990042/rs61750126 | ||
| ABCA4 | 36 | c.5186T>C | p.Leu1729Pro | Het | Mut | 1 | CM990062/rs61750567 | ||
| ABCA4 | 13 | c.1933G>A | p.Asp645Asn | Het | Mut | 1 | rs617494181933 | ||
| ABCA4 | 23 | c.3364G>A | p.Glu1122Lys | Het | Mut | 1 | CM990041 | ||
| ABCA4 | 48 | c.6529G>A | p.Asp2177Asn | Het | Mut | 1 | CM970023/rs1800555 | ||
| ABCA4 | 35 | c.4918C>T | p.Arg1640Trp | Het | Mut | 2 | CM983728/rs61751404 | ||
| ABCA4 | 28 | c.4222T>C | p.Trp1408Arg | Het | Mut | 1 | CM990048/rs61750135 | ||
| GUCY2D | 13 | c.2512C>T | p.Arg838Cys | Het | Mut | 1 | rs61750172 | ||
| GUCY2D | 13 | c.2513G>A | p.Arg838His | Het | Mut | 5 | CM012606/rs61750173 | ||
| ABCA4 | IVS7 | c.859-9T>C | Unknown | Hom | vAR/us | NA | NA | 1 | |
| ABCA4 | 42 | c.5882G>A | p.Gly1961Glu | Hom | Mut | 1 | CM970016/rs1800553 | ||
| ABCA4 | 43 | c.5917delG | Deletion | Hom | Mut | 1 | RISN_ABCR: c.5917delG | ||
| PRPH2 | 1 | c.514C>T | p.Arg172Trp | Het | Mut | 1 | CM930639 | ||
| No specific clinical diagnosis | |||||||||
| ABCA4 | 35 | c.4919G>A | p.Arg1640Gln | Het | Mut | 1 | CM003577 | ||
| ABCA4 | 42 | c.5882G>A | p.Gly1961Glu | Het | Mut | 1 | CM970016/rs1800553 | ||
| ABCA4 | IVS42 | c.5898-11G>A | NA | Het | vAR/us | NA | NA | 1 | |
| ABCA4 | IVS48 | c.6729+21C>T | NA | Het | vAR/us | NA | NA | 1 | |
Frequency signifies number of times a mutation is observed within the data set.
Six patients with late-onset retinal pathology and drusen had well-characterized clinical data. Case 1 had two known mutations, c.4919 G>A (p.Arg1640Gln) and c.5882G>A (p.Gly1961Glu), in exons 35 and 42 of ABCA4. The presence of these two mutations in the compound heterozygous state in patients with a diagnosis of SD and CRD has been reported.18,19 Involvement of ABCA4 in causing pathology in this patient could not be confirmed, since additional family members were unavailable to evaluate if the two mutations occurred in the cis or trans configuration. Case 2 also had LORD with drusenoid deposits and carried a single ABCA4 mutation in the heterozygous state. Although mutations in ABCA4 have been reported to be associated with LORD, the lack of data on additional family members and the absence of the second mutation in case 2 limited the ability to evaluate the association between genotype and phenotype. Three additional patients (cases 4, 5, and 6) had heterozygous mutations in PRPH2 (Table 2). Two of these patients (cases 5 and 6) had additional affected family members, and the mutations segregated with disease (Figs. 3, 4). These observations are consistent with earlier reports on association of PRPH2 mutations with a wide range of retinal dystrophy phenotypes including PD, late-onset drusen, and macular dystrophy.20 Overall, mutations in the ABCA4, PRPH2, and BEST1 genes were found in the six patients with a LORD phenotype. Selection of PRPH2 and BEST1 genes for testing may result in a higher success rate in providing a positive molecular diagnosis for patients with late-onset retinal pathology and positive family history of RD, whereas sporadic cases or patients with no family history are more likely to carry mutations in ABCA4.
The RD phenotype involving central vision loss is associated with a group of genes implicated in a broad range of overlapping clinical symptoms. In the current study, six patients were diagnosed with both BMD and PD; one patient (case 4) was found to carry a PRPH2 mutation, confirming the PD diagnosis, and a second patient (case 3) was found carrying a VUS in the BEST1 gene, supporting the BMD diagnosis. One patient (case 5) diagnosed with autosomal dominant CRD carried a PRPH2 mutation. Another patient (case 6) with late-onset PD also carried a PRPH2 mutation. One patient (case 1) with a primary diagnosis of DHRD and secondary diagnoses of STGD1 and SFD carried two heterozygous mutations in the ABCA4 gene. These cases demonstrate the heterogeneity in clinical phenotype of LORD and the challenge in establishing genotype–phenotype associations in retinal dystrophies. Analysis of a larger sample set with well-characterized phenotype data will assist in understanding the association between phenotypes and specific genotypes in known and novel genes. Inconsistencies in patient diagnosis from referring clinicians may have contributed to the discrepancies in findings since the genetic screening strategy first targeted genes that were associated with specific phenotypes. The small cohort size of patients with diseases such as SFD and LORD, eight and six, respectively, limited the ability to draw any significant conclusions on the outcome of genetic analysis. Furthermore, the lack of information on VUS also affected the ability to establish definitive molecular diagnosis. Although PolyPhen analysis was performed on each novel mutation, the results are computational predictions that require biological or experimental confirmation. Differing methodologies used for diagnostic genetic screening were also a limitation in this study. Some samples were screened for mutations in all the exons of the genes of interest, while others were screened for mutations in only a subset of genetic regions. Sequential genetic screening that examines the most common mutations first, followed by examination of the most common disease-associated genes and finally the less common disease-associated genes or all genes, is cost-effective and efficient if a causal mutation is identified. However, this strategy does not provide uniform genetic information on all samples. With the rapid decrease in sequencing costs, sequencing of whole genomes, exomes, or custom capture of all known retinal disease genes is currently the best approach to identifying the genetic basis for retinal diseases.
Acknowledgments
Supported by National Institutes of Health Grants EY021237, EY022589, P30EY022589; Research to Prevent Blindness; Foundation Fighting Blindness; and National Eye Institute intramural funds. The ClinicalTrials.gov identifier for eyeGENE is NCT00378742. More information is available at nei.nih.gov/eyeGENE (in the public domain).
Disclosure: A. Alapati, None; K. Goetz, None; J. Suk, None; M. Navani, None; A. Al-Tarouti, None; T. Jayasundera, None; S.J. Tumminia, None; P. Lee, None; R. Ayyagari, None
References
- 1. Blain D, Goetz KE, Ayyagari R. Tumminia SJ. eyeGENE®: a vision community resource facilitating patient care and paving the path for research through molecular diagnostic testing. Clin Genet. 2013; 84: 190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998; 19: 241–247 [DOI] [PubMed] [Google Scholar]
- 3. Caldwell GM, Kakuk LE, Griesinger IB, et al. Bestrophin gene mutations in patients with Best vitelliform macular dystrophy. Genomics. 1999; 58: 98–101 [DOI] [PubMed] [Google Scholar]
- 4. MacDonald IM, Hebert M, Yau RJ, et al. Effect of docosahexaenoic acid supplementation on retinal function in a patient with autosomal dominant Stargardt-like retinal dystrophy. Br J Ophthalmol. 2004; 88: 305–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stone EM, Lotery AJ, Munier FL, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999; 22: 199–202 [DOI] [PubMed] [Google Scholar]
- 6. Sorsby AMM, Gardener N. A fundus dystrophy with unusual features. Br J Ophthalmol. 1949; 33: 67–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weber BH, Vogt G, Pruett RC, Stohr H, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby's fundus dystrophy. Nat Genet. 1994; 8: 352–356 [DOI] [PubMed] [Google Scholar]
- 8. Ayyagari R, Mandal MN, Karoukis AJ, et al. Late-onset macular degeneration and long anterior lens zonules result from a CTRP5 gene mutation. Invest Ophthalmol Vis Sci. 2005; 46: 3363–3371 [DOI] [PubMed] [Google Scholar]
- 9. Hayward C, Shu X, Cideciyan AV, et al. Mutation in a short-chain collagen gene, CTRP5, results in extracellular deposit formation in late-onset retinal degeneration: a genetic model for age-related macular degeneration. Hum Mol Genet. 2003; 12: 2657–2667 [DOI] [PubMed] [Google Scholar]
- 10. Lodato G, Giuffre G. Unusual associations of pattern dystrophies [in French]. J Fr Ophtalmol. 1985; 8: 147–154 [PubMed] [Google Scholar]
- 11. Chen S, Wang QL, Nie Z, et al. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron. 1997; 19: 1017–1030 [DOI] [PubMed] [Google Scholar]
- 12. Gregory-Evans K, Kelsell RE, Gregory-Evans CY, et al. Autosomal dominant cone-rod retinal dystrophy (CORD6) from heterozygous mutation of GUCY2D, which encodes retinal guanylate cyclase. Ophthalmology. 2000; 107: 55–61 [DOI] [PubMed] [Google Scholar]
- 13. Kitiratschky VB, Wilke R, Renner AB, et al. Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Invest Ophthalmol Vis Sci. 2008; 49: 5015–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cremers FP, van de Pol DJ, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet. 1998; 7: 355–362 [DOI] [PubMed] [Google Scholar]
- 15. Jaakson K, Zernant J, Kulm M, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003; 22: 395–403 [DOI] [PubMed] [Google Scholar]
- 16. Seo TS, Bai X, Kim DH, et al. Four-color DNA sequencing by synthesis on a chip using photocleavable fluorescent nucleotides. Proc Natl Acad Sci U S A. 2005; 102: 5926–5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuntz CA, Jacobson SG, Cideciyan AV, et al. Sub-retinal pigment epithelial deposits in a dominant late-onset retinal degeneration. Invest Ophthalmol Vis Sci. 1996; 37: 1772–1782 [PubMed] [Google Scholar]
- 18. Webster AR, Heon E, Lotery AJ, et al. An analysis of allelic variation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2001; 42: 1179–1189 [PubMed] [Google Scholar]
- 19. Briggs CE, Rucinski D, Rosenfeld PJ, Hirose T, Berson EL, Dryja TP. Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone-rod degeneration. Invest Ophthalmol Vis Sci. 2001; 42: 2229–2236 [PubMed] [Google Scholar]
- 20. Travis GH, Christerson L, Danielson PE, et al. The human retinal degeneration slow (RDS) gene: chromosome assignment and structure of the mRNA. Genomics. 1991; 10: 733–739 [DOI] [PubMed] [Google Scholar]