

Abstract
β-mercaptoacetate (MA) is a drug known to block mitochondrial oxidation of medium- and long-chain fatty acids (FAs) and to stimulate feeding. Because MA-induced feeding is vagally dependent, it has been assumed that the feeding response is mediated by MA's antimetabolic action at a peripheral, vagally innervated site. However, MA's site of action has not yet been identified. Therefore, we used fluorescent calcium measurements in isolated neurons from rat nodose ganglia to determine whether MA has direct effects on vagal sensory neurons. We found that MA alone did not alter cytosolic calcium concentrations in nodose neurons. However, MA (60 μM to 6 mM) significantly decreased calcium responses to both linoleic acid (LA; 10 μM) and caprylic acid (C8; 10 μM) in all neurons responsive to LA and C8. GW9508 (40 μM), an agonist of the FA receptor, G protein-coupled receptor 40 (GPR40), also increased calcium levels almost exclusively in FA-responsive neurons. MA significantly inhibited this response to GW9508. MA did not inhibit calcium responses to serotonin, high K+, or capsaicin, which do not utilize GPRs, or to CCK, which acts on a different GPR. GPR40 was detected in nodose ganglia by RT-PCR. Results suggest that FAs directly activate vagal sensory neurons via GPR40 and that MA antagonizes this effect. Thus, we propose that MA's nonmetabolic actions on GPR40 membrane receptors, expressed by multiple peripheral tissues in addition to the vagus nerve, may contribute to or mediate MA-induced stimulation of feeding.
Keywords: calcium imaging, nodose ganglion, G-protein coupled receptor 40, fatty acids, β-mercaptoacetate
agents that reduce metabolism of fatty acids (FA), a major metabolic fuel for peripheral tissues, induce robust feeding responses (12, 64). These agents include β-mercaptoacetate (MA), which antagonizes fat oxidation by blocking mitochondrial acyl-CoA-dehydrogenase (2, 3), and methyl-palmoxirate, which inhibits fat oxidation by antagonizing the mitochondrial long-chain fatty acid transporter, carnitine palmitoyltransferase 1 (CPT1). Etomoxir, which also blocks CPT1 (72), has been shown to stimulate feeding in humans (33), as well as in rats (23). Moreover, MA-induced feeding is antagonized by infusion of nutrients, including lipids, glucose, and fructose, further suggesting a metabolic mechanism of action (67). These and other observations have led to the concept of a “lipoprivic” control of food intake (41, 43, 62), which has been widely accepted as an important control of ingestive behavior.
Feeding elicited by MA is dependent on vagal sensory neurons (5, 62, 63) and on higher-order neurons comprising a visceral afferent pathway in the brain that includes the nucleus of the solitary tract (63), lateral parabrachial nucleus (9), and central nucleus of the amygdala (60). Nevertheless, the peripheral site(s) at which MA exerts its actions to stimulate feeding remains unresolved. The liver has been suggested to be the detection site (39, 40), but not all data are consistent with this view (37, 38, 50, 59). An alternative hypothesis has been put forth recently suggesting the intestinal enterocyte as the receptor cell that communicates fatty acid oxidation status to vagal afferent nerve endings (38, 40). Data showing that intestinal or portal vein infusions of FAs alter the firing of vagal neurons (35, 54, 55) and reduce food intake by a vagally dependent mechanism (49, 75, 76) are consistent with this hypothesis.
In the present set of experiments, we examined an alternative or additional mechanism through which MA might alter vagal function; that is, by acting directly on vagal sensory neurons. We measured cytosolic calcium responses of isolated vagal afferent neurons from the nodose ganglia using calcium imaging techniques. The advantage of this approach is that the actions of applied compounds can unequivocally be attributed to a direct action on the cultured neuron. We found that while MA had no direct effect on calcium signaling in vagal neurons by itself, it was capable of blocking the calcium signal induced by FA. Our findings suggest that these FA-induced responses are mediated by G protein-coupled receptor 40 (GPR40), a recently described plasma membrane receptor responsive to FAs (7, 16, 27, 28). These findings suggest that MA has additional actions besides inhibition of FA oxidation and that these other actions need to be considered as potential mechanisms underlying MA's various physiological effects, including MA-induced feeding.
MATERIALS AND METHODS
Cell isolation and culture.
Nodose ganglia were collected from male Sprague-Dawley rats purchased from Simonsen Laboratories (Gilroy, CA). Rats were housed in a facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care. They were maintained on a 12:12-h light-dark schedule with ad libitum access to standard pelleted rodent chow and tap water prior to tissue collection. All experimental procedures were approved by Washington State University Institutional Animal Care and Use Committee, which conforms to the National Institutes of Health's guidelines.
Nodose ganglia were removed from anesthetized rats (ketamine 25 mg/100 g, xylazine 2.5 mg/100 g) under aseptic conditions and placed in ice-cold Hanks' balanced salt solution (HBSS). The ganglia were cleaned of connective tissue and desheathed. For each rat, both left and right ganglia were combined into a single isolation. Nodose cells were dissociated according to a procedure described by Zhao and Simasko (77). Briefly, the desheathed ganglia were washed once in HBSS, placed in ∼3 ml of digestion buffer (1 mg/ml dispase II and 1 mg/ml collagenase type Ia in Ca2+- and Mg2+-free-HBSS), sliced into ∼1-mm3 fragments using a sterile scalpel blade, and placed in an incubator at 37°C to digest for 120 min. At the end of the digestion, the cells were dispersed by gentle trituration through Pasteur pipettes coated with Sigmacote. The dispersed cells were then washed in HEPES-buffered Dulbecco's modified eagle's medium (HDMEM), supplemented with 10% FCS and antibiotic (penicillin-streptomycin). After the final resuspension, the cells were plated onto poly-l-lysine-coated coverslips (100 μg/ml polylysine) for 45 min and grown in HDMEM with 10% FCS at 37°C in an atmosphere of 5% CO2-95% air. All experimental procedures were conducted on the day following collection of the nodose ganglia.
Calcium measurements.
Intracellular calcium concentrations were determined by ratiometric measurements with Fura-2 AM. All manipulations and experiments were performed at room temperature in a physiological salt solution (in mM: 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 6 glucose, and 10 HEPES, with the pH adjusted to 7.4 with Tris base) (standard bath). Cells on coverslips were washed in standard bath and then allowed to take up Fura-2 AM (1 μM) for 30 min, followed by a 30-min wash period. Coverslips were then mounted in a closed chamber (∼0.3 ml) through which solutions were perfused by gravity (∼3 ml/min). Solution changes were made by switching inflow lines through a common manifold (new solutions reach cells ∼15 s after switch). Chambers were mounted on the stage of a Nikon TE2000-inverted microscope and examined with a 40× oil immersion lens. Cells were alternatively exposed to 340- and 380-nm light, and images were collected through a 510-nm filter with a charge-coupled device camera (CoolSnap HQ, Photometrics, Tucson, AZ). Image pairs were captured every 6 s, and ratios of fluorescent intensities at the two excitation wavelengths were obtained from regions over individual cells. Data collection and manipulations were performed with MetaFluor Software (Universal Imaging, West Chester, PA). Calcium concentrations were then determined by comparing ratio values to a standard curve obtained in a bath containing 10 μM Fura-2, 130 mM KCl, 10 mM MOPS buffered to pH 7.4 with KOH, 10 mM EGTA, and various concentrations of CaCl2 (0 to 10 mM). Free calcium concentrations were calculated using the computer program EQCAL (Biosoft, Ferguson, MO).
Cell selection and experimental protocols.
Nodose neurons are easily identified and selected on the basis of their large, round cell bodies. Nonneuronal cells have spindle or filamentous shapes. To be included in the data set for analysis, a neuron needed to maintain a basal calcium level below 200 nM, and induced calcium increases needed to be transient and return to near basal levels. Finally, at the end of every experiment, cells were depolarized with 55 mM K+ (high K+, reciprocal reduction in bath Na+). Any cells that did not exhibit a reversible calcium increase of at least 40 nM in response to high K+ were excluded from further analysis. Challenge solutions were applied for 1–3 min as indicated. Repeated challenges were separated by 2–3 min, unless recovery from a previous response required additional time or as noted. For each experimental protocol, nodose cells from at least three different isolations were used.
RT-PCR.
Total RNA was isolated from rat pancreas or whole nodose ganglia using RNAqueous-4PCR Kit (Ambion, Austin, TX). Genomic DNA contamination was removed by digestion with RNase-free DNase I at 37°C for 30 min. First-strand cDNAs were synthesized by incubating the total RNA (0.1 μg) with random decamers or oligo(dT) primers, RNase inhibitor, and Moloney murine leukemia virus RT for 1 h at 42°C. For single-cell PCR, cultured nodose neurons were collected into 11 μl of lysis buffer using sterile glass pipettes, and the single-cell lysate was then treated with DNase I (Cell-to-cDNA II kit; Ambion). After deactivating DNase I, RT reaction was initiated according to the instruction manual (77).
PCR was performed using specific primer pairs for rat GPR40 (gene accession no. AB095744). The forward primer was 5′-ACC AGT TCC CTG GGC ATC AAC ATA-3′ and the reverse primer was 5′-ACC AAG GGC AGA AAG AAG AGC AGA-3′. The PCR product size was 125 bp. Positive controls were total RNA from rat pancreas, which has a high expression of GPR40. All amplifications (40 μl) were performed in PCR buffer containing 1.5 mM MgCl2 and 125 μM dNTPs, 0.25 μM of each primer, and 25 units/ml Taq DNA polymerase (Invitrogen). Reactions were performed as follows: 5 min at 95°C, 45 cycles at 95°C for 45 s, annealing temperature at 60°C for 45 s, 72°C for 1 min, and final extension at 72°C for 5 min. PCR products were analyzed by electrophoresis on a 2% agarose gel containing ethidium bromide. The RT products without RT were used as a negative control. For single-cell PCR, PGP9.5 (Protein Gene Product 9.5; gene accession no. NM017237) was used as a neuronal marker. Only single-cell samples positive for PGP9.5 were included in the final analysis. The primers for PGP9.5 were 5′-CCT GCT GCT GCT GTT TCC-3′ (forward) and 5′-TGT CCC TTC AGT TCC TCA ATT T-3′ (reverse).
To confirm the DNA sequence of the PCR product, the resulting PCR products were extracted from agarose gel by using QIAquick gel extraction kit (Qiagen, Valencia, CA). Then the sequences were determined by a fluorescence labeled dye-terminator reaction using the BigDye terminator system.
Chemicals.
Media components (DMEM, FCS, and antibiotics), HBSS (and Ca2+- and Mg2+-free-HBSS), and Fura-2 AM came from Invitrogen (Carlsbad, CA). Dispase was purchased from Roche (Indianapolis, IN). NaCl, KCl, and CaCl2 are from JT Baker (Philipsburn, NJ). GW9508 and diazoxide were purchased from Tocris Bioscience (Ellisville, MO). CCK octapeptide 26–33 was purchased from Peptides International (Louisville, KY). All other agents [collagenase, mercaptoacetate (MA), caprylic acid (C8), linoleic acid (LA), capsaicin, serotonin (5HT), poly-l-lysine, and other buffer components] were purchased from Sigma (St. Louis, MO). The LA preparation contained methyl-β-cyclodextran (MbC) to improve water solubility (Sigma catalog no. L5900).
Data analysis.
Calcium responses to the various challenges are expressed as the change in cytosolic calcium from a basal value (Δcalcium). Basal values were determined from the period just prior to exposure of the challenge solution. In some cases, percent inhibition was based on the reduced level from a peak response (blocker added during the response) or, in other cases, it was based on percent inhibition compared with a control response (response with and without inhibitor present) Summarized results are averages ± SE. To be included as a responsive cell, the Δcalcium needed to be at least 20 nM, as this level of change greatly exceeded typical background fluctuations (<5 nM). Statistical comparisons of responses within the same neuron were performed by a paired t-test. Comparisons between neurons or effects of different agents were tested by a t-test if only two results were compared, or an ANOVA with the Holm-Sidak test was used for post hoc comparisons when more than two responses were compared. Tests for the significance of distributions of responsive neurons were done with a χ2 analysis. Statistical significance was considered if P < 0.05.
RESULTS
LA and C8 increase cytosolic calcium in nodose neurons via an MA-sensitive pathway.
Acute application of the long-chain FA, LA (10 μM), and the medium-chain FA, C8 (10 μM), significantly increased cytosolic calcium from basal levels (Fig. 1). The calcium response had a rapid onset, reached peak amplitude relatively slowly, and was reversible following washout of the FA (Fig. 1, A and B). Fig. 1C shows an example of a nodose neuron that was nonresponsive to LA, but that still exhibited the expected response to high K+, indicating that the nonresponsiveness was not due to an unhealthy neuron. On average, the responses to LA and C8 were not different (LA: 105 ± 10 nM; n = 78; C8: 83 ± 12 nM; n = 27), and the percentage (∼25%) of neurons responsive to C8 and LA did not differ (Fig. 1D). MbC was used in LA solutions to improve LA's water solubility. The highest concentration of MbC in LA solutions in the experiments was 8.4 μg/ml. We tested the effect of MbC at 10 μg/ml on calcium response. MbC (2 to 3 min) treatment had no effect on calcium influx, as the average of Δcalcium was 15 ± 6 nM after MbC treatment, while the same cells responded to 10 μM LA with an average of Δcalcium 200 ± 32 nM (n = 10 responding cells from a total of 31 nodose cells).
Fig. 1.
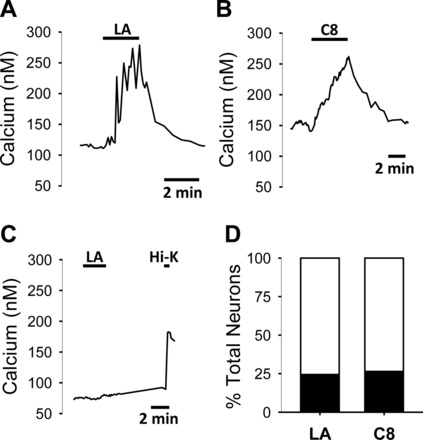
Representative calcium traces in isolated nodose neurons showing responses to linoleic acid (LA; A), caprylic acid (C8; B), and a neuron not responsive to LA, but that did respond to depolarization with a high K+ bath (C). The labeled lines above the data trace indicate when LA (10 μM), C8 (10 μM), or the high KCl bath (55 mM K+) was applied to the neurons. D: percent distribution of neurons responding (black) or not responding (open) to LA (83 responding and 243 insensitive) and C8 (29 responding and 77 insensitive). There was no significant difference between the distribution of LA- and C8-responsive neurons (χ2 analysis).
Bath application of MA alone (6 mM) did not alter calcium in 65 neurons tested (representative responses in Fig. 2, A and C). However, when MA was applied during a response to LA (Fig. 2A) or C8 (Fig. 2C) or coadministered with LA (Fig. 2E), there was a rapid and rapidly reversible decline of calcium levels (summarized in Fig. 2, B and D). MA suppressed the ongoing calcium response to LA or C8 only while present in the medium. MA inhibited the calcium increase induced by either LA or C8 in every neuron in which MA was tested. A dose-response study of the effects of MA indicated that 60 μM MA had a significant effect on the response induced by LA (Fig. 2E). Since responses to C8 and LA did not differ, we conducted the remaining experiments using LA as the stimulus. In addition, we used 6 mM MA in the remaining work, to ensure that we were observing maximal effects of MA on the induced responses.
Fig. 2.
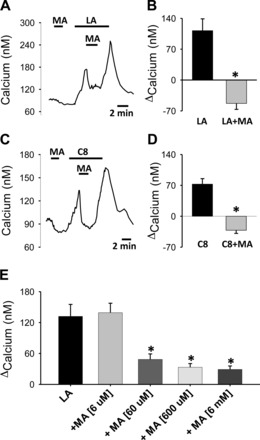
Effects of mercaptoacetate (MA) on the calcium responses induced by LA and C8. A: representative trace of inhibition by MA (6 mM) of a calcium response induced by LA (10 μM). B: summary of the effects of MA on the calcium response induced by LA (LA: response to LA alone; LA+MA: reported as the calcium decrease induced by MA from the response induced by LA). MA induced a decrease in every neuron tested (n = 13, *P = 0.008, paired t-test). C: representative trace of inhibition of a C8 (10 μM)-induced calcium response by MA (6 mM). D: summary of the effects of MA on responses induced by C8 (reported as in B). Inhibition was seen in all C8-responsive neurons tested (n = 14, *P < 0.001, paired t-test). E: dose-response study of the effect of MA on calcium responses induced by LA (LA was 10 μM in every challenge; responses are reported as the increase in calcium above a baseline obtained just prior to challenge with LA). LA alone (n = 26); + MA [6 μM] (n = 2), + MA [60 μM] (n = 15), + MA [600 μM] (n = 25), and + MA [6 mM] (n = 13). *P <0.001 compared with LA alone and LA+MA (6 μM), Holm-Sidak post hoc test.
Effect of diazoxide on the LA response.
Often, the link between metabolism and cell activation is closure of the ATP-regulated K+ channel (KATP), when the cell is given a metabolic substrate (31). To determine whether this mechanism plays a role in the calcium response to LA, we examined the effect of the KATP channel opener, diazoxide (78) on this response. We found that diazoxide did not significantly reduce the calcium response induced by LA (Fig. 3, A and B), indicating that the response of nodose neurons to LA does not require closure of the KATP channel.
Fig. 3.
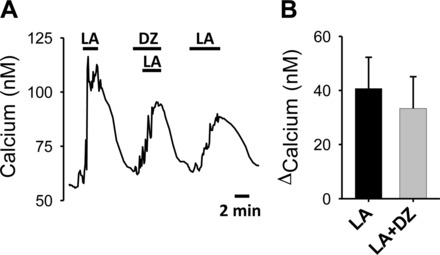
The effect of the KATP channel agonist, diazoxide (DZ; 10 μM) on LA-induced calcium increases. A: representative calcium trace of an LA-induced response in the presence of DZ. B: summarized results from DZ experiments (n = 7; P = 0.4, paired t-test).
GW9508 stimulates LA-responsive cells and the response to GW9508 is inhibited by MA.
We next investigated the possibility that nodose neuron responses to FAs are mediated by GPR40, a cell surface receptor that has been shown to mediate FA responses in other systems (7, 11, 28). We found that the GPR40 agonist, GW9508 (6), stimulated a rapid and reversible increase in cytosolic calcium (Fig. 4A). The average response to GW9508 was approximately the same as the response to LA (Fig. 4B). Responsiveness to LA and GW9508 was highly correlated (21 of 26 neurons responsive to GW9508 also responded to LA, and 21 of 23 neurons responsive to LA also responded to GW9508; Fig. 4C). This further suggests that LA and GW9508 share a common mechanism, the GPR40 receptor, for stimulation of nodose neurons.
Fig. 4.
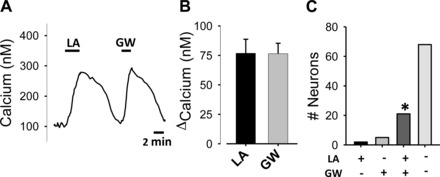
The effect of the GPR40 agonist, GW9508 (GW) on calcium levels in nodose neurons. A: representative trace from an LA (10 μM) and GW (40 μM) responsive neuron. B: average calcium increase induced by LA and GW in LA and GW responsive neurons (n = 23 and 26, respectively). C: number of neurons responsive to LA and GW. Symbols on the x-axis indicate responsiveness (+) or unresponsiveness (−) to LA (top line) or GW (bottom line) only and those responsive to both LA and GW. Most LA-responsive neurons (about 20% of the 96 neurons tested) were also responsive to GW. *Occurrence of GW and LA responsiveness significantly overlapped (P < 0.001, χ2 analysis).
Because overlap between LA and GW9508 responses suggested a common mechanism of action between these agents, and MA inhibited the response to LA, we next tested whether MA could also inhibit the response to GW9508. We found that MA significantly inhibited calcium responses induced by GW9508 (Fig. 5, A and B) and that this inhibition occurred in every neuron tested with GW9508 and MA. This suggests that MA inhibits calcium responses to LA and GW9508 by acting at the GPR40 receptor. In a separate experiment, the effects of repeated GW9508 challenges on calcium responses were measured. In cells that responded to GW9508 (12 of 39) there was no sign of densensitization between responses (40 μM, 2 min each; Fig. 5, C and D).
Fig. 5.
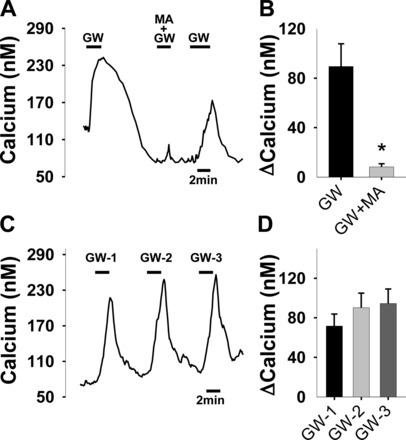
The effect of MA (6 mM) on the calcium response induced by GW (40 μM) (A and B) and the effect of repeated GW treatments on calcium response (C and D). A: representative calcium trace. B: summary of the effect (n = 16, *P < 0.001 paired t-test). C: representative calcium trace by three GW treatments (40 μM; 2 min each). B: summary of the effect (n = 12; P > 0.4, one-way ANOVA).
Identification of GPR40 gene in nodose neurons.
As shown in Fig. 6, the presence of PCR product corresponding to rat GPR40-encoding sequence was detected in both whole nodose ganglia and single-cell cultured nodose neurons, as well as in the pancreas. The GPR40 gene was positively identified in 2 of 30 cells (7%) analyzed with single-cell PCR, confirming its presence in a subpopulation of nodose neurons. The sequences of PCR products were confirmed by DNA sequencing after extraction of PCR products from the agorose gel.
Fig. 6.
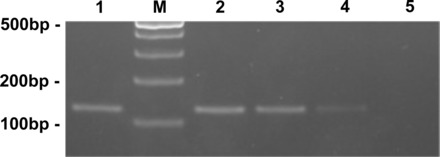
Expression of GPR40 mRNA detected by RT-PCR in different tissues. Lanes 1 and 4 show the expression of GPR40 in the pancreas (lane 4 is from a 1/10 dilution of RT product from the lane 1 sample). Lane 2 is from whole nodose ganglia. Lane 3 is from a single cultured nodose neuron. Lane 5 is from whole nodose ganglia total RNA without RT reaction, as a negative control. Lane M, DNA size marker.
Effects of MA on other agents that induce calcium responses.
The ability of MA to reduce GW9508-induced responses suggested that the effect of MA is likely mediated by disrupting GPR40-induced activation of nodose neurons. To determine whether the effect of MA is selective for this pathway, we examined MA's effects on the calcium response induced by agents that cause cytosolic calcium to increase via a different G protein receptor (other than GPR40) or by a non-G protein-mediated pathway. We found that MA did not alter the calcium response to CCK (Fig. 7A), which is mediated by a different G protein receptor (71). Similarly, MA had no effect on the response to capsaicin (Fig. 7B), which is mediated by the transient receptor potential vanilloid 1 (TRPV1) receptor (17, 51) and did not impair the calcium response to a Hi-K+, which opens voltage-gated ion channels (Fig. 7C). 5HT3 receptors, which are ligand-gated ion channels (69), are expressed by vagal neurons (26, 34), and previous data have shown an increase in calcium in response to 5HT in the isolated nodose neurons (66). MA did not significantly impair the 5HT-induced calcium response (Fig. 7D). The percent inhibition by MA of these various agents on cytosolic calcium responses to these agents is summarized in Fig. 7E.
Fig. 7.
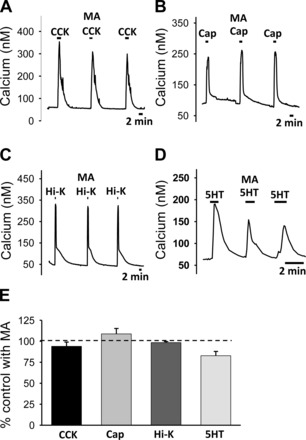
The effect of MA (6 mM) on calcium responses induced in nodose neurons by CCK (10 nM), capsaicin (100 nM), and high K+ and 5HT (10 μM). Panels show representative traces of the calcium responses elicited by each individual drug alone, by the drug in the presence of MA, and by the drug alone after recovery for CCK (A; n = 7), capsaicin (B; n = 5), high K+ (C; n = 16), and 5HT (D; n = 9). Summary of effects are shown in E. MA did not significantly alter the calcium response to any of these drugs alone.
DISCUSSION
Mercaptoacetate has been widely used to investigate control of food intake. This compound stimulates feeding by mechanisms dependent on an intact abdominal vagal afferent innervation. Because MA blocks mitochondrial acyl-CoA dehydrogenase (2, 3), it has been assumed that blockade of FA oxidation is the action responsible for its stimulatory effect on feeding. Indeed, this assumption is supported by a variety of evidence (43). We found, however, that both FAs and MA are capable of altering the responses of a subset of vagal sensory neurons by actions that appear to be mediated at the level of the cell membrane. Both LA and C8, as well as the GPR40 selective agonist, GW9508, increased cytosolic calcium levels in isolated nodose neurons. MA alone did not alter calcium levels. However, MA blocked FA-induced responses in all FA-responsive neurons tested, as well as blocking responses to GW9508. Use of cultured neurons in this work makes it likely that effects that we observed result from a direct action of these compounds on the neurons themselves, rather than from the release or action of other bioactive substances present in tissue or whole animal preparations or from activation of nonvagal cells.
Although it appears that certain neurons are able to oxidize fat (32, 36), neurons in general do not utilize fat as a substrate for energy metabolism. For this reason, a direct metabolic action of MA on nodose neurons would not necessarily be expected. However, we explored this possibility in our experiments by the use of diazoxide. Alterations in membrane potential following metabolic perturbations are often mediated by changes in the cellular ATP/ADP ratio. Mitochondrial oxidation of fuels, including fat, increases the ATP/ADP ratio within the cell, thereby closing the KATP channels, depolarizing the cell, and opening voltage-dependent calcium channels (1, 31). As a KATP channel opener, diazoxide would be expected to produce a hyperpolarizing effect and inhibit or reduce FA-induced calcium influx in nodose neurons, as it has been shown to do in pancreatic β-cells (57). However, diazoxide had no effect on the FA-induced increase in cytosolic calcium in nodose neurons. This result is not due to the absence of this channel in nodose neurons, since effects of glucose on glucose-excited nodose neurons have been shown to be mediated by the KATP channel (15). Our finding that FA effects in nodose neurons are independent of this channel suggests that these effects do not require changes in ATP levels in these neurons.
The MA-induced blockade of FA-induced calcium signals did not appear to be attributable to nonspecific or toxic effects on nodose neurons. MA did not inhibit calcium responses to 5HT, capsaicin, or high K+ concentrations, all of which increase cytosolic calcium via receptors or mechanisms that are not G protein-coupled. Serotonin increases calcium in nodose neurons via the 5HT3 receptor, a ligand-gated ion channel (66). High K+ concentrations open voltage-sensitive Ca2+ channels, and capsaicin activates TRPV1 channels (17, 51). Furthermore, MA did not attenuate calcium responses of nodose neurons to CCK, a hormone that binds to a G protein receptor, but not GPR40 (71). This result suggests that MA acts on a specific and limited population of G protein-coupled receptors that are responsive to FAs.
The compound GW9508 is an agonist of GPR40 (6), a member of a larger family of G protein-coupled receptors that responds directly to FAs (7, 8, 28, 52, 66). GPR40 (7, 28) and GPR120 (20, 28, 53) respond to both medium- and long-chain FAs, as well as saturated FAs, and have been reported in pancreatic islets, intestinal enteroendocrine cells (19), taste cells (10), and brain (4, 47, 48). Receptors responsive to short-chain FAs include GPR41 and GPR43 (8, 52), which are expressed in adipocytes and immune cells, respectively. FA stimulation of GPR40 in pancreatic cells (13, 27, 28) causes an increase in cytosolic calcium levels. Similarly, we found that GW9508 increased calcium levels in nodose neurons, mimicking the effect of LA, and this action was almost exclusively on a mutual population of neurons. The effects of GW9508 and LA on cytosolic calcium levels were both blocked by MA. Further evidence for this conclusion comes from our observation that we could measure messenger RNA for GPR40 in the nodose and that via single-cell RT-PCR measurements, we found GRP40 message in cells positively identified as neurons via expression of PGP9.5. Together, these results suggest that MA reduces the calcium response to FA in nodose neurons by actions at the level of the GPR40 membrane receptor, an effect that would not require fat metabolism for signaling. However, at this time we are not able to exclude the possibility that MA may also exert effects on the related FA receptor, GPR120, since the available agonists and antagonists for GPR40 and GPR120 receptors are not totally selective. Both of these receptors interact with long- and medium-chain FAs, and both are classified as Gq receptor types (25).
We found that MA alone does not induce a Ca2+ influx in nodose neurons; rather, it blocks FA-induced Ca2+ influx, which appears to contradict previous electrophysiological results showing that MA activates vagal neurons in vivo (46). However, in the in vivo experiments, intraportal administration of MA at doses within the range of those used in our tissue cultures produced a very slowly developing response (a delay of 10–30 min), while the effect of MA that we observed was immediate. This delay may indicate that the stimulation they observed was a result of indirect effects of MA that would not be present in cultured nodose neurons. Indeed, the novel action of MA at GPR40 receptors that we describe here indicates a probable widespread action of MA, not only on nodose neurons, but also on GPR40 receptors in other cell types. Significantly, recent work has shown that GPR40 mediates CCK (44) and GLP-1 (73) secretion in response to FA, and the enhancement of glucose stimulated insulin secretion by FA (27, 28). In other words, the results reported here strongly suggest that an important mechanism underlying MA-induced feeding is likely to arise from its effects on FA signaling at enteroendocrine and pancreatic GPR40 receptors that normally stimulate secretion of peptides that powerfully alter food intake, as well as by its action on the vagus nerve. We are currently pursuing this hypothesis using both in vivo and in vitro approaches. Our results to date have shown that MA blocks insulin secretion almost completely, even in the context of elevated blood glucose (70) and have found that MA blocks FA-induced GLP-1 secretion both in vivo and in vitro (61).
It may be significant that effects of MA that we describe are opposite to the effects of FAs at the cellular level, as they are at the behavioral level. Fatty acids reduce food intake, in part, by capsaicin-sensitive vagal sensory mechanisms (22, 29, 49, 75, 76). In agreement, while MA stimulates a feeding response, this response is also mediated by capsaicin-sensitive vagal sensory neurons (62, 63). Interestingly, we found in related work using the same techniques employed in the present study that all neurons responsive to LA or GW9508 (n = 96) were also responsive to capsaicin, although not all capsaicin-responsive neurons were responsive to LA or GW9508. The possibility that MA and FAs have opposite actions mediated by a common cellular mechanism and on a common neuronal population, as suggested here, is, therefore, consistent with what is currently known regarding the physiological substrates for these agents. Of additional interest is that decreased food intake produced by CCK is also dependent on capsaicin-sensitive neurons (58), thus defining an important subpopulation of neurons within the vagus nerve that are critical to nutrient and hormone-induced feeding cues.
Some laboratories have reported that MA does not stimulate feeding effectively in rats unless they are maintained on fat-enriched diets (e.g., 64), raising the question of whether we would obtain different results in nodose neurons if our rats had been maintained on a fat-enriched diet. Although fat-enriched diets do enhance MA-induced feeding, we and others have previously reported robust effects of MA on food intake in rats maintained on a standard rodent diet (24). Most importantly, MA was not without effects in the present work, but rather, produced very clear and potent effects on FA-induced Ca2+ influx, even using the standard diet. Thus, it is possible that the apparently more robust effect of MA in animals fed fat-enriched diets could result from the possible higher level of expression of GPR40 in animals on these fat-enriched diets.
The identification of FA receptors and elucidation of their participation in metabolic functions is a rapidly expanding field of investigation. In addition to GPR40, FA receptors of other types have been shown recently to participate in control of food intake and energy homeostasis (for reviews, see Refs. 25 and 68). For example, GPR40 and GPR120 both interact with long- and medium-chain FAs. GPR120 has been shown to mediate FA-induced suppression of gastric ghrelin secretion (30, 45). GPR84 interacts with medium-chain, but not long-chain FAs, while GPR41 and GPR43 interact with short-chain FAs, mediating a broad array of functions related to metabolism, inflammation, and disease (68). GPR43 is expressed in adipocytes and functions during adipogenesis (14, 42). Our present findings expand and complement this growing awareness of the diverse functions and mechanisms of FA signaling underlying energy homeostasis.
Results reported here show that calcium influx is increased in ∼20–25% of cultured nodose neurons by long- and medium-chain FAs and by GW9508, a GPR40 agonist. Although MA did not have an effect of its own on Ca2+ influx, MA blocked the Ca2+ response to both FA and GW9508 in nearly all neurons responsive to these agents. PCR results indicate that GPR40 is expressed in nodose ganglia. We draw three conclusions from these results: 1) that vagal afferent neurons are physiological targets for FA signaling; 2) that the direct effects of both FA and MA on vagal afferent neurons are mediated, at least in part, at the level of the cell membrane by GPR40; 3) that effects of MA and FA at GPR40 are directly antagonistic. Since GPR40 receptors influence insulin secretion and control secretion of gastrointestinal enteroendocrine peptides with vagally mediated effects that alter food intake, these findings support the novel hypothesis that MA may stimulate feeding by cumulative inhibitory effects on FA signaling at widely distributed GPR40 receptors. These results are foundational for a revised understanding of the “lipoprivic control” of feeding, which, since its discovery in 1986 (64), has been viewed as being based solely on reduced mitochondrial FA oxidation. Although the use of MA in previous studies of food intake has provided significant insights into the effects of fatty acid availability on appetite, it is clear from our present results that its mechanisms of action have not been completely understood and that interpretations of MA effects on food intake must now shift from oxidative metabolism to include G protein receptor-coupled signaling.
Perspectives and Significance
Studies examining the effect of fat on body weight homeostasis, food intake, and insulin sensitivity have largely focused on stored fat and on leptin's important role as an endocrine signal of adiposity. In contrast, in the present work, we examine FAs themselves as signaling agents and the possibility that MA interferes with that signaling function at the level of the cell membrane. Although further work is required, the present results expand our understanding of possible mechanisms and sites of action that may mediate MA-induced food intake. Fatty acid receptors, especially GPR40, are being aggressively pursued as therapeutic targets for treating obesity and diabetes (18, 21, 56, 65, 74). Consequently, as a relatively inexpensive FA antagonist, MA will continue to be useful as a tool with which to study mechanisms through which circulating and intestinal FAs alter neuronal, enteroendocrine, pancreatic, and behavioral responses. Furthermore, this unexpected pharmacological effect of MA may point to the development of a new series of compounds that have antagonistic effects on GPR40 receptors, compounds that may find numerous therapeutic uses in diabetes, obesity, and other metabolic disease states.
GRANTS
This work was supported by National Institutes of Health Grants DK-040498, DK-081546 and DK-097437 awarded to S. Ritter and DK-067146 awarded to S. M. Simasko.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: R.A.D., S.M.S., and S.R. conception and design of research; R.A.D., H.Z., D.K., and A.-J.L. performed experiments; R.A.D., S.M.S., and S.R. analyzed data; R.A.D., S.M.S., and S.R. interpreted results of experiments; R.A.D. and A.-J.L. prepared figures; R.A.D. and S.R. drafted manuscript; R.A.D., H.Z., D.K., A.-J.L., S.M.S., and S.R. approved final version of manuscript; A.-J.L., S.M.S., and S.R. edited and revised manuscript.
ACKNOWLEDGMENTS
Present address for R. Darling: School of Pharmacy, University of Wyoming, Laramie, Wyoming 72071 (e-mail: badarling03@yahoo.com).
REFERENCES
- 1.Ashcroft FM, Rorsman P. ATP-sensitive K+ channels: a link between B-cell metabolism and insulin secretion. Biochem Soc Trans 18: 109–111, 1990 [DOI] [PubMed] [Google Scholar]
- 2.Bauche F, Sabourault D, Giudicelli Y, Nordmann J, Nordmann R. 2-Mercaptoacetate administration depresses the β-oxidation pathway through an inhibition of long-chain acyl-CoA dehydrogenase activity. Biochem J 196: 803–809, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauche F, Sabourault D, Giudicelli Y, Nordmann J, Nordmann R. Inhibition in vitro of acyl-CoA dehydrogenases by 2-mercaptoacetate in rat liver mitochondria. Biochem J 215: 457–464, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boneva NB, Kaplamadzhiev DB, Sahara S, Kikuchi H, Pyko IV, Kikuchi M, Tonchev AB, Yamashima T. Expression of fatty acid-binding proteins in adult hippocampal neurogenic niche of postischemic monkeys. Hippocampus 21: 162–171, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Brandt K, Arnold M, Geary N, Langhans W, Leonhardt M. Vagal afferents mediate the feeding response to mercaptoacetate but not to the β3 adrenergic receptor agonist CL 316,243. Neurosci Lett 411: 104–107, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, McCoy DC, Kenakin TP, Andrews JL, Ammala C, Fornwald JA, Ignar DM, Jenkinson S. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol 148: 619–628, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR, Jr, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI. The orphan G protein-coupled receptor GPR40 is activated by medium and long-chain fatty acids. J Biol Chem 278: 11303–11311, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, Murdock PR, Holder JC, Marshall FH, Szekeres PG, Wilson S, Ignar DM, Foord SM, Wise A, Dowell SJ. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 278: 11312–11319, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Calingasan NY, Ritter S. Lateral parabrachial subnucleus lesions abolish feeding induced by mercaptoacetate but not by 2-deoxy-d-glucose. Am J Physiol Regul Integr Comp Physiol 265: R1168–R1178, 1993 [DOI] [PubMed] [Google Scholar]
- 10.Cartoni C, Yasumatsu K, Ohkuri T, Shigemura N, Yoshida R, Godinot N, le Coutre J, Ninomiya Y, Damak S. Taste preference for fatty acids is mediated by GPR40 and GPR120. J Neurosci 30: 8376–8382, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57: 2280–2287, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman MI, Tordoff MG, Ramirez I. Integrated metabolic control of food intake. Brain Res Bull 17: 855–859, 1986 [DOI] [PubMed] [Google Scholar]
- 13.Fujiwara K, Maekawa F, Yada T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: mediation by PLC and L-type Ca2+ channel and link to insulin release. Am J Physiol Endocrinol Metab 289: E670–E677, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Ge H, Li X, Weiszmann J, Wang P, Baribault H, Chen JL, Tian H, Li Y. Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology 149: 4519–4526, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Grabauskas G, Song I, Zhou S, Owyang C. Electrophysiological identification of glucose-sensing neurons in rat nodose ganglia. J Physiol 588: 617–632, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gromada J. The free fatty acid receptor GPR40 generates excitement in pancreatic β-cells. Endocrinology 147: 672–673, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Gunthorpe MJ, Benham CD, Randall A, Davis JB. The diversity in the vanilloid (TRPV) receptor family of ion channels. Trends Pharmacol Sci 23: 183–191, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Hara T, Hirasawa A, Ichimura A, Kimura I, Tsujimoto G. Free fatty acid receptors FFAR1 and GPR120 as novel therapeutic targets for metabolic disorders. J Pharm Sci 100: 3594–3601, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Hirasawa A, Hara T, Katsuma S, Adachi T, Tsujimoto G. Free fatty acid receptors and drug discovery. Biol Pharm Bull 31: 1847–1851, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med 11: 90–94, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Holliday ND, Watson SJ, Brown AJ. Drug discovery opportunities and challenges at g protein coupled receptors for long chain free Fatty acids. Front Endocrinol (Lausanne) 2: 112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holzer HH, Turkelson CM, Solomon TE, Raybould HE. Intestinal lipid inhibits gastric emptying via CCK and a vagal capsaicin-sensitive afferent pathway in rats. Am J Physiol Gastrointest Liver Physiol 267: G625–G629, 1994 [DOI] [PubMed] [Google Scholar]
- 23.Horn CC, Ji H, Friedman MI. Etomoxir, a fatty acid oxidation inhibitor, increases food intake and reduces hepatic energy status in rats. Physiol Behav 81: 157–162, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Hudson BD, Emanuel AJ, Wiater MF, Ritter S. The lipoprivic control of feeding is governed by fat metabolism, not by leptin or adipose depletion. Endocrinology 151: 2087–2096, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichimura A, Hirasawa A, Hara T, Tsujimoto G. Free fatty acid receptors act as nutrient sensors to regulate energy homeostasis. Prostaglandins Other Lipid Mediat 89: 82–88, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Ireland SJ, Tyers MB. Pharmacological characterization of 5-hydroxytryptamine-induced depolarization of the rat isolated vagus nerve. Br J Pharmacol 90: 229–238, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itoh Y, Hinuma S. GPR40, a free fatty acid receptor on pancreatic beta cells, regulates insulin secretion. Hepatol Res 33: 171–173, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422: 173–176, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Jambor de Sousa UL, Benthem L, Arsenijevic D, Scheurink AJ, Langhans W, Geary N, Leonhardt M. Hepatic-portal oleic acid inhibits feeding more potently than hepatic-portal caprylic acid in rats. Physiol Behav 89: 329–334, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Janssen S, Laermans J, Iwakura H, Tack J, Depoortere I. Sensing of fatty acids for octanoylation of ghrelin involves a gustatory G-protein. PLoS One 7: e40168, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang C, Haddad GG. Modulation of K+ channels by intracellular ATP in human neocortical neurons. J Neurophysiol 77: 93–102, 1997 [DOI] [PubMed] [Google Scholar]
- 32.Jordan SD, Konner AC, Bruning JC. Sensing the fuels: glucose and lipid signaling in the CNS controlling energy homeostasis. Cell Mol Life Sci 67: 3255–3273, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kahler A, Zimmermann M, Langhans W. Suppression of hepatic fatty acid oxidation and food intake in men. Nutrition 15: 819–828, 1999 [DOI] [PubMed] [Google Scholar]
- 34.Kilpatrick GJ, Jones BJ, Tyers MB. Binding of the 5-HT3 ligand, [3H]GR65630, to rat area postrema, vagus nerve and the brains of several species. Eur J Pharmacol 159: 157–164, 1989 [DOI] [PubMed] [Google Scholar]
- 35.Lal S, Kirkup AJ, Brunsden AM, Thompson DG, Grundy D. Vagal afferent responses to fatty acids of different chain length in the rat. Am J Physiol Gastrointest Liver Physiol 281: G907–G915, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat Neurosci 8: 579–584, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Langhans W. The enterocyte as an energy flow sensor in the control of eating. Forum Nutr 63: 75–84, 2010 [DOI] [PubMed] [Google Scholar]
- 38.Langhans W. Fatty acid oxidation in the energostatic control of eating—a new idea. Appetite 51: 446–451, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Langhans W, Egli G, Scharrer E. Regulation of food intake by hepatic oxidative metabolism. Brain Res Bull 15: 425–428, 1985 [DOI] [PubMed] [Google Scholar]
- 40.Langhans W, Leitner C, Arnold M. Dietary fat sensing via fatty acid oxidation in enterocytes: possible role in the control of eating. Am J Physiol Regul Integr Comp Physiol 300: R554–R565, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Langhans W, Scharrer E. Evidence for a vagally mediated satiety signal derived from hepatic fatty acid oxidation. J Auton Nerv Syst 18: 13–18, 1987 [DOI] [PubMed] [Google Scholar]
- 42.Lee T, Schwandner R, Swaminath G, Weiszmann J, Cardozo M, Greenberg J, Jaeckel P, Ge H, Wang Y, Jiao X, Liu J, Kayser F, Tian H, Li Y. Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol Pharmacol 74: 1599–1609, 2008 [DOI] [PubMed] [Google Scholar]
- 43.Leonhardt M, Langhans W. Fatty acid oxidation and control of food intake. Physiol Behav 83: 645–651, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Liou AP, Lu X, Sei Y, Zhao X, Pechhold S, Carrero RJ, Raybould HE, Wank S. The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology 140: 903–912, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu X, Zhao X, Feng J, Liou AP, Anthony S, Pechhold S, Sun Y, Lu H, Wank S. Postprandial inhibition of gastric ghrelin secretion by long-chain fatty acid through GPR120 in isolated gastric ghrelin cells and mice. Am J Physiol Gastrointest Liver Physiol 303: G367–G376, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lutz TA, Diener M, Scharrer E. Intraportal mercaptoacetate infusion increases afferent activity in the common hepatic vagus branch of the rat. Am J Physiol Regul Integr Comp Physiol 273: R442–R445, 1997 [DOI] [PubMed] [Google Scholar]
- 47.Ma D, Lu L, Boneva NB, Warashina S, Kaplamadzhiev DB, Mori Y, Nakaya MA, Kikuchi M, Tonchev AB, Okano H, Yamashima T. Expression of free fatty acid receptor GPR40 in the neurogenic niche of adult monkey hippocampus. Hippocampus 18: 326–333, 2008 [DOI] [PubMed] [Google Scholar]
- 48.Ma D, Tao B, Warashina S, Kotani S, Lu L, Kaplamadzhiev DB, Mori Y, Tonchev AB, Yamashima T. Expression of free fatty acid receptor GPR40 in the central nervous system of adult monkeys. Neurosci Res 58: 394–401, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Maggio CA, Koopmans HS. Food intake after intragastric meals of short-, medium-, or long-chain triglyceride. Physiol Behav 28: 921–926, 1982 [DOI] [PubMed] [Google Scholar]
- 50.Mansouri A, Koss MD, Brandt K, Geary N, Langhans W, Leonhardt M. Dissociation of mercaptoacetate's effects on feeding and fat metabolism by dietary medium- and long-chain triacylglycerols in rats. Nutrition 24: 360–365, 2008 [DOI] [PubMed] [Google Scholar]
- 51.Montell C, Birnbaumer L, Flockerzi V. The TRP channels, a remarkably functional family. Cell 108: 595–598, 2002 [DOI] [PubMed] [Google Scholar]
- 52.Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem Biophys Res Commun 303: 1047–1052, 2003 [DOI] [PubMed] [Google Scholar]
- 53.Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687–698, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Randich A, Spraggins DS, Cox JE, Meller ST, Kelm GR. Jejunal or portal vein infusions of lipids increase hepatic vagal afferent activity. Neuroreport 12: 3101–3105, 2001 [DOI] [PubMed] [Google Scholar]
- 55.Randich A, Tyler WJ, Cox JE, Meller ST, Kelm GR, Bharaj SS. Responses of celiac and cervical vagal afferents to infusions of lipids in the jejunum or ileum of the rat. Am J Physiol Regul Integr Comp Physiol 278: R34–R43, 2000 [DOI] [PubMed] [Google Scholar]
- 56.Rayasam GV, Tulasi VK, Davis JA, Bansal VS. Fatty acid receptors as new therapeutic targets for diabetes. Expert Opin Ther Targets 11: 661–671, 2007 [DOI] [PubMed] [Google Scholar]
- 57.Remizov O, Jakubov R, Dufer M, Krippeit Drews P, Drews G, Waring M, Brabant G, Wienbergen A, Rustenbeck I, Schofl C. Palmitate-induced Ca2+-signaling in pancreatic β-cells. Mol Cell Endocrinol 212: 1–9, 2003 [DOI] [PubMed] [Google Scholar]
- 58.Ritter RC, Ladenheim EE. Capsaicin pretreatment attenuates suppression of food intake by cholecystokinin. Am J Physiol Regul Integr Comp Physiol 248: R501–R504, 1985 [DOI] [PubMed] [Google Scholar]
- 59.Ritter S, Calingasan NY. Neural substrates for metabolic controls of feeding. In: Appetite and Body Weight Regulation: Sugar, Fat, and Macronutrient Substitutes, edited by Fernstrom J, Miller G. D. Boca Raton, FL: CRC Press, 1994, p. 77–94 [Google Scholar]
- 60.Ritter S, Hutton B. Mercaptoacetate-induced feeding is impaired by central nucleus of the amygdala lesions. Physiol Behav 58: 1215–1220, 1995 [DOI] [PubMed] [Google Scholar]
- 61.Ritter S, Li AJ, Wang Q, Simasko SM. Novel non-metabolic mechanism of action underlying 2-mercaptocacetate (MA)-induced feeding. In: 2013 Neuroscience Meeting Planner, San Diego, CA: Society for Neuroscience, 2013 [Google Scholar]
- 62.Ritter S, Taylor JS. Capsaicin abolishes lipoprivic but not glucoprivic feeding in rats. Am J Physiol Regul Integr Comp Physiol 256: R1232–R1239, 1989 [DOI] [PubMed] [Google Scholar]
- 63.Ritter S, Taylor JS. Vagal sensory neurons are required for lipoprivic but not glucoprivic feeding in rats. Am J Physiol Regul Integr Comp Physiol 258: R1395–R1401, 1990 [DOI] [PubMed] [Google Scholar]
- 64.Scharrer E, Langhans W. Control of food intake by fatty acid oxidation. Am J Physiol Regul Integr Comp Physiol 250: R1003–R1006, 1986 [DOI] [PubMed] [Google Scholar]
- 65.Schioth HB. G protein-coupled receptors in regulation of body weight. CNS Neurol Disord Drug Targets 5: 241–249, 2006 [DOI] [PubMed] [Google Scholar]
- 66.Simasko SM, Peters J, Weins J, Ritter RC. Co-activation of nodose ganglion neurons by cholecystokinin and serotonin. In: 2002 Neuroscience Meeting Planner, Orlando, FL: Society for Neuroscience, 2002 [Google Scholar]
- 67.Singer LK, Ritter S. Differential effects of infused nutrients on 2-deoxy-d-glucose- and 2-mercaptoacetate-induced feeding. Physiol Behav 56: 193–196, 1994 [DOI] [PubMed] [Google Scholar]
- 68.Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol 121: 91–119, 2014 [DOI] [PubMed] [Google Scholar]
- 69.Tutwiler GF, Brentzel HJ, Kiorpes TC. Inhibition of mitochondrial carnitine palmitoyl transferase A in vivo with methyl 2-tetradecylglycidate (methyl palmoxirate) and its relationship to ketonemia and glycemia. Proc Soc Exp Biol Med 178: 288–296, 1985 [DOI] [PubMed] [Google Scholar]
- 70.van Dijk G, Scheurink A, Ritter S, Steffens A. Glucose homeostasis and sympathoadrenal activity in mercaptoacetate-treated rats. Physiol Behav 57: 759–764, 1995 [DOI] [PubMed] [Google Scholar]
- 71.Wank SA. G protein-coupled receptors in gastrointestinal physiology. I. CCK receptors: an exemplary family. Am J Physiol Gastrointest Liver Physiol 274: G607–G613, 1998 [DOI] [PubMed] [Google Scholar]
- 72.Wolf HP. Possible new therapeutic approach in diabetes mellitus by inhibition of carnitine palmitoyltransferase 1 (CPT1). Horm Metab Res Suppl 26: 62–67, 1992 [PubMed] [Google Scholar]
- 73.Xiong Y, Swaminath G, Cao Q, Yang L, Guo Q, Salomonis H, Lu J, Houze JB, Dransfield PJ, Wang Y, Liu JJ, Wong S, Schwandner R, Steger F, Baribault H, Liu L, Coberly S, Miao L, Zhang J, Lin DC, Schwarz M. Activation of FFA1 mediates GLP-1 secretion in mice. Evidence for allosterism at FFA1. Mol Cell Endocrinol 369: 119–129, 2013 [DOI] [PubMed] [Google Scholar]
- 74.Yonezawa T, Kurata R, Yoshida K, Murayama MA, Cui X, Hasegawa A. Free fatty acids-sensing G protein-coupled receptors in drug targeting and therapeutics. Curr Med Chem 20: 3855–3871, 2013 [DOI] [PubMed] [Google Scholar]
- 75.Yox DP, Ritter RC. Capsaicin attenuates suppression of sham feeding induced by intestinal nutrients. Am J Physiol Regul Integr Comp Physiol 255: R569–R574, 1988 [DOI] [PubMed] [Google Scholar]
- 76.Yox DP, Stokesberry H, Ritter RC. Vagotomy attenuates suppression of sham feeding induced by intestinal nutrients. Am J Physiol Regul Integr Comp Physiol 260: R503–R508, 1991 [DOI] [PubMed] [Google Scholar]
- 77.Zhao H, Simasko SM. Role of transient receptor potential channels in cholecystokinin-induced activation of cultured vagal afferent neurons. Endocrinology 151: 5237–5246, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zunkler BJ, Lenzen S, Manner K, Panten U, Trube G. Concentration-dependent effects of tolbutamide, meglitinide, glipizide, glibenclamide and diazoxide on ATP-regulated K+ currents in pancreatic B-cells. Naunyn Schmiedebergs Arch Pharmacol 337: 225–230, 1988 [DOI] [PubMed] [Google Scholar]