

Abstract
Alcohol abuse with/without cirrhosis is associated with an impaired gut barrier and inflammation. Gut microbiota can transform primary bile acids (BA) to secondary BAs, which can adversely impact the gut barrier. The purpose of this study was to define the effect of active alcohol intake on fecal BA levels and ileal and colonic inflammation in cirrhosis. Five age-matched groups {two noncirrhotic (control and drinkers) and three cirrhotic [nondrinkers/nonalcoholics (NAlc), abstinent alcoholic for >3 mo (AbsAlc), currently drinking (CurrAlc)]} were included. Fecal and serum BA analysis, serum endotoxin, and stool microbiota using pyrosequencing were performed. A subgroup of controls, NAlc, and CurrAlc underwent ileal and sigmoid colonic biopsies on which mRNA expression of TNF-α, IL-1β, IL-6, and cyclooxygenase-2 (Cox-2) were performed. One hundred three patients (19 healthy, 6 noncirrhotic drinkers, 10 CurrAlc, 38 AbsAlc, and 30 NAlc, age 56 yr, median MELD: 10.5) were included. Five each of healthy, CurrAlc, and NAlc underwent ileal/colonic biopsies. Endotoxin, serum-conjugated DCA and stool total BAs, and secondary-to-primary BA ratios were highest in current drinkers. On biopsies, a significantly higher mRNA expression of TNF-α, IL-1β, IL-6, and Cox-2 in colon but not ileum was seen in CurrAlc compared with NAlc and controls. Active alcohol use in cirrhosis is associated with a significant increase in the secondary BA formation compared with abstinent alcoholic cirrhotics and nonalcoholic cirrhotics. This increase in secondary BAs is associated with a significant increase in expression of inflammatory cytokines in colonic mucosa but not ileal mucosa, which may contribute to alcohol-induced gut barrier injury.
Keywords: alcohol, cytokines, farnesoid X receptor, microbiome
changes in the gut milieu can predispose patients with cirrhosis to increased bacterial translocation and serious systemic infections such as spontaneous bacterial peritonitis (36). Alcohol, with or without accompanying cirrhosis, can negatively impact the gut barrier and potentially propagate the cycle of bacterial translocation and systemic inflammation (22). Bile acids (BAs) are key modulators of the gut microbiota and intestinal permeability and can activate nuclear receptors in the intestinal wall such as farnesoid X receptor (FXR), which can regulate inflammation and hepatic BA synthesis, and are affected by cirrhosis progression (12–14, 26). There is evidence of increased intestinal inflammation and diminished intestinal barrier function in animal models of alcoholic injury and in patients (22). However, the interaction of BAs and alcohol as comodulators of intestinal inflammation in human cirrhosis has not been previously studied (31, 33). The aim was to study the interactions between alcohol, microbiota, and BAs, which could increase our insight into mechanisms behind intestinal inflammation in cirrhosis and encourage focused therapies directed at ameliorating these negative effects.
Our approach was to determine 1) the effect of current and prior alcohol use in cirrhotic and noncirrhotic subjects on serum and fecal BA profiles and 2) effects of current alcohol use in the setting of cirrhosis on expression of specific nuclear receptors [FXR, small heterodimer partner (SHP)] and mediators of inflammation that occur downstream [IL-1β, IL-6, TNF-α, monocyte chemoattractant protein-1 (MCP), and cyclooxygenase-2 (COX2)] in colonic and ileal mucosa.
MATERIALS AND METHODS
We prospectively enrolled five groups of subjects: three cirrhotic groups that were 1) cirrhotics without alcohol as an etiology, 2) those with alcohol as an etiology but who were abstinent for >3 mo, 3) those who had an alcoholic etiology and continued to drink and two noncirrhotic groups, age-matched, 4) healthy controls, and 5) active drinkers without liver disease. The diagnosis of cirrhosis was made using a combination of clinical [low platelets, high aspartate aminotransferase (AST)-to-alanine aminotransferase (ALT) ratio, or frank decompensation in the setting of chronic liver disease], radiological (evidence of cirrhosis and portal hypertension on abdominal imaging), histological (biopsy evidence), or endoscopic (presence of varices) evidence. We excluded subjects who were not able to give informed consent, had alcoholic hepatitis currently or within 6 mo of the study, or had an unclear diagnosis of cirrhosis. Active drinkers without liver disease had normal AST, ALT, liver function, and imaging. Patients underwent written informed consent and a detailed questionnaire regarding their stool habits, medications, and past and current alcohol use.
Sample Collection
Stool.
All subjects underwent stool collection; in those who agreed to undergo a colonoscopy for colonic and ileal biopsies, their stool collection was performed at least 5 days from the colonoscopy. The stool was divided into two aliquots and frozen at −80°C. One sample was used for the BA determinations and the other for microbiome analysis.
Blood.
All cirrhotic patients underwent blood collection for analysis of MELD score (validated logarithmic score of serum bilirubin, creatinine, and international normalized ratio of the prothrombin time) and serum albumin. A portion of the serum was analyzed for BAs (Junshin Clinic Bile Acid Institute, Tokyo) and endotoxin (Assaygate, Ijamsville, MD) (5, 14).
Ileal and colonic biopsies.
A subset of these eligible subjects (controls, cirrhotics without alcoholic etiology, and those who were currently drinking) underwent colonoscopy using a standard colonic preparation that was performed by experienced endoscopists. Consent was obtained to get pinch biopsy samples of the terminal ileum and sigmoid colon while performing the colonoscopy. During the colonoscopy, the pinch biopsy samples were snap-frozen on retrieval and stored at −80°C until the analysis was performed.
Specimen Analysis
BA analysis. The fecal BA analysis was performed using previously published HPLC and LC-MS techniques (14) (see appendix). Specific BAs quantitated included: primary [cholic (CA) and chenodeoxycholic (CDCA)] and secondary [lithocholic (LCA) and deoxycholic (DCA)] BA acids. The median primary, secondary BA concentrations, and their ratios (CA + CDCA/LCA + DCA) were compared between groups. Because this was a one-time collection of stool, the ratios were considered more important compared with the individual BA concentrations. LC-MS using published techniques was also used to perform serum BA measurements, which included conjugated (tauro or glycol N-acyl amidated), double conjugated (N-acyl amidated and C-3 sulfated), and unconjugated (14).
Stool Microbiome Analysis
We performed multitagged pyrosequencing according to previously published techniques (5) (in appendix).
Tissue Analysis
Tissue analysis was performed on ileal and colonic biopsy samples to determine mRNA levels of 1) bile acid modulators: FXR, SHP, and fibroblast growth factor (FGF)-19 and 2) potential mediators of intestinal inflammation: IL-1β, IL-6, TNF-α, MCP-1, and COX-2. Total RNA was isolated from samples using QIAzol Lysis Reagent (from QIAGEN). Total RNA (1 μg) was used for the first-strand cDNA synthesis using the High-Capacity cDNA Archive kit (from Applied Biosystems). The mRNA levels of TNF-α, IL-6, IL-1β, FXRα, and COX-2 were quantified using specific primers for each gene. iQ SYBR Green Supermix was used as a fluorescent dye to detect the presence of double-stranded DNA, and the mRNA levels for each gene were normalized using β-actin as an internal control. The ratio of normalized mean value for each treatment group to the vehicle control group was calculated.
Statistical Analysis
Demographics, cirrhosis severity, medication use, serum inflammatory cytokines, and BAs, including ratios, were compared between groups using ANOVA, Chi square, and Kruskall-Wallis tests where appropriate. Kruskal-Wallis tests and Metastats were used to compare fecal microbiota abundance between groups, and Pearson's correlation analyses were used to correlate microbiota, MELD score, and BA concentrations (35). Tissue analytes were compared between the selected controls, drinkers, and nonalcoholic cirrhotic patients using Kruskal-Wallis tests.
This protocol was approved by the Institutional Review Board at the Hunter Holmes McGuire Veterans Affairs Medical Center in Richmond.
RESULTS
The study had the following two parts: 1) study of the entire group with fecal and serum BA analysis and 2) study of the subgroup undergoing mucosal biopsies.
Analysis of the Entire Group
We enrolled a total of 103 subjects: 19 healthy, 6 drinkers without liver disease, and 78 cirrhotic patients of which 10 were currently drinking, 30 had never drank, and 38 had an alcoholic etiology but were abstinent for >6 mo. The pattern of alcohol abuse was similar among the three alcoholic groups with the difference of alcohol intake only occurring within the past 6 mo (median 21 yr of alcohol intake). The last intake of alcohol was a median of 1 day before enrollment in both groups of current drinkers. Groups were statistically similar on median age [controls 49, drinkers without liver disease, 51, nondrinkers/nonalcoholics (NAlc) 53, abstinent alcoholic for >3 mo (AbsAlc) 52, currently drinking (CurrAlc) 50 yr, P = 0. 61], percentage of men (controls 74%, drinkers without liver disease 67%, NAlc 93%, AbsAlc 92%, CurrAlc 100%, P = 0.84), or MELD score (NAlc 8.5, AbsAlc 10.5, CurrAlc 8.5, P = 0.24).
There was a significantly higher mean endotoxin level in CurrAlc compared with all groups (CurrAlc 0.75 ± 0.91 vs. NAlc 0.49 ± 0.68, AbsAlc 0.30 ± 0.56, noncirrhotic drinker 0.33 ± 0.21, and controls 0.0 ± 0.1 EU/ml, P = 0.021). Within the noncirrhotic group, drinkers' endotoxin levels were significantly higher compared with controls (P = 0.007).
Fecal BA Analysis
We combined the fecal BA profile of the two currently drinking groups (CurrAlc cirrhotics and those without liver disease) and compared them against the other groups using medians because of nonnormal BA distributions. The combined current drinkers (cirrhotics and noncirrhotics) had the highest median total fecal BAs (median 10.5, P < 0.0001), total secondary BAs (6.5, P < 0.0001), DCA (4.7, P < 0.0001), and LCA (2.5, P = 0.005) compared with all other groups, including controls. This group also had the highest secondary-to-primary BA ratio (median 26.4, P = 0.002) compared with the remaining cirrhotics; this ratio was similar to the control values. Primary BAs were not significantly different between cirrhotic groups (Table 1) but were the lowest in the controls. When groups were individually compared, the highest levels of total, secondary, and secondary/primary BA were in active drinkers without liver disease.
Table 1.
Median fecal bile acid concentrations between groups
| Cirrhosis |
Noncirrhotic |
||||
|---|---|---|---|---|---|
| Not currently drinking |
|||||
| Median Fecal Concentrations, μmol/g | Nonalcoholic (n = 30) | Abstinent (n = 38) | Currently drinking (n = 10) | Currently drinking (n = 6) | Controls (n = 19) |
| Total BA*** | 2.9 | 2.2 | 8.9 | 18.0†‡ | 5.4 |
| CA | 0.1 | 0.12 | <0.05 | <0.05 | <0.05 |
| CDCA* | 0.1 | 0.21 | 0.16 | 0.25 | 0.01 |
| LCA* | 1.0 | 0.4 | 1.9 | 3.6†‡ | 1.6 |
| DCA*** | 0.4 | 0.4 | 3.3 | 7.2†‡ | 2.3 |
| Total primary BAs* | 0.23 | 0.46 | 0.16 | 0.25 | 0.01 |
| Total secondary BAs*** | 1.5 | 1.0 | 5.7 | 10.7†‡ | 4.1 |
| Secondary/primary BA ratio* | 7.4 | 1.1 | 23.7 | 25.6† | 19.5 |
n, No. of subjects. Ratios were calculated between chenodexoycholic (CDCA) + cholic (CA) and lithocholic (LCA) + deoxycholic (DCA). Medians were used because of the nonnormal distribution of the bile acid (BA) profiles, which were compared using Kruskal-Wallis test.
P < 0.001 and
P < 0.05 between all five groups.
P < 0.05 in currently drinking groups (cirrhotic and noncirrhotic subjects) compared with the rest of the groups.
P < 0.05 in currently drinking noncirrhotic patients compared with the other groups.
Serum BA Analysis
There was a significantly higher concentration of conjugated DCA and a lower concentration of conjugated CDCA in current drinkers (both cirrhotic and noncirrhotic) compared with other groups (Table 2).
Table 2.
Serum BA concentrations between groups
| Cirrhosis |
Noncirrhotic |
|||||
|---|---|---|---|---|---|---|
| Nonalcoholic | Abstinent alcoholic | Currently drinking | Current drinkers | Healthy controls | P Value | |
| Conjugated | ||||||
| GDCA | 0.6 ± 1.2 | 1.3 ± 2.1 | 1.2 ± 2.2 | 2.4 ± 3.1 | 0.5 ± 0.5 | 0.05 |
| TDCA | 0.2 ± 0.5 | 0.4 ± 0.6 | 1.0 ± 2.6 | 0.8 ± 1.8 | 0.1 ± 0.1 | 0.04 |
| GCA | 6.1 ± 7.5 | 5.2 ± 5.3 | 4.3 ± 4.3 | 2.4 ± 3.3 | 0.3 ± 0.5 | 0.01 |
| TCA | 5.0 ± 6.4 | 5.2 ± 6.5 | 3.3 ± 5.8 | 0.6 ± 1.2 | 0.1 ± 0.1 | 0.01 |
| GCDCA | 17.1 ± 21.9 | 20.2 ± 25.2 | 12.4 ± 15.1 | 4.1 ± 4.8 | 0.9 ± 0.7 | 0.01 |
| TCDCA | 18.4 ± 23.6 | 23.4 ± 33.3 | 11.5 ± 18.1 | 2.0 ± 3.7 | 0.1 ± 0.2 | 0.01 |
| GUDCA | 11.8 ± 44.2 | 0.8 ± 1.0 | 0.4 ± 0.4 | 0.2 ± 0.3 | 0.0 ± 0.2 | 0.42 |
| TUDCA | 1.4 ± 4.7 | 0.2 ± 0.21 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.31 |
| Double conjugated | ||||||
| GDCA3S | 0.3 ± 0.6 | 0.1 ± 0.2 | 0.3 ± 0.3 | 0.2 ± 0.3 | 0.1 ± 0.2 | 0.64 |
| TDCA3S | 0.02 ± 0.06 | 0.01 ± 0.04 | 0.1 ± 0.2 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.25 |
| TCDCA3S | 1.1 ± 1.8 | 0.8 ± 1.1 | 1.1 ± 1.8 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.05 |
| GCDCA3S | 1.4 ± 1.4 | 0.9 ± 1.1 | 1.8 ± 3.0 | 0.2 ± 0.2 | 0.0 ± 0.0 | 0.005 |
| TUDCA3S | 0.3 ± 1.1 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.42 |
| GUDCA3S | 1.6 ± 5.8 | 0.1 ± 0.1 | 0.6 ± 0.9 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.45 |
| Unconjugated | ||||||
| CA | 0.4 ± 0.8 | 1.2 ± 2.9 | 0.1 ± 0.1 | 0.9 ± 1.8 | 0.0 ± 0.1 | 0.14 |
| CDCA | 0.9 ± 1.4 | 1.9 ± 3.9 | 0.1 ± 0.1 | 0.2 ± 0.3 | 0.6 ± 1.5 | 0.20 |
| UDCA | 1.0 ± 4.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.2 | 0.0 ± 0.0 | 0.43 |
| DCA | 0.4 ± 0.8 | 0.7 ± 1.3 | 0.2 ± 0.2 | 0.9 ± 0.8 | 0.5 ± 0.4 | 0.33 |
Values are means ± SD. Units are μmol/l. There was a significantly higher glycoconjugated deoxycholic acid in the serum of current drinking alcoholic noncirrhotic subjects and highest tauroconjugated deoxycholic acid in the currently drinking cirrhotic subject' serum compared with the rest. As expected, conjugated and 3-sulfate conjugated CDCA was the highest in cirrhotic subjects' serum compared with noncirrhotic subjects and relatively lower in actively drinking cirrhotic patients compared with the other cirrhosis groups. No difference in unconjugated serum BAs or any form of UDCA was seen between groups, and lithocholic acid was not detected in serum. G, glycine conjugated; T, taurine conjugated; −3S, sulfated forms; UDCA, ursodeoxycholic acid.
Microbiota, BAs, and MELD Score
In the entire group, MELD score was negatively correlated with total fecal BAs (r = −0.4, P = 0.004), LCA (r = −0.4, P = 0.001), DCA (r = −0.32, P = 0.009), total secondary BAs (r = −0.4, P = 0.004), and the secondary-to-primary BA ratio (P = −0.4, P = 0.002) but not CA or CDCA. Incertae sedis XIV abundance was positively correlated with total BAs (r = 0.4, P = 0.001), LCA (r = 0.2, P = 0.05), DCA (r = 0.3, P = 0.03), CA (r = 0.3, P = 0.005), CDCA (r = 0.2,P = 0.04), and total primary (r = 0.3,P = 0.01) and secondary (r = 0.2, P = 0.03) BAs. Enterobacteriaceae abundance was positively correlated with primary BAs (r = 0.3, P = 0.03) and with CA (r = 0.2, P = 0.024) but not with other BAs. MELD score was negatively correlated with Incertae Sedis XIV (r = −0.4, P = 0.003), Ruminococcaceae (r = −0.5, P < 0.0001), and Lachnospiraceae (r = −0.3, P = 0.03) and positively with Porphyromonadaceae (r = 0.3, P = 0.01).
Subgroup Analysis of Those with Mucosal Biopsies
Fifteen subjects, 5 controls, 5 active drinkers, and 5 cirrhotics without alcoholic etiology, agreed to have biopsies taken. They were statistically similar in age (median 50 vs. 51 vs. 53 yr), all were men, and there was no significant difference in the median MELD score between the cirrhotic patients (drinkers 8.5 vs. nonalcoholic cirrhosis 8).
Stool Microbiome Comparison
Stool microbiome comparison between the five drinkers and the five nonalcoholic cirrhotics showed a significant increase in a taxon from phylum Firmicutes, Veillonellaceae (5 vs. 2%, P = 0.02) and reduction in two Bacteroidetes taxa, Bacteroidaceae (4 vs. 2%, P = 0.03) and Porphyromonadaceae reduction trend (2 vs. 4%, P = 0.09) in drinkers. No changes were seen in components of phyla Actinobacteria or other taxa of Firmicutes, Lachnospiraceae (2 vs. 2%), Ruminococcaceae (0.7 vs. 1%), Incertae Sedis XIV (5 vs. 5%), other taxa of Bacteroidetes Prevotellaceae (6 vs. 6%), Rikenellaceae (0.4 vs. 1%), or phylum Proteobacteria, Enterobacteriaceae (4 vs. 3%).
Ileal and Colonic Tissue mRNA Expression
We found a significantly higher expression of mRNA of FXRα and COX-2 in the ileal and sigmoid colonic mucosa in drinkers compared with nonalcoholic cirrhotics and controls (Figs. 1–8; primers in Table 3). There was also a significantly higher expression of TNF-α, IL-1β, IL-6, MCP-1, and FGF-19 in drinkers compared with the other groups in the colon but not in the ileal mucosa. SHP expression was not different between groups in the colon or ileum. Although there was a trend toward an increasing FGF-19, TNF-α, MCP-1, IL-6, and IL-1β expression in the ileum in drinkers more than in nonalcoholic cirrhotics more than controls, it did not reach statistical significance.
Fig. 1.
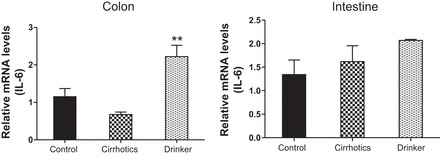
IL-6. Comparison of the fold change in the mRNA expression levels between the controls (control) compared with nonalcoholic cirrhotic subjects (cirrhotic) and actively drinking cirrhotic patients (drinker). Significant differences, **P < 0.01.
Fig. 8.
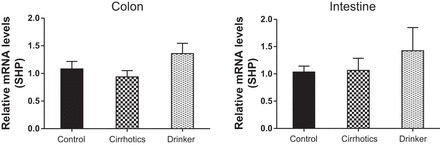
Small heterodimer partner (SHP). Comparison of the fold change in the mRNA expression levels between the controls (control) compared with nonalcoholic cirrhotic subjects (cirrhotic) and actively drinking cirrhotic patients (drinker).
Table 3.
Primers used for mRNA analysis
| Forward | Reverse | |
|---|---|---|
| IL-6 | CAGATTTGAGAGTAGTGAGGAAC | CGCAGAATGAGATGAGTTGTC |
| FGF-19 | CTCGGAGGAAGACTGTGC | GAAATGAGAGAGTGGAAGAAAGC |
| FXR-α | ATGAACTCAGGCGTATGC | CCACAAACAACACACAGC |
| IL-1β | TGGCTTATTACAGTGGCAATG | GTGGTGGTCGGAGATTCG |
| SHP | ACTGGGTGCTGTGAAG | GGTTCGAATGGACTTGAGG |
| TNF-α | CGAGTCTGGGCAGGTCTAC | GGGAGGCGTTTGGGAAGG |
| COX-2 | CCCTGAGCATCTACGGTTTG | TCGCATACTCTGTTGTGTTCC |
| MCP-1 | CCCAGTCACCTGCTGTTAT | TGCTGCTGGTGATTCTTTCT |
FGF-19, fibroblast growth factor-19; FXR-α, farnesoid X receptor-α; SHP, small heterodimer partner; COX-2, cyclooxygenase-2; MCP-1, monocyte chemoattractant protein-1.
Fig. 2.
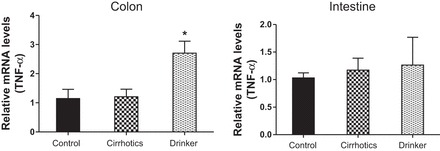
TNF-α. Comparison of the fold change in the mRNA expression levels between the controls (control) compared with nonalcoholic cirrhotic subjects (cirrhotic) and actively drinking cirrhotic patients (drinker). Significant differences, *P < 0.05.
Fig. 3.
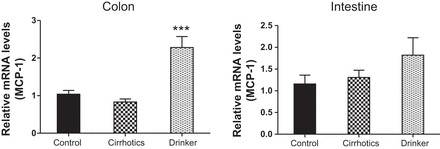
Monocyte chemoattractant protein-1 (MCP-1). Comparison of the fold change in the mRNA expression levels between the controls (control) compared with nonalcoholic cirrhotic subjects (cirrhotic) and actively drinking cirrhotic patients (drinker). Significant differences, ***P < 0.001.
Fig. 4.
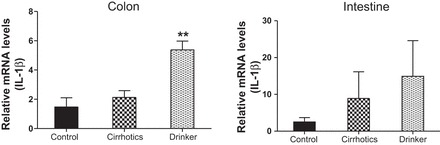
IL-1β. Comparison of the fold change in the mRNA expression levels between the controls (control) compared with nonalcoholic cirrhotic subjects (cirrhotic) and actively drinking cirrhotic patients (drinker). Significant differences, **P < 0.01.
Fig. 5.
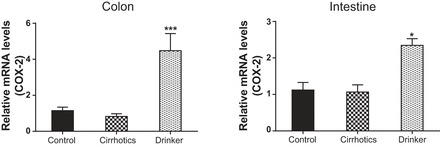
Cyclooxygenase-2 (COX-2). Comparison of the fold change in the mRNA expression levels between the controls (control) compared with nonalcoholic cirrhotic subjects (cirrhotic) and actively drinking cirrhotic patients (drinker). Significant differences, *P < 0.05 and ***P < 0.001.
Fig. 6.
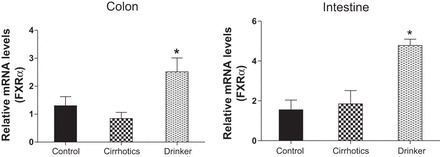
Farnesoid X receptor (FXR-α). Comparison of the fold change in the mRNA expression levels between the controls (control) compared with nonalcoholic cirrhotic subjects (cirrhotic) and actively drinking cirrhotic patients (drinker). Significant differences, *P < 0.05.
Fig. 7.
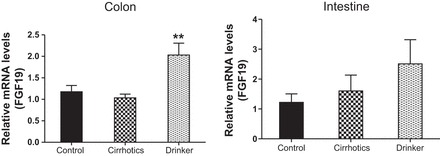
Fibroblast growth factor 19 (FGF-19). Comparison of the fold change in the mRNA expression levels between the controls (control) compared with nonalcoholic cirrhotic subjects (cirrhotic) and actively drinking cirrhotic patients (drinker). Significant differences, **P < 0.01.
DISCUSSION
Patients with cirrhosis are prone to a proinflammatory milieu that has consequences in the form of altered inflammatory responses to infections and complications of cirrhosis such as hepatic encephalopathy (29). This systemic inflammation often portends a worse prognosis, and the role of alcohol intake, ongoing or past, is important to delineate since alcohol is associated with a higher rate of infections and worse outcome in cirrhotic patients (27). However, the role of alcohol intake on fecal BA levels and their effect on intestinal and colonic inflammation is not completely understood (14). Our data show a highly significant increase in total and secondary fecal BA concentrations in current drinkers, regardless of cirrhosis status, compared with other cirrhotic patients and healthy controls that are not consuming alcohol. Whereas the abstinent alcoholics had relatively advanced cirrhosis, although statistically similar, which could partly explain the lower total BA, the nonalcoholics and active alcoholic cirrhotics had similar MELD scores; therefore, it is unlikely that the BA profile results would be a function of cirrhosis severity alone. This was accompanied by dysbiosis that was linked with liver disease severity.
There are a number of reports in the literature that alcohol increases the rate of synthesis of bile acids in humans and animal models (2, 9, 19, 37). This was corroborated by the highest fecal BA levels of total, secondary, and the secondary-to-primary BA ratio in current drinkers without cirrhosis compared with the rest. There was also a significant increase in the mRNA levels of genes encoding proinflammatory cytokines (TNF-α, IL-6, IL-1β, MCP-1) in colonic, but not ileal, tissue in cirrhotic patients consuming alcohol. Cox2 was highly elevated in cirrhotic patients consuming alcohol in both ileum and colon. However, Cox2 mRNA levels were higher in the colon. Cirrhotic patients and healthy controls not consuming alcohol did not have a significant increase in the mRNA levels of proinflammatory cytokines in either ileal or colonic tissue samples. The dichotomy between ileal and colonic tissue changes is interesting and may be because of the higher abundance of gut microbiota in the colon, which could modulate the BA 7α-dehydroxylation. This could also potentiate the impaired intestinal barrier function that has been shown to occur largely in the colon and not the small bowel in humans with compensated cirrhosis (similar to our population) (21). A potential functional test of this barrier function is serum endotoxemia, which was increased in currently drinking cirrhotics compared with the others and in noncirrhotics, was increased in drinkers compared with nondrinkers (39). This replicates prior studies and extends this functional gut barrier dysfunction to actively drinking cirrhotic patients (3).
Older studies in noncirrhotic humans, human hepatocytes, and animal models all report that alcohol increases rates of bile acid synthesis (2, 19). Animal model studies (mice and rats) suggest that both chronic and acute alcohol consumption increase bile acid synthesis and upregulate cholesterol 7α-hydroxylase (Cyp7A1). In noncirrhotic rats consuming ethanol, there was a decrease in intestinal FGF-15 possibly accounting for the upregulation of Cyp7A1 and bile acid synthesis (37). Recent studies in mice suggest that alcohol increases the expression of Cyp7A1 and Cyp27A1 via activation of hepatic cannabinoid receptor type 1 and CREBH in the liver (9). Our study found an increase in FXR and FGF-19, but not SHP, in ileum and sigmoid colon in drinkers but not in nonalcoholic cirrhotics and controls. This suggests that, while the feedback mechanism of a high FGF-19 mRNA expression due to a high intestinal BA load is intact, it is probably ineffective in reducing the accelerated BA synthesis induced by alcohol in the liver in cirrhosis. This is different from the findings in noncirrhotic rodents that were fed alcohol in which there was a lower FGF-15 that could in turn allow stimulation of BA synthesis in the liver. This is also supported by the increased level of conjugated serum DCA in current drinkers, which indicates that a higher proportion of DCA was absorbed through the enterohepatic cycle and reconjugated in the liver, but despite that these subjects continued to have higher fecal BA levels. We speculate that, in the setting of cirrhosis with ongoing alcohol consumption, there could be a differential regulation of the enterohepatic cycle, i.e., that CYP7A1 is not downregulated by FGF-19. If the cannabinoid receptor type 1 is activated by alcohol in the human liver, it might override the suppressive effects of FGF-19 on Cyp7A1 allowing for increased bile acid synthesis.
The increase in proinflammatory cytokine mRNA expression in the setting of FXR activation is interesting since prior animal studies and ileal biopsy analysis in inflammatory bowel disease showed that FXR activation can reduce inflammation by suppressing NF-kB (10, 32, 34). Our results, however, indicate a significantly higher inflammatory gene expression in the colon, but not the ileum, in actively drinking cirrhotics despite FXR activation in both locations of the gastrointestinal tract. The interactions of FXR with inflammation vary with disease and tissue, and FXR is associated with higher inflammation in Barrett's esophagus (6). Therefore our results indicate an inflammatory mechanism in the colon in alcoholic cirrhotics that is not suppressed by FXR activation. A strong candidate species for this could be the secondary BAs that were found in actively drinking cirrhotics and noncirrhotic subjects in concentrations even higher than healthy controls. Secondary BAs, especially deoxycholic acid, are found in high concentrations in fecal water and can cross epithelial cell membranes by simple diffusion. DCA also has been reported to activate various cell signaling pathways (EGFR, AKT, ERK 1/2, PKC, β-catenin, Cox-2) in primary cells, intestinal cell lines, and animal models at lower concentrations than CDCA (7, 8, 17, 20, 23, 24). Moreover, secondary bile acids activate cellular pathways producing ROS (NADPH oxidase, PLA2, mitochondria) that may activate stress pathways leading to the activation of NF-κB and proinflammatory cytokine synthesis (30).
The current results strongly suggest that an increased level of fecal secondary bile acids is associated with increased inflammation and COX-2 induction in colonic tissue. This may be because of the direct effects of secondary bile acids on colonic epithelial cells and/or changes in the colonic microbiota. Increased levels of bile acids are known to cause changes in the composition of the intestinal microbiota (25). Chronic inflammation is highly associated with an increased risk of colon cancers, including patients with ulcerative colitis and Crohn's disease (11). In cirrhotic patients, this intestinal inflammation could lead to an impaired gut-liver axis through barrier dysfunction, endotoxemia, and bacterial translocation (18). This has been shown to result in hepatic inflammation, which can reduce BA secretion from the liver and potentiate negative survival outcomes (1, 15, 16, 22, 38). Indeed we found the highest serum endotoxin level in CurrAlc patients. There could be several mechanisms for this increase in the secondary BA ratio that presents itself to the colon, including increased conversion of primary BAs to secondary BAs by alteration in microbiome, a decrease in colonic transit time caused by current alcohol intake, or an increased “spillover” of secondary BAs into the colon because of increased total BA presentation in the small bowel as a result of the heighted BA synthesis by alcohol intake. When germ-free mice were compared with conventionally raised ones, they had a significantly lower fecal BA concentration that corresponded to the colonic tissue BA profile, a reduced whole body BA pool, and no secondary BA due to absence of microbiota (28). Another study showed that supplementing mice diet with 1% cholate increased their 7α-dehydroxylation capacity by severalfold, indicating that the function of gut microbiota is modulated by the amount of BAs presenting to the intestine (25). In our study, when the two cirrhotic groups who ultimately had the biopsies taken (the active drinkers and those who did not have alcohol as an etiology) were compared, there was only a modest difference in the stool microbiome, especially in Veillonellaceae and Bacteroidaceae, which are not associated with bile acid 7α-dehydroxylation capacity but may promote growth of bacteria that do. Veillonellaceae specifically is linked with higher systemic inflammation and endotoxemia in cirrhotics; the association with secondary BAs in alcoholic cirrhotics could potentially be a mechanism (4). Despite these modest changes in bacterial composition, we ultimately found that the microbiota function, i.e., 7α-dehydroxylation demonstrated by the highest fecal secondary BAs, was significantly different in the current drinkers (cirrhotic and not). Therefore, it is plausible that a higher total BA pool in alcoholic cirrhotics could lead to a higher substrate for microbiota that can perform 7α-dehydroxylation and can survive in an environment of relatively higher secondary BA concentration or that alcohol use could lead to an enhanced 7α-dehydroxylating capacity of the microbiota. Further metagenomic studies are required to explore these mechanisms.
We conclude that the consumption of alcohol in noncirrhotic and cirrhotic subjects increases the levels of total fecal bile acids and secondary bile acids compared with other cirrhotic patients and healthy controls. This was accompanied by a significant increase in serum endotoxin, serum conjugated secondary BAs, and several markers of colonic inflammation in cirrhotic patients consuming alcohol that were not suppressed by concomitant FXR activation. The fecal total and secondary BA increase in actively drinking alcoholics was also not susceptible to the increase in FGF-19 expression in the colon and ileum and may represent a new dimension of the pathogenesis of inflammation and gut barrier injury in alcoholic liver disease.
GRANTS
This work was supported by RO1AA020203 from the National Institute on Alcohol Abuse and Alcoholism, by grant RO1DK087913 from the National Institute of Diabetes and Digestive and Kidney Diseases, and by the McGuire Research Institute.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: G.K., P.B.H., H.Z., W.M.P., D.J.K., E.C.G., Y.W., R.L., and P.M.G. performed experiments; G.K., H.Z., W.M.P., H.T., H.N., Y.W., and R.L. analyzed data; G.K., P.B.H., H.Z., W.M.P., D.M.H., D.J.K., H.T., H.N., J.M.R., M.F., E.C.G., Y.W., R.L., P.M.G., and J.S.B. interpreted results of experiments; G.K., P.B.H., W.M.P., H.N., M.F., E.C.G., Y.W., R.L., A.J.S., P.M.G., and J.S.B. edited and revised manuscript; G.K., P.B.H., H.Z., W.M.P., D.M.H., D.J.K., H.T., H.N., J.M.R., M.F., P.M.G., and J.S.B. approved final version of manuscript; P.B.H., H.Z., W.M.P., D.M.H., and J.S.B. conception and design of research; D.J.K., H.T., M.F., and J.S.B. drafted manuscript.
Appendix: DETAILED METHODS
Fecal Bile Acid Analysis
Chemicals.
Bile acid standards were purchased from Steraloids. (Newport, RI). Phenacyl bromide and triethylamine, which were used for derivatization of bile acids, were obtained from Sigma-Aldrich (St. Louis, MO). All other reagents were used of highest grade except for water and methanol for mobile phase, which were of HPLC grade.
Extraction of bile acids from stools.
Freeze-dried stool ∼50 mg, to which 20 μg of 24-Nor-3α,12α-dihydroxy-5β-cholan-23-oic acid (nor-DCA) was added, was homogenized with 0.1 N NaOH (5 ml) by ultrasonication for 30 min in a screw-cap tube. After centrifugation (3,000 rpm, 10 min),supernatant was taken out to the clean tube (this procedure was repeated two times). The residue was further extracted with methanol (500 μl) with ultrasonication for 15 min, and all of the extracts were combined and subjected to solid-phase extraction by a Waters Sep-Pak tC18 column (Milford, MA). The column was successively washed with water (10 ml), 10% acetone (5 ml), and water (5 ml) and then retained bile acids were eluted with methanol (7 ml), which was evaporated to dryness under N2 stream.
24-Phenacyl esterification for extracted bile acids.
After sample was completely dried in a vacuum oven overnight, 200 μl of TEA (10 mg/ml in acetone) and 200 μl phenacyl bromide (12 mg/ml in acetone) were added, and the mixture was ultrasonicated for 1 h and then heated at 50°C for 1 h in a screw-cap tube. The reaction mixture was diluted with acetone (2 ml) and was passed through a Waters Sep-Pak silica cartridge. Bile acid phenacyl ester derivatives were eluted by acetone (5 ml). The sample was dried under nitrogen stream, resuspended in 100 μl of acetone, and filtered through with a 0.45-μm Millipore membrane, and an aliquot (10 μl) was injected to the following HPLC system. To assign the retention times of bile acid phenacyl esters, bile acid standards (each 1 mg) were also derivatized by the above method.
Analyses of bile acid 24-phenacyl esters by the HPLC-UV method.
The apparatus used was a Waters alliance series 2695 separation module equipped with a 2487 dual λ absorbance detector that is controlled by Empower Pro software. A Nova-Pak C18 column (300 mm x 3.9 mm ID, particle size 4 μm; Waters) was used for analytical purpose, which was kept at 32°C during analysis. Methanol (84%) was used for the mobile phase, and its flow rate was kept constant at 0.72 ml/min. Bile acid 24-phenacyl esters were monitored by the absorption at 254 nm. Peak assignment was done by direct comparison with the retention times of authentic standards, and their concentration was determined by the ratio of the peak area with that of internal standard (nor-DCA).
Microbiome.
We first use Length Heterogeneity PCR (LH-PCR) fingerprinting of the 16S rRNA to rapidly survey our samples and standardize the community amplification. We then interrogated the microbial taxa associated with the gut fecal microbiome using Multitag Pyrosequencing (MTPS). This technique allows for rapid sequencing of multiple samples at one time yielding thousands of sequence reads per sample.
Microbiome community fingerprinting.
LH-PCR was done to standardize the community analysis as previously published. Briefly, total genomic DNA was extracted from tissue using a Bio101 kit from MP Biomedicals (Montreal, Quebec) as per the manufacturer's instructions. About 10 ng of extracted DNA were amplified by PCR using a fluorescently labeled forward primer 27F [5'-(6FAM) AGAGTTTGATCCTGGCTCA G-3'] and unlabeled reverse primer 355R' (5'-GCTGCCTCCCGTAGGAGT-3'). Both primers are universal primers for bacteria. The LH-PCR products were diluted according to their intensity on agarose gel electrophoresis and mixed with ILS-600 size standards (Promega) and HiDi Formamide (Applied Biosystems, Foster City, CA). The diluted samples were then separated on a ABI 3130xl fluorescent capillary sequencer (Applied Biosystems) and processed using the Genemapper software package (Applied Biosystems). Normalized peak areas were calculated using a custom PERL script, and operational taxonomic units constituting <1% of the total community from each sample were eliminated from the analysis to remove the variable low-abundance components within the communities.
MTPS.
We employed the MTPS process to characterize the microbiome from the fecal and biopsy samples. Specifically, we have generated a set of 96 emulsion PCR fusion primers that contain the 454 emulsion PCR linkers on the 27F and 355R primers and a different eight base “barcode” between the A adapter and 27F primer. Thus, each fecal sample was amplified with unique bar-coded forward 16S rRNA primers, and then up to 96 samples were pooled and subjected to emulsion PCR and pyrosequenced using a GS-FLX pyrosequencer (Roche). Data from each pooled sample were “deconvoluted” by sorting the sequences into bins based on the barcodes using custom PERL scripts. Thus, we were able to normalize each sample by the total number of reads from each barcode. We have noted that ligating tagged primers to PCR amplicons distorts the abundances of the communities, and thus it is critical to incorporate the tags during the original amplification step.
Microbiome community analysis.
We identified the taxa present in each sample using the Bayesian analysis tool in Version 10 of the Ribosomal Database Project. The abundances of the bacterial identifications were then normalized using a custom PERL script, and genera present at >1% of the community were tabulated. We chose this cutoff because of our a priori assumption that genera present in <1% of the community vary between individuals and have minimal contribution to the functionality of that community, and 2,000 reads/sample will only reliably identify community components that are >1% in abundance.
REFERENCES
- 1.Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108: 218–224, 1995 [DOI] [PubMed] [Google Scholar]
- 2.Axelson M, Mork B, Sjovall J. Ethanol has an acute effect on bile acid biosynthesis in man. FEBS Lett 281: 155–159, 1991 [DOI] [PubMed] [Google Scholar]
- 3.Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, Noble NA, Unser AB, Daita K, Fisher AR, Sikaroodi M, Gillevet PM. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol 60: 940–947, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bajaj JS, Hylemon PB, Ridlon JM, Heuman DM, Daita K, White MB, Monteith P, Noble NA, Sikaroodi M, Gillevet PM. The colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am J Physiol Gastrointest Liver Physiol 306: G675–G685, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bajaj JS, Ridlon JM, Hylemon PB, Thacker LR, Heuman DM, Smith S, Sikaroodi M, Gillevet PM. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am J Physiol Gastrointest Liver Physiol 302: G168–G175, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baptissart M, Vega A, Maqdasy S, Caira F, Baron S, Lobaccaro JM, Volle DH. Bile acids: from digestion to cancers. Biochimie 95: 504–517, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Barrasa JI, Olmo N, Lizarbe MA, Turnay J. Bile acids in the colon, from healthy to cytotoxic molecules. Toxicol In Vitro 27: 964–977, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Chadwick VS, Gaginella TS, Carlson GL, Debongnie JC, Phillips SF, Hofmann AF. Effect of molecular structure on bile acid-induced alterations in absorptive function, permeability, and morphology in the perfused rabbit colon. J Lab Clin Med 94: 661–674, 1979 [PubMed] [Google Scholar]
- 9.Chanda D, Kim YH, Li T, Misra J, Kim DK, Kim JR, Kwon J, Jeong WI, Ahn SH, Park TS, Koo SH, Chiang JY, Lee CH, Choi HS. Hepatic cannabinoid receptor type 1 mediates alcohol-induced regulation of bile acid enzyme genes expression via CREBH. PLoS One 8: e68845, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gadaleta RM, Oldenburg B, Willemsen EC, Spit M, Murzilli S, Salvatore L, Klomp LW, Siersema PD, van Erpecum KJ, van Mil SW. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kappaB signaling in the intestine. Biochim Biophys Acta 1812: 851–858, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol 35: 229–244, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofmann AF, Eckmann L. How bile acids confer gut mucosal protection against bacteria. Proc Natl Acad Sci USA 103: 4333–4334, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 141: 1773–1781, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, Takei H, Muto A, Nittono H, Ridlon JM, White MB, Noble NA, Monteith P, Fuchs M, Thacker LR, Sikaroodi M, Bajaj JS. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 58: 949–955, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kosters A, Karpen SJ. The role of inflammation in cholestasis: clinical and basic aspects. Semin Liver Dis 30: 186–194, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Vee M, Lecureur V, Stieger B, Fardel O. Regulation of drug transporter expression in human hepatocytes exposed to the proinflammatory cytokines tumor necrosis factor-alpha or interleukin-6. Drug Metab Dispos 37: 685–693, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Mekjian HS, Phillips SF, Hofmann AF. Colonic secretion of water and electrolytes induced by bile acids: perfusion studies in man. J Clin Invest 50: 1569–1577, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res 33: 1836–1846, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nilsson LM, Sjovall J, Strom S, Bodin K, Nowak G, Einarsson C, Ellis E. Ethanol stimulates bile acid formation in primary human hepatocytes. Biochem Biophys Res Commun 364: 743–747, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Pai R, Tarnawski AS, Tran T. Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell 15: 2156–2163, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pijls KE, Koek GH, Elamin EE, de Vries H, Masclee AA, Jonkers DM. Large intestine permeability is increased in patients with compensated liver cirrhosis. Am J Physiol Gastrointest Liver Physiol 306: G147–G153, 2014 [DOI] [PubMed] [Google Scholar]
- 22.Purohit V, Bode JC, Bode C, Brenner DA, Choudhry MA, Hamilton F, Kang YJ, Keshavarzian A, Rao R, Sartor RB, Swanson C, Turner JR. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol 42: 349–361, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiao L, Studer E, Leach K, McKinstry R, Gupta S, Decker R, Kukreja R, Valerie K, Nagarkatti P, El Deiry W, Molkentin J, Schmidt-Ullrich R, Fisher PB, Grant S, Hylemon PB, Dent P. Deoxycholic acid (DCA) causes ligand-independent activation of epidermal growth factor receptor (EGFR) and FAS receptor in primary hepatocytes: inhibition of EGFR/mitogen-activated protein kinase-signaling module enhances DCA-induced apoptosis. Mol Biol Cell 12: 2629–2645, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao YP, Stravitz RT, Vlahcevic ZR, Gurley EC, Sando JJ, Hylemon PB. Activation of protein kinase C alpha and delta by bile acids: correlation with bile acid structure and diacylglycerol formation. J Lipid Res 38: 2446–2454, 1997 [PubMed] [Google Scholar]
- 25.Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids, and gut microbiota: Unraveling a complex relationship. Gut Microbes In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47: 241–259, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Rosa H, Silverio AO, Perini RF, Arruda CB. Bacterial infection in cirrhotic patients and its relationship with alcohol. Am J Gastroenterol 95: 1290–1293, 2000 [DOI] [PubMed] [Google Scholar]
- 28.Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyotylainen T, Oresic M, Backhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17: 225–235, 2013 [DOI] [PubMed] [Google Scholar]
- 29.Shawcross DL, Davies NA, Williams R, Jalan R. Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J Hepatol 40: 247–254, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Stenman LK, Holma R, Eggert A, Korpela R. A novel mechanism for gut barrier dysfunction by dietary fat: epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol 304: G227–G234, 2013 [DOI] [PubMed] [Google Scholar]
- 31.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol 16: 1321–1329, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 183: 6251–6261, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Liu Y, Sidhu A, Ma Z, McClain C, Feng W. Lactobacillus rhamnosus GG culture supernatant ameliorates acute alcohol-induced intestinal permeability and liver injury. Am J Physiol Gastrointest Liver Physiol 303: G32–G41, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48: 1632–1643, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol 5: e1000352, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiest R, Krag A, Gerbes A. Spontaneous bacterial peritonitis: recent guidelines and beyond. Gut 61: 297–310, 2012 [DOI] [PubMed] [Google Scholar]
- 37.Xie G, Zhong W, Li H, Li Q, Qiu Y, Zheng X, Chen H, Zhao X, Zhang S, Zhou Z, Zeisel SH, Jia W. Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB J 27: 3583–3593, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zapater P, Frances R, Gonzalez-Navajas JM, de la Hoz MA, Moreu R, Pascual S, Monfort D, Montoliu S, Vila C, Escudero A, Torras X, Cirera I, Llanos L, Guarner-Argente C, Palazon JM, Carnicer F, Bellot P, Guarner C, Planas R, Sola R, Serra MA, Munoz C, Perez-Mateo M, Such J. Serum and ascitic fluid bacterial DNA: a new independent prognostic factor in noninfected patients with cirrhosis. Hepatology 48: 1924–1931, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Zhong W, Li Q, Xie G, Sun X, Tan X, Sun X, Jia W, Zhou Z. Dietary fat sources differentially modulate intestinal barrier and hepatic inflammation in alcohol-induced liver injury in rats. Am J Physiol Gastrointest Liver Physiol 305: G919–G932, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]