

Abstract
In aging societies increasing cases of neurodegenerative protein deposit diseases urge for the identification of the underlying mechanisms. Expectations are that in 2050 the percentage of population over age 60 is 42% in Japan, 34% in China, and 27% in the US. The cell nucleus is a major target of amyloid-like protein fibrillation in a variety of disorders that are characterized by widespread aggregation of proteins with instable homopolymeric amino acid repeats, ubiquitin, and other proteinaceous components. Additionally, accumulation of insoluble, SDS-resistant proteins has been identified as an intrinsic property of organismal aging. This review collects current knowledge about the composition and function of insoluble, nuclear protein inclusions from the protein homeostasis perspective. It discusses the occurrence and role of nuclear amyloid in the diseased as well as the healthy cell. Features of nuclear inclusions such as protein composition and locally active protein degradation may predict neural fitness and survival in a variety of health or disease settings.
Keywords: aging, amyloid, Congo red, fibrillation, Huntington disease, neurodegeneration, nucleus, nuclear inclusions, prion disease, polyQ, protein aggregation
Introduction
Stepwise fibrillation of proteins to amyloid structures and amyloid-like protein aggregates occurs naturally or is disease associated. The hallmark of amyloid aggregation is conversion of otherwise soluble proteins into β-sheet conformations that locate extracellularly, in the cytoplasm or in the cell nucleus.1 Formation of amyloid-like protein aggregates unifies superficially unrelated human conditions such as Alzheimer, Parkinson, or Huntington disease that are therefore consolidated as neurodegenerative deposition diseases.2 In brains of patients with Alzheimer disease amyloid β peptides (Aβs) deposit in extracellular plaques or fibrils, whereas tau protein is enriched in intraneuronal neurofibrillary tangles.3 Protein deposition in Parkinson disease occurs as characteristic Lewy body inclusions in the cytoplasm of residual substantia nigra and dopaminergic striatal neurons.4,5 Huntington disease constitutes one out of nine neurodegenerative disorders that are characterized by aberrant deposition of signature proteins containing unstable homopolymeric repeats of the amino acid glutamine (polyQ). Neuronal cells of patients with polyQ-repeat-length diseases are characterized by occurrence of microscopically discernible nuclear inclusions (NIs) that contain proteins with expanded polyQ stretches, components of the ubiquitin-proteasome system (UPS), and other cellular proteins.6,7 Aggregates can likewise be observed in other neural compartments such as the cytoplasm, dendrites, and axon terminals.8 A common feature of such inclusions is the recruitment of mutated, misfolded, and waste proteins that leave solution because of the tendency of normally buried hydrophobic domains to associate with one another.9 The presence of proteasome-dependent proteolytic activity in NIs indicates that misfolded or excess polypeptides form intracellular aggregates before their degradation.10 Yet, the role of nuclear protein aggregates in disease pathology is still unknown, and their assumed function ranges from being cytotoxic to benign or even neuroprotective.11-13 This review compiles current knowledge about nuclear protein aggregation, e.g., amyloid deposition with special emphasis on their protein composition mandating the idea that this composition reflects perturbed protein interaction networks and holds the key for a better understanding of underlying pathologic as well as physiologic events.14-16
Amyloid: Structure and Detection
The term amyloid was first used in the 19th century by German pathologist Rudolph Virchow to describe starch-like deposits within mammalian cells based on similar tinctorial properties with polysaccharides from plants. However, Virchow in fact observed waxy protein structures and the term amyloid currently designates proteinaceous deposits that are defined by one or more histochemical and biophysical features such as (1) an ultrastructure of long, unbranched fibrils of approximately 10 nm in width, (2) a green birefringence under cross polarized light after staining with the azo dye Congo red, (3) a shift of fluorescence wavelength after staining with the dye Thioflavin T, (4) a peak at around 1620 by Fourier transform infrared spectroscopy indicating the presence of β-sheet, (5) a crossed β-pleated sheet structure, and (6) binding to amyloid-specific antibodies, peptides or compounds.17-20
In vitro characterization of amyloid fiber structure and the actual processes of amyloid protein fibrillation has largely progressed by usage of high-resolution structural studies, i.e., solid-state nuclear magnetic resonance (NMR). The process constitutes a nucleated growth mechanism that initiates stepwise fibrillation of otherwise soluble proteins with a lag phase of “nuclei” formation and proceeds by further association of monomers or oligomers. In the in vitro scenario fibril growth either occurs by transition of oligomeric species to spherical, chain-like fibrils or by sequential fibrillation of soluble oligomers, unstructured aggregates, and short curly protofibrils that finally form mature amyloid fibrils.1 It is important to note that structural intermediates of this fibrillation process may yet have varying amyloid features such as β-structure, Thioflavin T-binding or Congo red-binding.
Formation of intracellular amyloid deposits, e.g., in vivo protein fibrillation, may be initiated by local unfolding of native proteins through physiological fluctuations and subsequent fibrillation.21 Examples are changes of thermal conditions as observed in heat shock responses or experimentally induced fluctuations. Consistently, several reports describe controlled nucleation of protein fibrillation by heat shock, metals, or nanoparticles in vitro, in mammalian cell culture as well as in whole organisms.10,22-25 The analysis of intracellular amyloid formation has advanced in recent years due to the diagnostic value of amyloid fibril localization within cells and tissues.26 In line with this conformation-specific antibodies, peptides and luminescent compounds have proven useful to monitor amyloid distribution in fixed and living specimens; however, methods for the discrimination of specific fibrillation intermediates in vivo are still in their infancy.17,27,28
Nuclear Amyloid Depositions in Disease
Amyloid-like NIs in neurodegenerative aggregation diseases are predominately characterized by histological methods and biochemical purification of SDS-insoluble protein fractions. At least nine CAG-repeat diseases including Huntington disease (HD), the spinocerebellar ataxias SCA1, SCA2, SCA3, SCA6, SCA7, SCA17, and dentatorubral pallidoluysian atrophy (DRPLA), as well as spinal and bulbar muscular atrophy (SBMA) show NIs in specific sections of postmortem brains.29,30
In postmortem brains of patients with HD it was discovered that mutated huntingtin forms mostly spherical or ovoid NIs in cortical neurons (Table 1; Fig. 1).31 These NIs were positioned throughout the nucleoplasm with a subfraction located in proximity to nucleoli. In a third of the observed neurons NIs covered nearly 50% of the cross-sectional nuclear area. Additionally to huntingtin NIs frequently contained ubiquitin. Electron microscopy revealed that NIs in cortical HD neurons are composed of granules, filaments, and randomly or parallel oriented fibrils.31 Huntingtin and ubiquitin also occupy spherical NIs in the striatum of mice transgenic for the huntingtin mutation.32 Interestingly, striatal neurons of symptomatic transgenic mice showed significant alterations of the nuclear envelope structure, i.e., numerous indentations of the nuclear membrane and aberrant clustering of nuclear pore complexes. In contrast, cross-sectional size of nuclei, nucleoli, and Cajal bodies were described as unchanged. Notably, nuclear huntingtin occurs as an early pathological hallmark 1–2 wk before any murine phenotypes such as brain or body weight loss and approximately 5 wk before the first disease-specific symptoms.32
Table 1. Components of nuclear protein inclusions with listing of their histochemical and biophysical amyloid features.
| Amyloid component | IHC | NI | PolyQ- induced |
PolyQ disease |
Animal model |
Cell culture |
CR/ThT | CR bi | AP | anti Aβ | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| huntingtin | ● | ● | ● | ● | ● | ● | ● | ● | 31, 32, 71, 72 | ||
| ubiquitin | ● | ● | ● | ● | ● | ● | 31, 32, 43, 72 | ||||
| androgen receptor | ● | ● | ● | ● | ● | ● | 36, 73 | ||||
| ataxin-1 | ● | ● | ● | ● | ● | ● | 38, 74 | ||||
| 20S proteasome | ● | ● | ● | ● | ● | 38, 41 | |||||
| HDJ-2/HSDJ | ● | ● | ● | ● | ● | 38 | |||||
| HSP-70 | ● | ● | ● | ● | ● | 38, 43, 75 | |||||
| p53 | ● | ● | ● | 43 | |||||||
| mdm-2 | ● | ● | ● | 43 | |||||||
| TATA binding protein | ● | ● | ● | ● | ● | ● | 43, 72, 76, 77 | ||||
| NPC proteins | ● | ● | ● | ● | 43 | ||||||
| ataxin-3 | ● | ● | ● | ● | ● | 78, 79 | |||||
| atrophin-1 | ● | ● | ● | ● | 80 | ||||||
| CREB binding protein | ● | ● | ● | ● | ● | 45, 80 | |||||
| PML | ● | ● | ● | ● | ● | ● | 34, 74, 80 | ||||
| SUMO-1 | ● | ● | ● | ● | ● | ● | 34, 81 | ||||
| SUMO-2 | ● | ● | ● | ● | ● | ● | 81 | ||||
| ataxin-7 | ● | ● | ● | ● | ● | ● | 81 | ||||
| p80 coilin | ● | ● | ● | ● | ● | 34, 74 | |||||
| histone H3 | ● | ● | ● | ● | 82 | ||||||
| hnRNP F/H | ● | ● | ● | ● | 82 | ||||||
| smn1 | ● | ● | ● | ● | 15 | ||||||
| gemin4 | ● | ● | ● | ● | 15 | ||||||
| SR140 | ● | ● | ● | ● | 15 | ||||||
| WDR3 | ● | ● | ● | ● | 15 | ||||||
| ranBP1 | ● | ● | ● | ● | 15 | ||||||
| laminB1 | ● | ● | ● | ● | 15 | ||||||
| NuMA | ● | ● | ● | ● | 15 | ||||||
| nuclear amyloid structure | ● | ● | ● | ● | ● | ● | ● | 24, 25, 40 |
Anti Aβ, detection by amyloid specific antibodies; AP, amyloid peptide binding; CR bi, Congo red birefringence; CR/ThT, Congo red and/or Thioflavin T binding; IHC, nuclear inclusions defined by immunohistochemistry; NI, nuclear inclusion.
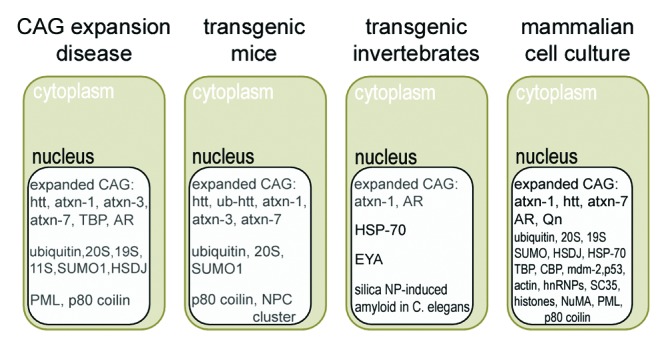
Figure 1. Schematic representation of proteins in nuclear inclusions (NIs) from patients with neurodegenerative polyQ (CAG) expansion diseases and respective animal or mammalian cell culture models. 20S, 20S proteasome subunits; 19S, 19S proteasome regulator subunits; 11S, 11S proteasome regulator subunits; AR, androgen receptor; atxn-1, ataxin-1; atxn-3, ataxin-3; atxn-7, ataxin-7; CBP, CREB binding protein; EYA, eyes absent protein; hnRNPs, heterogeneous nuclear ribonucleoproteins; HSDJ, DNAJ protein homolog; HSP-70, heat shock protein 70; htt, huntingtin; mdm-2, mouse double minute protein; NP, nanoparticle; NPC, nuclear pore complex; NuMA, nuclear mitotic apparatus protein; PML, promyelocytic leukemia protein; Qn, different length polyQ repeat; SUMO, small ubiquitin-related modifier; ub, ubiquitinated; TBP, TATA binding protein.
In SCA1 NIs are observable in neurons of the cerebral cortex, striatum, substantia nigra, and pontine nuclei.33 They have a diameter of 1 to 3 μm and contain ataxin-1 and ubiquitin as well as unspecified proteins with polyQ repeats (Table 1; Figs. 1 and 2). By transmission electron microscopy inclusions appear as round, spherical bodies in the nucleoplasm consisting of mixtures of granular and filamentous structures that are either organized in random or parallel arrays.30 Postmortem brains of SCA3 patients display a similar morphology of NIs in the spinocerebellum, cerebellar dentate nucleus, cerebral cortex, and striatum. They are immunoreactive with ataxin-3, ubiquitin, polyQ repeat domains, and a variety of transcription factors (Table 1; Fig. 1).30 Additionally, NIs of SCA3 patients and respective mouse models have been shown to associate with PML and Cajal nuclear bodies in nucleoplasmic microdomains.34 The incidence of NIs in the polyQ repeat diseases SCA2 and DRPLA is low with 1–3% of affected neurons; however these NIs are widely distributed within different brain regions. In SBMA, also termed Kennedy disease, the androgen receptor (AR) is mutated concerning expansions of its instable polyQ repeats and accumulates in NIs that appear in motor neurons of the brainstem, the spinal cord, but also in skin, testis, and additional visceral tissues (Table 1; Fig. 1).35,36
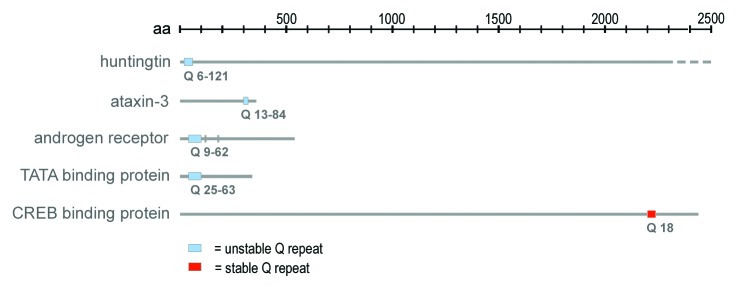
Figure 2. Schematic representation of homopolymeric glutamine repeat positioning in primary protein sequences of NI components. Unstable polyQ repeats vary considerably with respect to their location in the primary protein sequence and length. Aa, scale of amino acid positions; Q, glutamine.
It has been acknowledged very early that the distribution of NIs within specific brain regions is not exactly correlated with degeneration of the affected neurons. While the striatum represents a brain region that undergoes extensive neurodegeneration in HD, only 1–4% of striatal neurons contain nuclear aggregates.37 Long-term observation of live neurons by automated microscopy revealed a higher viability of neurons with mutated huntingtin accumulating in NIs compared with neurons with diffuse nuclear localization of huntingtin.11 These observations suggested a protective role of nuclear protein aggregation, i.e., a coping response of the nucleus to excess fibrillation of misfolded proteins. Consistent with this and as already mentioned above, ubiquitin, a main player of proteasome-dependent protein degradation, is identified by histopathology as a frequent component of NIs. Likewise are heat shock proteins, 20S proteasomes and the 19S proteasome regulator units (Table 1).38-41 With all the players on the field and the demonstration of proteasome-dependent proteolytic activity in NIs a picture is emerging where at least a subpopulation of intranuclear protein aggregates operate as proteolytic centers.10 Here, formation of NIs may serve recruitment of excess, mutated, or misfolded nuclear proteins to their degradation. Consistent with this idea it was shown that neurons have different capacities to clear mutated huntingtin, and that those with greater clearance capacities live longer.42 In this respect NIs may have a protective function by sequestering and clearing excess or misfolded nuclear proteins from the nucleoplasm and thus maintaining protein homeostasis within the cell nucleus.
While it seems appropriate to boost this cellular defense mechanism, e.g., nuclear proteolysis, with therapeutic interventions it must also be considered that protein components of NIs are part of a nuclear proteome network and respective maintenance of cellular functions such as gene expression or progression of the cell cycle. Intracellular inclusions that were (1) biochemically isolated from cells transfected with expanded huntingtin polyQ tracts and (2) subjected to mass spectrometry, contained among other components ubiquitin, HSP-70, cell cycle regulators p53 and mdm-2 as well as the general transcription factor TATA binding protein (TBP).43,44 The transcriptional coactivator and histone acetylase CREB-binding protein (CBP) that carries a stable polyQ repeat was similarly identified as a component of NIs (Table 1; Fig. 1 and 2).45
Moreover, proteins that interact with intracellular amyloid seem to participate in an amyloid interactome (Table 1).15 This amyloid interactome is characterized by large proteins rich in predicted unstructured regions that hence may be targeted to amyloidogenic aggregation and depleted from their cellular functions. Based on the identified amyloidogenic candidate proteins affected nuclear functions may include ribosome biogenesis, RNA processing, chromatin organization, transcription, and nuclear structure.15 Evidence for interactions between gene expression, protein aggregation, and altered gene expression has been provided previously; however, a global perspective on the role of such interactions in physiologic settings as well as in disease progression is clearly missing.46,47 Systematic profiling of protein aggregate compositions with unbiased systems biology approaches may provide for a promising concept to identify and eventually modulate the intracellular amyloid landscape in health and disease.
Besides neurodegenerative protein deposition diseases aberrant fibrillation of proteins to intranuclear inclusion bodies has likewise been observed as a pathological feature in certain systemic autoimmune disorders such as scleroderma, in rodent animal models of xenobiotic-induced autoimmunity and in breast cancer.48-50 While these observations currently are of anecdotal nature, they suggest that amyloid protein fibrillation may play a role in other cells than neurons36 and in more pathologic disease conditions.
Functional Amyloid Fibrillation
Besides its disease association mounting evidence suggests that amyloid protein fibrillation also occurs naturally, i.e., in a healthy cellular environment. An increasing number of proteins with no link to protein aggregation diseases have been found to form, under defined in vitro conditions, fibrillar aggregates that have structural properties of amyloid fibrils, including binding to reporter dyes such as Congo red and Thioflavin T. This is consistent with the notion that the ability to convert into an amyloid structure is a generic property of polypeptide chains and virtually every protein has the potential to undergo amyloid fibrillation.1 Moreover, due to the advance of respective techniques functional amyloid is being increasingly identified in both, bacteria and eukaryotes. Examples are the mammalian protein Pmel17 that forms amyloid structures and thereby accelerates covalent polymerization of reactive small molecules into melanin or peptide hormones that are stored as amyloid-like cross-β-sheet-rich conformations in pituitary glands.51,52 Here, the amyloid fold is clearly used in a non-disease-associated manner, namely in normal mammalian cell physiology.
Consistent with this, bioinformatics-based approaches demonstrate that proteins containing stable polyQ stretches promote polymerization of functional macromolecular complexes. A global genome survey from Whisstock and coworkers revealed common functional patterns of repeat-containing proteins.53 The bulk of proteins with homopolymeric amino acid sequences, including polyQ, perform roles in processes that are characterized by assembly of multiprotein or nucleoprotein complexes and involve RNA and DNA. Nuclear processes that account for gene expression are driven by dynamic formation and disassembly of macromolecules at the promoters of active genes: Proteins containing stable polyQ repeats such as CREB-binding protein (CBP) and transcription factor SP1, or unstable homopolymeric polyQ repeats such as the androgen receptor (AR), glucocorticoid receptor (GR), ataxin-7, and TATA-binding protein (TBP) enable assembly of transcriptional initiation complexes (Table 1; Fig. 2).14,54 Here, polyQ stretches might act as adaptor motifs that facilitate the formation of the transcription initiation machinery. In line with this, it was shown that homopolymeric glutamine (Q)- and proline (P)-stretches activate transcription in vitro when fused to the DNA-binding domain of yeast activator protein GAL4.55 Transcriptional activity correlated with the repeat length and was determined as being maximal with stretches of 10 to 30 Q or 10 P residues.
Given that polyQ stretches seem to be associated with both, amyloid fibrillation in neurodegenerative aggregation diseases, and functional organization of the cell nucleus the question arises which features of polyQ repeats contribute to either pathology or biological function. Is there a critical threshold of homopolymeric amino acid stretch length that decides if polyQ plays a role in nuclear self-organization or acts as a prion? Indeed, a critical threshold concerning the length of consequent Q residues exists in polyQ deposition disorders that correlates with disease onset, and severity of disease.56 This critical threshold has been reproduced in biophysical in silico experiments, Saccharomyces cerevisiae and animal models including Caenorhabditis elegans, Drosophila melanogaster, mice, zebrafish, and non-human primates.56-60 Since the length threshold is not uniform and may vary between 35–40 Q-residues in Huntington disease and 20–30 Q-residues in spinocerebellar ataxia type 6 it is anticipated that the critical length of a polyQ repeat is defined by additional context (Fig. 2).61 Context dependency may concern the whole protein context, as well as sequences directly neighboring the polyQ stretch. Confirmedly, a polyP repeat flanking the polyQ stretches has been identified in huntingtin that acts as a cis-inhibitor of polyQ aggregation.62,63
In line with results that polyQ stretches of 10 to 20 Q-residues maximally promote transcriptional activity55 it is tempting to speculate that shorter homopolymeric Q-repeats sustain nuclear processes such as gene expression by their propensity to induce mild protein scaffolding, whereas the intrinsic capacity to form organized fibrillar structures with polyQ length above a certain threshold disturbs the functional organization of the cell nucleus by excess fibrillation that culminates in formation of insoluble, amyloid-like protein aggregates in the nucleoplasm (Table 1; Fig. 1). Such yin and yang of polyQ fibrillation may have prohibited evolutionary eradication of unstable, homopolymeric polyQ repeats from genomes.14,16 Consistent with these ideas is the notion that the nucleus represents a highly crowded environment which on one hand essentially enables the self-organization of macromolecular, multiprotein complexes, however, likewise potentially promotes aberrant amyloid protein fibrillation through inordinate volume exclusion.14,64-67 Along these lines it was shown that xenobiotics such as certain heavy metals and nanoparticles sustain amyloid protein fibrillation, accumulate in specific nuclear microenvironments and locally induce nuclear amyloid.22-25 Thus, a fine balance between nuclear crowding and overcrowding may be required to maintain functional protein aggregation.
Conclusion
Nuclear protein aggregation and amyloid deposition are a prominent hallmark of neurodegenerative protein deposit diseases. While it was initially thought that nuclear amyloid is in any case responsible for neural cell death, time-resolved experiments that correlate nuclear amyloid and neurodegeneration on the single cell level rather suggest a cell protective role. Aggregation of misfolded or excess nuclear proteins in spherical nucleoplasmic microenvironments may segregate dysfunctional proteins and recruit them for degradation by the ubiquitin-proteasome system. The frequent presence of ubiquitin, proteasomes and heat shock proteins in NIs and the discovery of proteasomal activity in amyloid-like nucleoplasmic protein aggregates is consistent with this idea. Confirmedly, the nucleus establishes itself as a major cellular compartment for protein degradation, as it was shown recently in yeast that misfolded proteins are transported from the cytoplasm to nuclear degradation by chaperone Sis1p.68,69
In addition to a protective role of NIs in neurodegeneration evidence accumulates for functional amyloid in the nucleus. Amyloid-indicating features such as Congo red and Thioflavin T-binding as well as reactivity with amyloid-specific antibodies and peptides can be located in distinct subnuclear microenvironments under physiological conditions.18,24,25 Moreover, 55% of eukaryotic proteins are predicted to contain unstructured protein regions that are intrinsically amyloidogenic.21,70 To a certain degree the fibrillation capacity of amyloidogenic proteins may be required for nuclear function that is characterized by dynamic assembly and disassembly of large multiprotein and ribonucleoprotein complexes. An example is provided by the role of homopolymeric polyQ repeats in the initiation of transcription.53,55
A better understanding of amyloid fibrillation in the nucleus is a prerequisite for promising therapeutic interventions in neurodegenerative protein deposition diseases. Before boosting the protein degradation machinery the function and topology of NIs, nuclear protein homeostasis and nuclear amyloid should be fully characterized. This requires development of methods that allow for the identification of different amyloid fibrillation steps in living cells and whole organisms. Comparative definition of protein aggregation landscapes by systems biology in different disease models as well as under physiological conditions has the potential to provide for global aggregome networks as sources for informed, personalized therapeutic interventions.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
I thank Peter Hemmerich for helpful comments on the manuscript and members of the von Mikecz laboratory for critical support. The author acknowledges financial support from the German Science Foundation (DFG) through grants (MI 486/7-1) and GRK 1033, and from graduate college iBRAIN at the Heinrich-Heine-University Duesseldorf.
Glossary
Abbreviations:
- HD
Huntington disease
- NI
nuclear inclusion
- polyQ
polyglutamine
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–66. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601–23. doi: 10.1146/annurev-genet-110711-155524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holdorff B, Rodrigues e Silva AM, Dodel R. Centenary of Lewy bodies (1912-2012) J Neural Transm. 2013;120:509–16. doi: 10.1007/s00702-013-0984-2. [DOI] [PubMed] [Google Scholar]
- 5.Beitz JM. Parkinson’s disease: a review. Front Biosci (Schol Ed) 2014;6:65–74. doi: 10.2741/S415. [Schol Ed] [DOI] [PubMed] [Google Scholar]
- 6.Wanker EE. Protein aggregation and pathogenesis of Huntington’s disease: mechanisms and correlations. Biol Chem. 2000;381:937–42. doi: 10.1515/BC.2000.114. [DOI] [PubMed] [Google Scholar]
- 7.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 8.Vonsattel JP. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:55–69. doi: 10.1007/s00401-007-0306-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–9. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 10.Chen M, Singer L, Scharf A, von Mikecz A. Nuclear polyglutamine-containing protein aggregates as active proteolytic centers. J Cell Biol. 2008;180:697–704. doi: 10.1083/jcb.200708131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 12.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 13.Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–8. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Mikecz A. PolyQ fibrillation in the cell nucleus: who’s bad? Trends Cell Biol. 2009;19:685–91. doi: 10.1016/j.tcb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer-Hartl M, Hartl FU, Vabulas RM. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 16.Schaefer MH, Wanker EE, Andrade-Navarro MA. Evolution and function of CAG/polyglutamine repeats in protein-protein interaction networks. Nucleic Acids Res. 2012;40:4273–87. doi: 10.1093/nar/gks011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci U S A. 2002;99:1485–90. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiesehan K, Buder K, Linke RP, Patt S, Stoldt M, Unger E, Schmitt B, Bucci E, Willbold D. Selection of D-amino-acid peptides that bind to Alzheimer’s disease amyloid peptide abeta1-42 by mirror image phage display. Chembiochem. 2003;4:748–53. doi: 10.1002/cbic.200300631. [DOI] [PubMed] [Google Scholar]
- 19.Marshall KE, Serpell LC. Structural integrity of beta-sheet assembly. Biochem Soc Trans. 2009;37:671–6. doi: 10.1042/BST0370671. [DOI] [PubMed] [Google Scholar]
- 20.Butterfield S, Hejjaoui M, Fauvet B, Awad L, Lashuel HA. Chemical strategies for controlling protein folding and elucidating the molecular mechanisms of amyloid formation and toxicity. J Mol Biol. 2012;421:204–36. doi: 10.1016/j.jmb.2012.01.051. [DOI] [PubMed] [Google Scholar]
- 21.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 22.Uversky VN, Li J, Fink AL. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem. 2001;276:44284–96. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- 23.Linse S, Cabaleiro-Lago C, Xue WF, Lynch I, Lindman S, Thulin E, Radford SE, Dawson KA. Nucleation of protein fibrillation by nanoparticles. Proc Natl Acad Sci U S A. 2007;104:8691–6. doi: 10.1073/pnas.0701250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnhold F, von Mikecz A. Quantitative feature extraction reveals the status quo of protein fibrillation in the cell nucleus. Integr Biol (Camb) 2011;3:761–9. doi: 10.1039/c0ib00146e. [DOI] [PubMed] [Google Scholar]
- 25.Scharf A, Piechulek A, von Mikecz A. Effect of nanoparticles on the biochemical and behavioral aging phenotype of the nematode Caenorhabditis elegans. ACS Nano. 2013;7:10695–703. doi: 10.1021/nn403443r. [DOI] [PubMed] [Google Scholar]
- 26.Jack CR, Jr., Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80:1347–58. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Legleiter J, Lotz GP, Miller J, Ko J, Ng C, Williams GL, Finkbeiner S, Patterson PH, Muchowski PJ. Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J Biol Chem. 2009;284:21647–58. doi: 10.1074/jbc.M109.016923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aslund A, Sigurdson CJ, Klingstedt T, Grathwohl S, Bolmont T, Dickstein DL, Glimsdal E, Prokop S, Lindgren M, Konradsson P, et al. Novel pentameric thiophene derivatives for in vitro and in vivo optical imaging of a plethora of protein aggregates in cerebral amyloidoses. ACS Chem Biol. 2009;4:673–84. doi: 10.1021/cb900112v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vonsattel JP, Keller C, Cortes Ramirez EP. Huntington’s disease - neuropathology. Handb Clin Neurol. 2011;100:83–100. doi: 10.1016/B978-0-444-52014-2.00004-5. [DOI] [PubMed] [Google Scholar]
- 30.Yamada M, Sato T, Tsuji S, Takahashi H. CAG repeat disorder models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:71–86. doi: 10.1007/s00401-007-0287-5. [DOI] [PubMed] [Google Scholar]
- 31.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 32.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–48. doi: 10.1016/S0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 33.Duyckaerts C, Dürr A, Cancel G, Brice A. Nuclear inclusions in spinocerebellar ataxia type 1. Acta Neuropathol. 1999;97:201–7. doi: 10.1007/s004010050975. [DOI] [PubMed] [Google Scholar]
- 34.Yamada M, Sato T, Shimohata T, Hayashi S, Igarashi S, Tsuji S, Takahashi H. Interaction between neuronal intranuclear inclusions and promyelocytic leukemia protein nuclear and coiled bodies in CAG repeat diseases. Am J Pathol. 2001;159:1785–95. doi: 10.1016/S0002-9440(10)63025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, Doyu M, Hashizume Y, Fischbeck KH, Sobue G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998;44:249–54. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- 36.Li M, Nakagomi Y, Kobayashi Y, Merry DE, Tanaka F, Doyu M, Mitsuma T, Hashizume Y, Fischbeck KH, Sobue G. Nonneural nuclear inclusions of androgen receptor protein in spinal and bulbar muscular atrophy. Am J Pathol. 1998;153:695–701. doi: 10.1016/S0002-9440(10)65612-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19:2522–34. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–54. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 39.Wyttenbach A, Carmichael J, Swartz J, Furlong RA, Narain Y, Rankin J, Rubinsztein DC. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington’s disease. Proc Natl Acad Sci U S A. 2000;97:2898–903. doi: 10.1073/pnas.97.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abel A, Walcott J, Woods J, Duda J, Merry DE. Expression of expanded repeat androgen receptor produces neurologic disease in transgenic mice. Hum Mol Genet. 2001;10:107–16. doi: 10.1093/hmg/10.2.107. [DOI] [PubMed] [Google Scholar]
- 41.Jana NR, Zemskov EA, Wang Gh, Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. 2001;10:1049–59. doi: 10.1093/hmg/10.10.1049. [DOI] [PubMed] [Google Scholar]
- 42.Tsvetkov AS, Arrasate M, Barmada S, Ando DM, Sharma P, Shaby BA, Finkbeiner S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat Chem Biol. 2013;9:586–92. doi: 10.1038/nchembio.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153:283–94. doi: 10.1083/jcb.153.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitsui K, Doi H, Nukina N. Proteomics of polyglutamine aggregates. Methods Enzymol. 2006;412:63–76. doi: 10.1016/S0076-6879(06)12005-4. [DOI] [PubMed] [Google Scholar]
- 45.Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:11404–9. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nucifora FC, Jr., Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–8. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 47.Bithell A, Johnson R, Buckley NJ. Transcriptional dysregulation of coding and non-coding genes in cellular models of Huntington’s disease. Biochem Soc Trans. 2009;37:1270–5. doi: 10.1042/BST0371270. [DOI] [PubMed] [Google Scholar]
- 48.Chen M, von Mikecz A. Specific inhibition of rRNA transcription and dynamic relocation of fibrillarin induced by mercury. Exp Cell Res. 2000;259:225–38. doi: 10.1006/excr.2000.4923. [DOI] [PubMed] [Google Scholar]
- 49.Chen M, Dittmann A, Kuhn A, Ruzicka T, von Mikecz A. Recruitment of topoisomerase I (Scl-70) to nucleoplasmic proteasomes in response to xenobiotics suggests a role for altered antigen processing in scleroderma. Arthritis Rheum. 2005;52:877–84. doi: 10.1002/art.20962. [DOI] [PubMed] [Google Scholar]
- 50.Harada O, Hoe R, Lin J, Thike AA, Jara-Lazaro AR, Petersson F, Tan PH. Intranuclear inclusions in epithelial cells of benign proliferative breast lesions. J Clin Pathol. 2011;64:776–80. doi: 10.1136/jclinpath-2011-200094. [DOI] [PubMed] [Google Scholar]
- 51.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid--from bacteria to humans. Trends Biochem Sci. 2007;32:217–24. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 52.Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KP, Simon R, Schubert D, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–32. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faux NG, Bottomley SP, Lesk AM, Irving JA, Morrison JR, de la Banda MG, Whisstock JC. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res. 2005;15:537–51. doi: 10.1101/gr.3096505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He Y, Fang J, Taatjes DJ, Nogales E. Structural visualization of key steps in human transcription initiation. Nature. 2013;495:481–6. doi: 10.1038/nature11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gerber HP, Seipel K, Georgiev O, Höfferer M, Hug M, Rusconi S, Schaffner W. Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science. 1994;263:808–11. doi: 10.1126/science.8303297. [DOI] [PubMed] [Google Scholar]
- 56.Landrum E, Wetzel R. Biophysical underpinnings of the repeat length dependence of polyglutamine amyloid formation. J Biol Chem. 2014;289:10254–60. doi: 10.1074/jbc.C114.552943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 58.Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–22. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet. 2005;39:153–71. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- 60.Klein FA, Pastore A, Masino L, Zeder-Lutz G, Nierengarten H, Oulad-Abdelghani M, Altschuh D, Mandel JL, Trottier Y. Pathogenic and non-pathogenic polyglutamine tracts have similar structural properties: towards a length-dependent toxicity gradient. J Mol Biol. 2007;371:235–44. doi: 10.1016/j.jmb.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 61.La Spada AR, Taylor JP. Polyglutamines placed into context. Neuron. 2003;38:681–4. doi: 10.1016/S0896-6273(03)00328-3. [DOI] [PubMed] [Google Scholar]
- 62.Bhattacharyya A, Thakur AK, Chellgren VM, Thiagarajan G, Williams AD, Chellgren BW, Creamer TP, Wetzel R. Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol. 2006;355:524–35. doi: 10.1016/j.jmb.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 63.Darnell G, Orgel JP, Pahl R, Meredith SC. Flanking polyproline sequences inhibit beta-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J Mol Biol. 2007;374:688–704. doi: 10.1016/j.jmb.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 64.Ellis RJ, Minton AP. Protein aggregation in crowded environments. Biol Chem. 2006;387:485–97. doi: 10.1515/BC.2006.064. [DOI] [PubMed] [Google Scholar]
- 65.Zhou HX, Rivas G, Minton AP. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–97. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hemmerich P, Schmiedeberg L, Diekmann S. Dynamic as well as stable protein interactions contribute to genome function and maintenance. Chromosome Res. 2011;19:131–51. doi: 10.1007/s10577-010-9161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hancock R. The crowded nucleus. Int Rev Cell Mol Biol. 2014;307:15–26. doi: 10.1016/B978-0-12-800046-5.00002-3. [DOI] [PubMed] [Google Scholar]
- 68.von Mikecz A. The nuclear ubiquitin-proteasome system. J Cell Sci. 2006;119:1977–84. doi: 10.1242/jcs.03008. [DOI] [PubMed] [Google Scholar]
- 69.Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, Hayer-Hartl M, Hartl FU. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013;154:134–45. doi: 10.1016/j.cell.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 70.Uversky VN. Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: another illustration of the D(2) concept. Expert Rev Proteomics. 2010;7:543–64. doi: 10.1586/epr.10.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–58. doi: 10.1016/S0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 72.Huang CC, Faber PW, Persichetti F, Mittal V, Vonsattel JP, MacDonald ME, Gusella JF. Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat Cell Mol Genet. 1998;24:217–33. doi: 10.1023/B:SCAM.0000007124.19463.e5. [DOI] [PubMed] [Google Scholar]
- 73.Palazzolo I, Nedelsky NB, Askew CE, Harmison GG, Kasantsev AG, Taylor JP, Fischbeck KH, Pennuto M. B2 attenuates polyglutamine-expanded androgen receptor toxicity in cell and fly models of spinal and bulbar muscular atrophy. J Neurosci Res. 2010;88:2207–16. doi: 10.1002/jnr.22389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Skinner PJ, Koshy BT, Cummings CJ, Klement IA, Helin K, Servadio A, Zoghbi HY, Orr HT. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature. 1997;389:971–4. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- 75.Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine protein aggregates are dynamic. Nat Cell Biol. 2002;4:826–31. doi: 10.1038/ncb863. [DOI] [PubMed] [Google Scholar]
- 76.Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol. 1998;143:1457–70. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–8. doi: 10.1093/hmg/10.14.1441. [DOI] [PubMed] [Google Scholar]
- 78.Bichelmeier U, Schmidt T, Hübener J, Boy J, Rüttiger L, Häbig K, Poths S, Bonin M, Knipper M, Schmidt WJ, et al. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence. J Neurosci. 2007;27:7418–28. doi: 10.1523/JNEUROSCI.4540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seidel K, den Dunnen WF, Schultz C, Paulson H, Frank S, de Vos RA, Brunt ER, Deller T, Kampinga HH, Rüb U. Axonal inclusions in spinocerebellar ataxia type 3. Acta Neuropathol. 2010;120:449–60. doi: 10.1007/s00401-010-0717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamada M, Wood JD, Shimohata T, Hayashi S, Tsuji S, Ross CA, Takahashi H. Widespread occurrence of intranuclear atrophin-1 accumulation in the central nervous system neurons of patients with dentatorubral-pallidoluysian atrophy. Ann Neurol. 2001;49:14–23. doi: 10.1002/1531-8249(200101)49:1<14::AID-ANA5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 81.Janer A, Werner A, Takahashi-Fujigasaki J, Daret A, Fujigasaki H, Takada K, Duyckaerts C, Brice A, Dejean A, Sittler A. SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum Mol Genet. 2010;19:181–95. doi: 10.1093/hmg/ddp478. [DOI] [PubMed] [Google Scholar]
- 82.Hazeki N, Tsukamoto T, Yazawa I, Koyama M, Hattori S, Someki I, Iwatsubo T, Nakamura K, Goto J, Kanazawa I. Ultrastructure of nuclear aggregates formed by expressing an expanded polyglutamine. Biochem Biophys Res Commun. 2002;294:429–40. doi: 10.1016/S0006-291X(02)00498-9. [DOI] [PubMed] [Google Scholar]