

Abstract
RNAs play pivotal roles in the cell, ranging from catalysis (e.g., RNase P), acting as adaptor molecule (tRNA) to regulation (e.g., riboswitches). Precise understanding of its three-dimensional structures has given unprecedented insight into the molecular basis for all of these processes. Nevertheless, structural studies on RNA are still limited by the very special nature of this polymer. The most common methods for the determination of 3D RNA structures are NMR and X-ray crystallography. Both methods have their own set of requirements and give different amounts of information about the target RNA. For structural studies, the major bottleneck is usually obtaining large amounts of highly pure and homogeneously folded RNA. Especially for X-ray crystallography it can be necessary to screen a large number of variants to obtain well-ordered single crystals. In this mini-review we give an overview about strategies for the design, in vitro production, and purification of RNA for structural studies.
Keywords: in vitro transcription, structural biology, X-ray, NMR, ribozymes
Introduction
Structural studies on RNA molecules began in the late 1960s with the work on tRNA. Structures of the yeast phenylalanine-tRNA gave for the first time a detailed atomic insight into the complex folding of RNA.1-3
Interest in RNA structures was renewed when the first catalytically active RNAs were discovered.4,5 However, it required two decades of investigation by several groups to gather structural information on catalytic RNAs.6-9 In the 1990s, (DNA and) RNA-molecules (termed aptamers) were designed by systematic evolution approaches (SELEX) for highly specific binding to proteins and small molecules.10,11 NMR and X-ray structures of these aptamers in complex with their respective target molecule revealed that RNA and DNA can adopt sophisticated folds to bind a far wider range of molecules than what was known at that time (reviewed in ref. 12). RNAs with similar function were later discovered to occur naturally. These RNAs, termed riboswitches, are usually located in the 5′-UTR of mRNAs. They couple the binding of a small molecule to the transcription or translation of the mRNA, consequently inducing or preventing gene expression.13-16 In addition, riboswitches have been identified that bind to tRNAs or allow the cells to react rapidly to temperature changes (reviewed in refs. 17 and 18). Recent progress in structural biology has led to a dramatic increase in structures deposited in the protein data bank (PDB). The PDB (a database for structures of macromolecules) contained at the time of writing this review 96 417 experimentally determined structures of macromolecules. However, only a small fraction of those are pure RNA structures (1061) determined either by NMR (548) or X-ray crystallography (513), owing to the inherent difficulties when working with RNA. In this review we will discuss state of the art techniques for the design, production, and purification of RNA for use in structural studies.
RNA Design and Production
Target RNAs that contain a large number of modified residues (e.g., tRNAs) or are part of a complex protein/RNA assembly (ribosomes, RNase P) and are not easily reconstituted in vitro are best purified from their native sources. The required methods as well as RNA synthesis by solid-phase chemical synthesis will not be discussed here.
Generally, handling RNA requires some extra precautions to ensure that the sample is kept intact. The additional 2’-OH group at the ribose moiety can catalyze cleavage of the backbone at basic pH, and thus, makes RNA a much less stable polymer than DNA. In addition, RNase contaminations are a big concern as they are quite stable and difficult to remove.
The size of the target RNA as well as its sequence directly dictate the optimal course of action and design. In general, in vitro production of RNA is performed by a phage RNA polymerase using a linear DNA template and nucleoside triphosphates. This review will focus on strategies based on T7 RNA polymerase (T7 RNAP), as it is the most commonly used enzyme. It has high processivity, is highly specific to the T7 RNAP promoter, and is easily produced recombinantly and well characterized.19 T7 RNAP functions as a monomer with a molecular weight of 98 kDa and contains the entire catalytic activity for initiation, elongation, and termination of the transcription process. There is no need for the assembly of a holo-enzyme, making it very easy to handle. Transcription starts at the +1 base after the T7 RNAP promoter and proceeds till a T7 terminator stem loop is encountered or the template ends and the T7 RNAP falls off (run-off transcription). The latter is usually preferred, as insertion of a restriction enzyme site at the 3′-end allows stopping the transcription at a defined place and avoids the synthesis of extraneous RNA. The starting sequence of the target RNA should be purine-rich, with the strongest transcription observed if three consecutive guanosines reside after the T7 RNAP promoter.20 A mutant variant of the T7 RNAP (P266L) has been described that relaxes these requirements for the starting codons and may be an option if the first nucleotide has to be a pyrimidine base.21
T7 RNAP is known to add non-templated nucleotides at the 5′- and 3′-end.22 Addition of non-templated nucleotides at the 3′-end (N+1, N+2, etc. products) is observed under almost all conditions. Since sample homogeneity is very important for NMR and crystallography, special care needs to be taken to remove N+1, N+2, etc. products to obtain > 95% pure samples. 5′-heterogeneity can be reduced with the use of an alternative promoter sequence or the addition of a self-cleaving hammerhead ribozyme upstream of the target RNA (Fig. 1A).23 Similarly a self-cleaving ribozyme may be fused to the 3′-end to reduce 3′-heterogeneity (Fig. 1B). Instead of fusing the ribozymes to the target RNA (cis) there is also the option to only add the recognition sites of the ribozyme at the 3′-end of the RNA. The separately prepared and purified ribozyme is then added to the transcription reaction in trans.24

Figure 1. Design of RNA–ribozyme constructs. The target RNA (green), with optional up- and downstream ribozymes, is cloned with the T7 RNA promoter (T7P) between EcoRI and XbaI sites of a high-copy plasmid (A). After linearization with XbaI, transcription by the T7 RNAP begins at +1 site (marked with an arrow). The synthesized RNA (highlighted in green) includes the hammerhead ribozyme (highlighted in blue) fused at the 3′-end; cleavage occurs after the GUC triplet (bold) indicated with a red arrowhead (B). The hammerhead ribozyme is easily adapted and fused to any RNA either at the 5′- or 3′-end (C). Alternatively, the HDV-ribozyme (D) or the recognition stem-loop of the VS-ribozyme may be fused to the 3′-end (E). Cleavage sites are marked with red arrowheads. In the case of the VS recognition stem-loop, cleavage occurs by addition of the VS-ribozyme in trans.
While the hammerhead ribozyme is quite small (50 nt) and easily adapted to the target RNA, it requires the addition of two specific nucleotides at the 3′-end that remain after cleavage (Fig. 1C). If low cleavage efficiency is observed or the addition of two nucleotides is unfavorable, then the HδV- or VS-ribozyme may be used (Fig. 1D and E).25 The HδV-ribozyme is only slightly bigger than the hammerhead ribozyme but does not require adaptation to the target RNA sequence (Fig. 1D). It will cleave efficiently after any base.26 The ribozyme construct can be reused if a restriction enzyme site has been inserted into its sequence.27,28 The Varkud satellite (VS) ribozyme can cleave efficiently as long as the base preceding the cleavage site is not a C.29 The small stem-loop that is recognized by the VS ribozyme is fused to the 3′-end of the target RNA (Fig. 1E) and the VS ribozyme is added in trans. Fusion of the VS-ribozyme to the target RNA is also possible, although, with a length of approximately 150 nt, it is considerably bigger than the HH- and HδV-ribozyme.
Instead of a linearized plasmid, a PCR product can also be used as a template. Such an approach is particularly useful to optimize yield, as multiple starting sequences can be quickly screened in parallel by use of different forward primers. In such a case, incorporation of two bases with a 2’-O-methyl-modification at the 3′-end can cause the transcription to stop without addition of non-templated nucleotides.30 Since no ribozyme is required, the in vitro transcription reaction will only yield the target RNA, thus making a more efficient use of rNTPs. This setup can increase yield and reduce costs especially when 13C/15N-labeled rNTPs are used for NMR studies. Once the sequence design is complete it can be cloned into a high-copy plasmid such as the pUC-series.31,32 The restriction enzyme chosen at the 3′-end should be robust, relatively inexpensive (as large amounts of DNA will need to be digested), heat-inactivatable, and should not leave a 3′-overhang after cleavage as this may inhibit the transcription.
Large-scale plasmid preparations can be performed either with commercial kits or by using a standard anion-exchange chromatography with a resin such as Q-Sepharose.33
After linearization of plasmids with a restriction enzyme, the reaction should be further processed in order to remove the restriction enzyme and to ensure that no RNase is carried over into the in vitro transcription. RNases in turn can be effectively removed either by performing a chloroform-phenol extraction or by treating the reaction with Proteinase K. We usually opt for the Proteinase K treatment only as it requires less hands-on time and avoids the handling of chloroform and phenol. Proteinase K is simply added to the restriction digest reaction to a final concentration of 0.05–0.1 mg/ml and the reaction mixture is incubated for 60 min at 50–55 °C. Proteinase K is inactivated by incubating at 95 °C for 10 min. This restriction digest can then be used for RNA production without any further purification. The general flow of sequence design to transcription and subsequent purification is summarized in Figure 2. It is recommended to perform 50 µl-test-reactions to optimize the concentration of MgCl2, rNTPs, and template to maximize yield and also cleavage efficiency before performing large-scale reactions. In vitro transcription is performed at 37 °C for 2–4 h. Transcription activity of the T7 RNAP is indicated by the production of inorganic pyrophosphate forming a white precipitate if concentration reaches a certain threshold. Addition of phosphatase may improve yield by removing the pyrophosphate, which can inhibit T7 RNAP. Low cleavage efficiency of the hammerhead ribozyme can be increased by incubation at 40–55 °C for 30–60 min. It has to be kept in mind that most RNAs will be denatured to a certain degree at this temperature. This is suitable if further purification will be done under denaturing conditions and a refolding protocol is established.
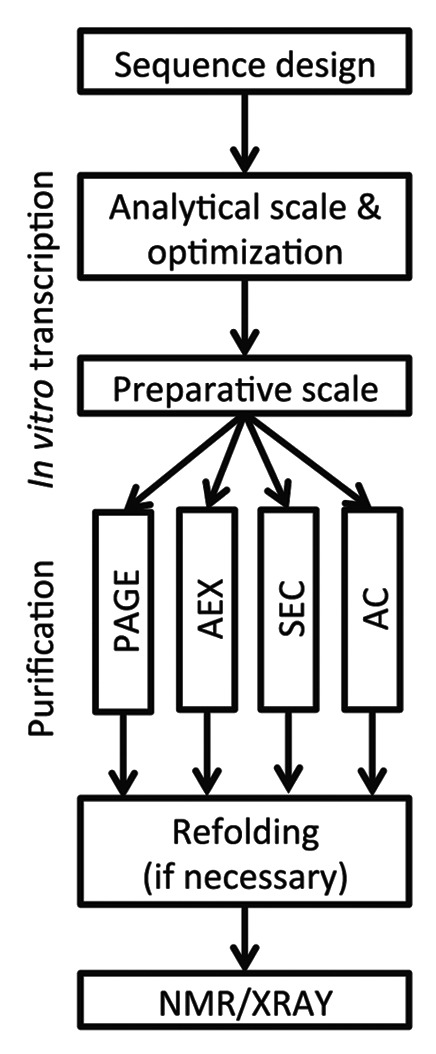
Figure 2. General flow from sequence design to NMR or X-ray structure analysis.
Ribozyme processed RNA will contain a cyclic 2’-3′-phosphate at the 3′-end. If the same RNA is used for labeling reactions the cyclic phosphate may be post-transcriptionally removed by treatment with maleic acid and calf intestine alkaline phosphatase (CIAP).24
Sample Purification
Purification of RNA can be done using several methods. In addition to the approach of excising bands of the target RNA from large preparative urea-PAGE gels, alternative methods have been developed. Some of these methods are performed under native conditions using size exclusion chromatography (SEC), anion exchange chromatography (AEX), or affinity chromatography (AC).
PAGE
The classic way is to separate target RNA from ribozymes or abortive transcripts, proteins, and nucleotides using large urea-PAGE gels with a polyacrylamide content of 4–20% depending on the RNA size.34 The target RNA band is detected via UV-shadowing and excised. Extraction of the RNA from the gel matrix is either done by electroelution or by the crush and soak method.35 Electroelution is a second electrophoresis step that allows the elution of the RNA from the excised band into a dialysis bag.36 Although commercially available equipment is relatively expensive, the advantage of this method is the very high recovery rate and small elution volume. Alternatively, the crush and soak method can be used. The excised gel piece is crushed and sliced into a fine slurry and soaked in elution buffer. Elution of the RNA, which occurs by simple diffusion, is usually performed overnight or even longer times. Even though the crush and soak method does not require expensive special hardware it suffers from low recovery rates and long incubation times.
Purification via preparative PAGE is still a very laborious method but it provides very high resolution, purity, and is very robust.
SEC
Size exclusion chromatography can also bypass the denaturing and refolding step and allows a very gentle purification of the RNA. Following in vitro transcription, the reaction is quenched with 50 mM EDTA. Optionally, proteins and non-incorporated rNTPs can be removed by phenol-chloroform extraction and buffer exchanged before being subjected to size exclusion chromatography.37 While properly folded RNA behaves similar to proteins in SEC, there is a noticeable difference in elution volume to similar sized proteins. The work of Kim et al. contains the elution volumes of RNAs with different length on standard SEC columns such as Superdex 75 and 200 10/300.37 A prerequisite for successful purification is that the in vitro transcription reaction will produce mostly a single and properly folded species. This was successfully shown for the purification of a truncated tRNA and subsequent crystallization with the general tRNA export factor exportin-t.38
AEX
In general, the achievable resolution in anion-exchange chromatography strongly depends on the amount of secondary structures, and therefore, accessible negative charge, making it a less favorable method for large RNAs.39 For small RNAs (< 30 nt), anion exchange can be performed under native conditions and gives sufficient resolution.40 Larger RNAs however tend to elute in multiple peaks and also at very high salt concentrations leading to large losses.40 Keeping the column at a constant temperature by a column heater is recommended to maintain adequate resolution. Weak ion exchange material such as DEAE has also successfully been used to purify RNA suitable for structural studies.41
RNA purification by SEC or AEX should be performed on an HPLC system dedicated to RNA work only, to prevent RNase-contamination. Both methods allow the purification of milligram amounts of RNA within one day following transcription and give sufficient purity required for NMR or crystallographic studies.
AC
Recently, several methods for affinity chromatographic purification of RNA for structural studies have been reported.28,42,43 Target RNA is fused to a ribozyme and an affinity sequence. The affinity sequence is bound by a specific protein and allows the immobilization of the transcribed target RNA-ribozyme fusion to a resin. After extensive washing, the target RNA is eluted by triggering the cleavage by a special ribozyme. The ribozymes employed here are triggered to cleave by addition of a small molecule, which they either require natively (glmS) or because they have been modified (HδV C75U variant) to do so. The drawback of these methods is that a substantial amount of the synthesized RNA is not the target RNA and that auxiliary proteins need to be purified. Nevertheless, for some of the RNAs in our lab this method proved quite useful and straightforward, especially since the RNA is not denatured.
Depending on the purification method, the RNA might require a refolding step before it can be used for structural and biochemical studies. A full review of refolding protocols is beyond the scope of this mini-review, but briefly the RNA solution is diluted into a refolding buffer, heated to 95 °C, and allowed to cool to room temperature either at a slow or fast rate (seconds to hours). The composition of the refolding buffer as well as the cooling protocol for each individual RNA has to be established by trial and error. This topic has been well covered in the literature.44-46 Properly refolded RNA should result in one distinct band on a native gel or a single peak on size exclusion chromatography.
Purified RNA can be concentrated by centrifugal concentrators or by Ethanol-precipitation and quantified by measuring the absorbance at 260 nm. Moreover, it is recommended to also measure absorbance at 280 nm or even perform a spectrum scan from 200 to 320 nm to detect possible contaminants such as Phenol. A 260/280 ratio, which is lower than 2.0, indicates significant contamination with protein or DNA.
Conclusions
Irrespective of the chosen purification method, a way to assess the folding state and integrity of the sample after purification is indispensable. Is it still possible to form a complex with a known binding partner? Do different refolding protocols give different migration behavior on native PAGE or SEC? If there is a catalytic activity—how well is it retained over the course of purification? It is better to think about these questions early on before investing time and money in the preparation of supposedly well-folded RNA that has however lost its biological activity.
RNA structural biology has come a long way since the first crystal structures of tRNAs. Methods to produce and purify large amounts of RNA of different sizes are now very sophisticated and robust, which has resulted in an average of 50 pure RNA structures being deposited per year in the PDB since 2000. Problems still arise when handling larger RNAs for NMR or obtaining and phasing well-diffracting crystals for X-ray analysis. Larger RNAs can be tackled by NMR, using nucleotide selective or segmental labeling (reviewed in refs. 47 and 48). Obtaining well diffracting crystals of RNA still remains a bottleneck and different techniques have been used to address this issue. Insertion of a tetra-loop/tetra-loop-receptor-pair can promote crystal packing.49,50 Another strategy is to incorporate a loop, which is specifically recognized by a protein and co-crystallize with that protein.51-53 Co-crystallization with a helper-protein can also provide initial phases for structure determination. If experimental phases are essential, then insertion of heavy-atom binding sequences at an early stage of the project is recommended.54 In contrast to the structural genomics approach for proteins there are, at the moment, no systematic and coordinated efforts underway targeting RNA molecules. For crystallographic studies, RNA requires a much more focused approach and is less amenable to an automated high-throughput procedure. NMR studies while potentially more straightforward, require the use of expensive labeled nucleotides making it a less appropriate choice for a high-throughput project. Nevertheless NMR and X-ray crystallography can accomplish structure determination of even larger RNAs such as the structure of the core encapsidation signal of the Moloney murine leukemia virus (32 kDa, NMR) or the self-spliced group II intron (133 kDa, X-ray).55,56
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Achim Dickmanns and Thomas Monecke for critical reading of the manuscript, and the DFG SPP1258 for funding.
Glossary
Abbreviations:
- NMR
nuclear magnetic resonance
- SELEX
systematic evolution of ligands by exponential enrichment
- PDB
protein data bank
- rNTP
ribonucleoside triphosphate
- SEC
size exclusion chromatography
- AEX
anion-exchange chromatography
- AC
affinity chromatography
- UTR
untranslated region, RNAP, RNA polymerase
- PAGE
poly-acrylamide gel electrophoresis
- CIAP
calf intestine alkaline phosphatase
- HH
hammer-head
- HδV
hepatitis δ-virus
- VS
Varkud satellite
References
- 1.Kim SH, Quigley GJ, Suddath FL, McPherson A, Sneden D, Kim JJ, Weinzierl J, Rich A. Three-dimensional structure of yeast phenylalanine transfer RNA: folding of the polynucleotide chain. Science. 1973;179:285–8. doi: 10.1126/science.179.4070.285. [DOI] [PubMed] [Google Scholar]
- 2.Suddath FL, Quigley GJ, McPherson A, Sneden D, Kim JJ, Kim SH, Rich A. Three-dimensional structure of yeast phenylalanine transfer RNA at 3.0angstroms resolution. Nature. 1974;248:20–4. doi: 10.1038/248020a0. [DOI] [PubMed] [Google Scholar]
- 3.Robertus JD, Ladner JE, Finch JT, Rhodes D, Brown RS, Clark BF, Klug A. Structure of yeast phenylalanine tRNA at 3 A resolution. Nature. 1974;250:546–51. doi: 10.1038/250546a0. [DOI] [PubMed] [Google Scholar]
- 4.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–57. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 5.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–57. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 6.Golden BL, Gooding AR, Podell ER, Cech TR. A preorganized active site in the crystal structure of the Tetrahymena ribozyme. Science. 1998;282:259–64. doi: 10.1126/science.282.5387.259. [DOI] [PubMed] [Google Scholar]
- 7.Guo F, Gooding AR, Cech TR. Structure of the Tetrahymena ribozyme: base triple sandwich and metal ion at the active site. Mol Cell. 2004;16:351–62. doi: 10.1016/j.molcel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Adams PL, Stahley MR, Kosek AB, Wang J, Strobel SA. Crystal structure of a self-splicing group I intron with both exons. Nature. 2004;430:45–50. doi: 10.1038/nature02642. [DOI] [PubMed] [Google Scholar]
- 9.Golden BL, Kim H, Chase E. Crystal structure of a phage Twort group I ribozyme-product complex. Nat Struct Mol Biol. 2005;12:82–9. doi: 10.1038/nsmb868. [DOI] [PubMed] [Google Scholar]
- 10.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–22. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 11.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–10. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 12.Patel DJ, Suri AK. Structure, recognition and discrimination in RNA aptamer complexes with cofactors, amino acids, drugs and aminoglycoside antibiotics. J Biotechnol. 2000;74:39–60. doi: 10.1016/s1389-0352(99)00003-3. [DOI] [PubMed] [Google Scholar]
- 13.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic control by a metabolite binding mRNA. Chem Biol. 2002;9:1043. doi: 10.1016/S1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 14.Mironov AS, Gusarov I, Rafikov R, Lopez LE, Shatalin K, Kreneva RA, Perumov DA, Nudler E. Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell. 2002;111:747–56. doi: 10.1016/S0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 15.Winkler W, Nahvi A, Breaker RR. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature. 2002;419:952–6. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- 16.Winkler WC, Cohen-Chalamish S, Breaker RR. An mRNA structure that controls gene expression by binding FMN. Proc Natl Acad Sci U S A. 2002;99:15908–13. doi: 10.1073/pnas.212628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green NJ, Grundy FJ, Henkin TM. The T box mechanism: tRNA as a regulatory molecule. FEBS Lett. 2010;584:318–24. doi: 10.1016/j.febslet.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kortmann J, Narberhaus F. Bacterial RNA thermometers: molecular zippers and switches. Nat Rev Microbiol. 2012;10:255–65. doi: 10.1038/nrmicro2730. [DOI] [PubMed] [Google Scholar]
- 19.Ellinger T, Ehricht R. Single-step purification of T7 RNA polymerase with a 6-histidine tag. Biotechniques. 1998;24:718–20. doi: 10.2144/98245bm03. [DOI] [PubMed] [Google Scholar]
- 20.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–98. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guillerez J, Lopez PJ, Proux F, Launay H, Dreyfus M. A mutation in T7 RNA polymerase that facilitates promoter clearance. Proc Natl Acad Sci U S A. 2005;102:5958–63. doi: 10.1073/pnas.0407141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pleiss JA, Derrick ML, Uhlenbeck OC. T7 RNA polymerase produces 5′ end heterogeneity during in vitro transcription from certain templates. RNA. 1998;4:1313–7. doi: 10.1017/S135583829800106X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coleman TM, Wang G, Huang F. Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 phi 2.5 promoter. Nucleic Acids Res. 2004;32:e14. doi: 10.1093/nar/gnh007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wichlacz A, Legiewicz M, Ciesiołka J. Generating in vitro transcripts with homogenous 3′ ends using trans-acting antigenomic delta ribozyme. Nucleic Acids Res. 2004;32:e39. doi: 10.1093/nar/gnh037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferré-D’Amaré AR, Doudna JA. Use of cis- and trans-ribozymes to remove 5′ and 3′ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic Acids Res. 1996;24:977–8. doi: 10.1093/nar/24.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferré-D’Amaré AR, Zhou K, Doudna JA. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395:567–74. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 27.Walker SC, Avis JM, Conn GL. General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res. 2003;31:e82. doi: 10.1093/nar/gng082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kieft JS, Batey RT. A general method for rapid and nondenaturing purification of RNAs. RNA. 2004;10:988–95. doi: 10.1261/rna.7040604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo HC, Collins RA. Efficient trans-cleavage of a stem-loop RNA substrate by a ribozyme derived from neurospora VS RNA. EMBO J. 1995;14:368–76. doi: 10.1002/j.1460-2075.1995.tb07011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kao C, Zheng M, Rüdisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA. 1999;5:1268–72. doi: 10.1017/S1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vieira J, Messing J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 1982;19:259–68. doi: 10.1016/0378-1119(82)90015-4. [DOI] [PubMed] [Google Scholar]
- 32.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–19. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 33.Chandra G, Patel P, Kost TA, Gray JG. Large-scale purification of plasmid DNA by fast protein liquid chromatography using a Hi-Load Q Sepharose column. Anal Biochem. 1992;203:169–72. doi: 10.1016/0003-2697(92)90060-K. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning : a laboratory manual. 2nd ed. New York: Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press, 1989.; 1989. [Google Scholar]
- 35.Barfod ET, Cech TR. Deletion of nonconserved helices near the 3′ end of the rRNA intron of Tetrahymena thermophila alters self-splicing but not core catalytic activity. Genes Dev. 1988;2:652–63. doi: 10.1101/gad.2.6.652. [DOI] [PubMed] [Google Scholar]
- 36.Petrov A, Wu T, Puglisi EV, Puglisi JD. RNA purification by preparative polyacrylamide gel electrophoresis. Methods Enzymol. 2013;530:315–30. doi: 10.1016/B978-0-12-420037-1.00017-8. [DOI] [PubMed] [Google Scholar]
- 37.Kim I, McKenna SA, Viani Puglisi E, Puglisi JD. Rapid purification of RNAs using fast performance liquid chromatography (FPLC) RNA. 2007;13:289–94. doi: 10.1261/rna.342607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cook AG, Fukuhara N, Jinek M, Conti E. Structures of the tRNA export factor in the nuclear and cytosolic states. Nature. 2009;461:60–5. doi: 10.1038/nature08394. [DOI] [PubMed] [Google Scholar]
- 39.Anderson AC, Scaringe SA, Earp BE, Frederick CA. HPLC purification of RNA for crystallography and NMR. RNA. 1996;2:110–7. [PMC free article] [PubMed] [Google Scholar]
- 40.Shields TP, Mollova E, Ste Marie L, Hansen MR, Pardi A. High-performance liquid chromatography purification of homogenous-length RNA produced by trans cleavage with a hammerhead ribozyme. RNA. 1999;5:1259–67. doi: 10.1017/S1355838299990945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Easton LE, Shibata Y, Lukavsky PJ. Rapid, nondenaturing RNA purification using weak anion-exchange fast performance liquid chromatography. RNA. 2010;16:647–53. doi: 10.1261/rna.1862210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batey RT, Kieft JS. Improved native affinity purification of RNA. RNA. 2007;13:1384–9. doi: 10.1261/rna.528007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pereira MJB, Behera V, Walter NG. Nondenaturing purification of co-transcriptionally folded RNA avoids common folding heterogeneity. PLoS One. 2010;5:e12953. doi: 10.1371/journal.pone.0012953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doudna JA. Preparation of homogeneous ribozyme RNA for crystallization. Methods Mol Biol. 1997;74:365–70. doi: 10.1385/0-89603-389-9:365. [DOI] [PubMed] [Google Scholar]
- 45.Ke A, Doudna JA. Crystallization of RNA and RNA-protein complexes. Methods. 2004;34:408–14. doi: 10.1016/j.ymeth.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 46.Baird NJ, Westhof E, Qin H, Pan T, Sosnick TR. Structure of a folding intermediate reveals the interplay between core and peripheral elements in RNA folding. J Mol Biol. 2005;352:712–22. doi: 10.1016/j.jmb.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 47.Lu K, Miyazaki Y, Summers MF. Isotope labeling strategies for NMR studies of RNA. J Biomol NMR. 2010;46:113–25. doi: 10.1007/s10858-009-9375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duss O, Lukavsky PJ, Allain FH-T. Isotope labeling and segmental labeling of larger RNAs for NMR structural studies. Adv Exp Med Biol. 2012;992:121–44. doi: 10.1007/978-94-007-4954-2_7. [DOI] [PubMed] [Google Scholar]
- 49.Ferré-D’Amaré AR, Zhou K, Doudna JA. A general module for RNA crystallization. J Mol Biol. 1998;279:621–31. doi: 10.1006/jmbi.1998.1789. [DOI] [PubMed] [Google Scholar]
- 50.Coonrod LA, Lohman JR, Berglund JA. Utilizing the GAAA tetraloop/receptor to facilitate crystal packing and determination of the structure of a CUG RNA helix. Biochemistry. 2012;51:8330–7. doi: 10.1021/bi300829w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferré-D’Amaré AR, Doudna JA. Crystallization and structure determination of a hepatitis delta virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J Mol Biol. 2000;295:541–56. doi: 10.1006/jmbi.1999.3398. [DOI] [PubMed] [Google Scholar]
- 52.Smith KD, Lipchock SV, Ames TD, Wang J, Breaker RR, Strobel SA. Structural basis of ligand binding by a c-di-GMP riboswitch. Nat Struct Mol Biol. 2009;16:1218–23. doi: 10.1038/nsmb.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koldobskaya Y, Duguid EM, Shechner DM, Suslov NB, Ye J, Sidhu SS, Bartel DP, Koide S, Kossiakoff AA, Piccirilli JA. A portable RNA sequence whose recognition by a synthetic antibody facilitates structural determination. Nat Struct Mol Biol. 2011;18:100–6. doi: 10.1038/nsmb.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keel AY, Rambo RP, Batey RT, Kieft JS. A general strategy to solve the phase problem in RNA crystallography. Structure. 2007;15:761–72. doi: 10.1016/j.str.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.D’Souza V, Dey A, Habib D, Summers MF. NMR structure of the 101-nucleotide core encapsidation signal of the Moloney murine leukemia virus. J Mol Biol. 2004;337:427–42. doi: 10.1016/j.jmb.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 56.Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]