

Abstract
While aberrant cell proliferation and differentiation may contribute to epileptogenesis, the mechanisms linking an initial epileptic insult to subsequent changes in cell fate remain elusive. Using both mouse and human iPSC-derived neural progenitor/stem cells (NPSCs), we found that a combined transient muscarinic and mGluR1 stimulation inhibited overall neurogenesis but enhanced NPSC differentiation into immature GABAergic cells. If treated NPSCs were further passaged, they retained a nearly identical phenotype upon differentiation. A similar profusion of immature GABAergic cells was seen in rats with pilocarpine-induced chronic epilepsy. Furthermore, live cell imaging revealed abnormal de-synchrony of Ca++ transients and altered gap junction intercellular communication following combined muscarinic/glutamatergic stimulation, which was associated with either acute site-specific dephosphorylation of connexin 43 or a long-term enhancement of its degradation. Therefore, epileptogenic stimuli can trigger acute and persistent changes in cell fate by altering distinct mechanisms that function to maintain appropriate intercellular communication between coupled NPSCs.
Introduction
Seizure incidence and susceptibility are highest in the developing brain, and the occurrence of a protracted seizure during infancy or early childhood is associated with a significantly elevated risk of refractory epilepsy in adulthood (Rakhade and Jensen, 2009). The relationship between an initial epileptic event and refractory seizures is likely multifactorial and involves multiple processes including, but not limited to, increased neurogenesis, epigenetic changes, and transcriptional effects that ultimately alter the synaptic connectivity and architecture of the developing brain (Rakhade and Jensen, 2009).
Studies in animal models of temporal lobe epilepsy (TLE) have demonstrated increased neurogenesis in the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) (Parent et al., 1997; Sankar et al., 2000). In addition, while many studies have noted a loss of GABAergic interneurons from the hilus of chronic TLE brains, others have shown an increase in GABAergic synapses within DG cells as well as increased expression of the GABAergic markers GAD67 and GAD65 (Esclapez and Houser, 1999; Sun et al., 2014; Thind et al., 2010). This seeming paradox may be partly attributable to either a compensatory response to over excitation or to the persistence of GABA(A) receptor-induced depolarization, normally a feature of GABA action early in neurodevelopment (Young et al., 2012). In fact, antagonizing GABA has been shown to prevent long-lasting seizures in hippocampal slices from immature brain, but has the opposite effect in the mature brain (Khalilov et al., 2005). Along the same lines, mRNA from surgical specimens of patients with chronic refractory TLE demonstrate significantly increased levels of NKCC1, the chloride channel associated with depolarizing GABAergic interneurons (Palma et al., 2006).
While aberrant cell proliferation and differentiation may be a driver of epileptogenesis, the mechanisms linking the initial epileptic insult to changes in cell fate and differentiation remain elusive (Danzer, 2008). Given the rapid proliferation and differentiation of neural progenitor stem cells (NPSCs) throughout the immature brain, pathological effects of an inciting epileptic event on NPSCs have the potential to have a particularly profound effect on the subsequent development of TLE and other refractory epilepsies.
We previously demonstrated that reduced proliferation of murine derived NPSCs is associated with alterations in gap junction intercellular communication (GJIC) through the gap junction protein connexin 43 (Cx43) (Samarasinghe et al., 2011). In particular, de-synchrony in Cx43 gap junction or changes in hemi-channel mediated spontaneous Ca2+ currents are associated with altered NPSC proliferation (Malmersjo et al., 2013; Samarasinghe et al., 2011; Weissman et al., 2004). In addition, inhibition of GJIC and Ca2+ signaling modulates the onset of a GABAergic phenotype during the differentiation of NPSCs (Cheng et al., 2004; Ciccolini et al., 2003). Taken together these data suggest a possible mechanistic link between an initial epileptogenic stimuli and gap junction mediated alterations in NPSC function and differentiation fate.
The aim of this study was to investigate cellular mechanisms of aberrant GABAergic NPSC differentiation under pro-epileptogenic conditions. For this purpose, we utilized an established rat model of TLE that employs high dose pilocarpine exposure at a young age. While the initial seizure in this model is dependent on M1 receptor stimulation, sustained seizure activity is thought to be dependent on subsequent glutamatergic activity triggered by enhanced hippocampal glutamate release (Curia et al., 2008; Smolders et al., 1997). In order to adapt this model to an in-vitro cell culture system, an initial pilocarpine exposure was followed by subsequent glutamate treatment of mouse telencephalon or human inducible pluripotent stem cell (IPSC) derived NPSCs. Of note, we have previously established the utility of in-vitro approaches to examine the impact of pro-epileptogenic stimuli. These published studies revealed altered calcium responses and enhanced oxidative stress in cultured primary hippocampal neurons exposed to pilocarpine (Di Maio et al., 2011; Di Maio et al., 2012).
We presently employed a paradigm of dual pilocarpine and glutamate stimulation to examine both the acute and long-term effects of an epileptogenic stimulus on the neurogenic potential of NPSCs. Uniquely, we were able to take advantage of the ability of NPSC cultures (i.e. neurospheres) to be expanded and passaged over weeks. This allowed us to ascertain whether transient activation of mAChRs and glutamate receptors could lead to alterations in differentiation or calcium signaling following multiple proliferative divisions. We show here that a combined transient activation of mAChR1 and metabotropic glutamate receptor 1 (mGluR1) in NPSCs can trigger changes in gap junction function and intracellular calcium release dynamics which, consistent with in vivo studies, enhance the generation of immature GABAergic cells both acutely and after a refractory period of weeks with many intervening proliferative divisions (Esclapez and Houser, 1999; Palma et al., 2006). Dual pilocarpine and glutamate (Pilo+Glut) stimulation of NPSCs leads to site-specific Cx43 dephosphorylation acutely, but increased Cx43 protein degradation chronically (i.e. following multiple chemoconvulsant free passages). In a chronic pilocarpine-induced TLE model in rats, Cx43 protein levels were decreased in the hippocampal area. Therefore, NPSCs appear to retain a “memory” of mAChR and mGluR1 activation that disrupts gap-junction dependent homeostatic mechanisms and alters their differentiation fate even when differentiation-inducing signals are applied weeks following an epileptogenic stimulus.
In summary, this study provides a mechanistic link between an epileptogenic stimulus of NPSCs and subsequent alterations in stem cell differentiation fate. In doing so we have identified a unique set of potential therapeutic targets that could limit the refractory nature of TLE and other epilepsies.
Results
Muscarinic and glutamatergic activation of NSPCs reduces overall neurogenesis but promotes GABA-ergic differentiation
Neurospheres derived from embryonic (E14.5) mouse telencephalon were used to examine the impact on neurogenesis of an epileptogenic-like stimulus consisting of dual mAChR and glutamatergic activation (Samarasinghe et al., 2011). Coupled proliferating NSPCs in neurospheres maintain the capacity to differentiate into distinct neural and glial lineages upon growth factor withdrawal (Louis et al., 2008). In order to enrich for stem and progenitor cells, neurospheres were passaged twice before use. In addition, to examine whether proliferating NSPCs acquired a “memory” of neurotransmitter exposure, passage 2 (P2) cultures were also subjected to three additional passages over three weeks (i.e. to attain “P5-memory” cultures) prior to differentiation or biochemical analysis.
P2 and P5-memory NPSCs maintain the capacity to differentiate into neurons as revealed by expression of tubulin III (clone TuJ1) detected in immunofluorescence images following 10 days of differentiation (Figs 1a, c, e, g). A fraction (~10% of total) of differentiated neurons derived from both P2 and P5-memory NPSCs are GABAergic as defined by the expression of GAD67 (Figs 1b, c, f, g). To investigate whether an epileptogenic stimulus alters NSPC differentiation, they were exposed to the mAChR agonist pilocarpine (Pilo) for 24 hours (4μM) and glutamate (Glut) (30μM) for the last 2 hours prior to initiating differentiation (Di Maio et al., 2012). This paradigm of pilocarpine followed by glutamate (Pilo+Glut) exposure was chosen to mimic conditions attained in the established in-vivo Pilo model of epileptogenesis, since exposure of animals to a high concentration of Pilo leads to paroxysmal glutamate release and NMDA-mediated hyper-excitability in primary hippocampal cultures (Smolders et al., 1997) (Di Maio et al., 2012).
Fig 1. Increased GABAergic differentiation and decreased neurogenesis in Pilo+Glut exposed NPSCs.

P2 NPSCs were subjected to Phosphate Buffer Saline (PBS; Veh), 4μM pilocarpine (Pilo), 30μM glutamate (Glut) or dual pilocarpine and glutamate (Pilo+Glut) treatment. Cells were incubated for 22 hours with Pilo followed by 2 hours of Glut. P2 NPSCs were then either immediately subjected to 10 days of differentiation (P2) or passaged an additional 3 times and then differentiated (P5-memory). (a–b) Mean cell counts +SEM from differentiated NPSCs expressing Tuj-1 (a) or GAD67 (b); cells treated as indicated while proliferating. (n=3 independent experiments, counts are an average obtained from four random fields per image; 1-way ANOVA, p=0.0032 for Tuj-1; P=0.0019 for GAD67; post hoc Tukey’s Multiple Comparison Test, *= P<0.05). (c) Representative IIF image of differentiated P2 NPSCs. (d) qRT-PCR analysis of GAD67 expression in differentiated P2 NPSCs (n=4; error bars=SEM, 1-way ANOVA, P<0.0001; post hoc Tukey’s Multiple Comparison Test, *=P<0.05). (e–f) Mean cell counts +SEM from P5-memory differentiated NPSCs expressing Tuj-1 (e) or GAD67 (f); cells treated as indicated while proliferating at P2. (n=3; 1-way ANOVA, P=0.0260 for Tuj-1; P=0.0280 for GAD67; post hoc Tukey’s Multiple Comparison Test, *=P<0.05). (g) Representative IIF images of differentiated P5-memory NPSCs. (h) qRT-PCR analysis of GAD67 expression in differentiated P5 memory NPSCs (n=4; error bars=SEM; 1-way ANOVA, P=0.0025; post hoc Tukey’s Multiple Comparison Test, *= P<0.05). (i–j) Mean cell counts +SEM from P5-memory differentiated NPSCs expressing Tuj-1 (i) or GAD67 (j); cells treated as indicated while proliferating at P2 (i.e. 3 μM AIDA or 10μM MK801 30 minutes prior to Glut). (n=4; error bars = SEM, 1-way ANOVA, P<0.0001; post hoc Tukey’s Multiple Comparison Test, *=P<0.05). (k) Representative IIF images of differentiated P5-memory NPSCs.
As shown in Figure 1, a combined (Pilo+Glut) treatment of both P2 and P5-memory NPSCs led to a reduction in overall neurogenesis (i.e. reduced Tuj-1 positive cells; Figs 1a, c, e, g), although the fraction of cells expressing GAD67 was dramatically increased (~4–5 fold) (Figs 1b, f). This was associated with a significant increase in GAD67 mRNA expression as well (Figs 1d, h). Further analysis of P5-memory NPSCs demonstrated that this differentiation effect was dependent on the mGluR1 receptor and not the N-methyl-D-aspartate (NMDA) receptor. As shown in Figures 1i, j, and k, pre-treatment at the P2 stage with the highly selective mGluR1 antagonist, AIDA, but not MK801-mediated NMDA receptor antagonism prevented the enhancement of GABAergic differentiation.
Dual muscarinic/glutamatergic activation promotes the production of immature GABAergic cells in the hippocampus of chronic TLE rats and in-vitro differentiated mouse NPSCs
In the dentate gyrus (DG) of rats subjected to Pilo-induced chronic TLE, the normal pattern of GAD67 immunoreactivity was altered and the number of mature (MAP2 positive) hilar neurons was reduced (Fig 2b). There was also a significant correlation (Pearson index) between the expression of GAD67 and the NPSC marker SOX2 (Figs 2a, b), suggesting that the epileptogenic process stimulates proliferation of immature GABAergic neurons. Consistently, in vitro differentiation studies conducted in NPSCs showed a significant increase of SOX2 expression both in cells derived from P2 and P5-memory NPSCs when exposed to Pilo+Glut treatment at P2 (Figs 2c, d, e, f). Further analysis demonstrates a high degree of overlap between GAD67 and SOX2 staining in differentiated Pilo+Glut treated P2 and P5-memory NPSCs (Fig 2g) not present following vehicle, Pilo, or Glut treatment alone. Real time qRT-PCR analysis for the chloride transporter NKCC1, highly expressed in immature GABAergic neurons and for KCC2, found in more mature GABA-ergic neurons, revealed higher expression of NKCC1 in cells differentiated from Pilo+Glut exposed P2 and P5-memory NSPCs (Fig 2h). These results suggest that combined muscarinic/glutamatergic activation might constitute a key epileptogenic trigger that drives neurogenesis to an abnormal GABAergic lineage that retains an immature phenotype. Furthermore, the Pilo+Glut differentiation phenotype derived from both P2 NPSCs is mGluR1-dependent since transient knockdown of the receptor with siRNA (Supplementary Fig 1) rescues the phenotype (Fig 2i).
Fig 2. Increased immature GABAergic phenotype in the hippocampus of chronic TLE rats and in differentiated Pilo+Glut stimulated NPSCs.

(a) SOX2/GAD67 co-localization, as measured by the Pearson’s Correlation Coefficient, from IIF images (b) of hippocampi of young adult rats (3 weeks old) injected IP with Veh or Pilo (Chronic TLE) (n=6; student’s t-test; *=p<0.05). (c) Mean cell counts +SEM from differentiated P2 NPSCs expressing Sox-2; cells treated as indicated while proliferating (n=3; 1-way ANOVA, P=0.0005; post hoc Tukey’s Multiple Comparison Test, *=P<0.05). (d) Representative IIF images of differentiated P2 NPSCs. (e) Mean cell counts +SEM from differentiated P5-memory NPSCs expressing Sox-2; cells treated as indicated at P2 while proliferating (n=3; 1-way ANOVA, P=0.0073; post hoc Tukey’s Multiple Comparison Test, *= P<0.05). (f) Representative IIF images of differentiated P5-memory NPSCs. (g) SOX2/GAD67 co-localization in P2 and P5-memory NPSCs treated as indicated, as measured by the Pearson’s Correlation Coefficient (inset, representative image from differentiated Pilo+Glut treated P2 and P5-memory NPSCs). (h) qRT-PCR analysis of NKCC1 and KCC2 expression in differentiated P2 and P5-memory NPSCs treated with Pilo and Glut (n=3). (i) IIF staining of differentiated P2 NPSCs transiently transfected while proliferating with either an siRNA directed against mGluR1 (si mGluR) or a scrambled siRNA (Scr si) and also treated as indicated while proliferating. Identical results were obtained in a separate biological replicate transfection.
Muscarinic and glutamatergic activation alters calcium responses of NSPCs to an acute glutamate exposure
Since calcium dynamics and homeostasis play a critical role in neurogenesis, we investigated whether combined muscarinic/glutamatergic stimulation altered calcium signaling in NSPCs (Ciccolini et al., 2003; Malmersjo et al., 2013; Samarasinghe et al., 2011; Weissman et al., 2004). Results from live cell imaging assays with FURA2 AM loaded P2 neurospheres revealed the presence of synchronized calcium transients between coupled, single cells within individual neurospheres (Figs 3a, c). Measurements were made for a continuous period lasting 10 minutes after the acute administration of glutamate in cells previously treated with Pilo, Glut, Pilo+Glut, or Veh (See Supplementary Figure 2 for sample data). Interestingly, pretreatment with Pilo+Glut together triggered a significant de-synchronization of glutamate-induced calcium release (Figs 3b, c). Furthermore, the contribution of prior Glut treatment to calcium de-synchrony was due to activation of mGluR1 and not inotropic glutamate receptors as synchrony was maintained in Pilo+Glut exposed neurospheres pretreated briefly with the mGluR1 antagonist AIDA but not with the NMDA receptor antagonist MK801 (Fig 3e).
Fig 3. mGluR1 dependent calcium desynchrony in Pilo+Glut stimulated NPSCs.

Representative plots (a,b) of live cell intracellular Ca2+ flux stimulated by treatment with glutamate where indicated (red arrow), measured using the ratiometric calcium indicator Fura-2 AM. Each line in the graphs represents a single NPSC within a neurosphere. Ca2+ traces from P2 NPSCs treated with Veh (a) or Pilo+Glut (b). (c) Pearson’s Correlation Coefficient +SEM from P2 NPSCs treated with Veh, Pilo, Glut or PIlo+Glut (n=3; 1-way ANOVA, P<0.0001; post hoc Tukey’s Multiple Comparison Test, *=P<0.05). (d) Pearson’s Correlation Coefficient +SEM from P5-memory NPSCs treated at P2 with Veh, Pilo, Glut or Pilo+Glut (n=3; 1-way ANOVA, P<0.0001; post hoc Tukey’s Multiple Comparison Test, *=P<0.05; **=P<0.01; ***=P<0.005). Pearson’s Correlation Coefficient +SEM from P2 NPSCs (e) or P5-memory NPSCs treated at P2 (f) with Pilo+Glut, MK801+Pilo+Glut or AIDA+Pilo+Glut (n=3; 1-way ANOVA, P<0.0001; post hoc Tukey’s Multiple Comparison Test, *=P<0.05). Pearson’s Correlation Coefficient of live cell calcium synchrony between individual cells in P2 NPSCs (g) or P5-memory NPSCs (h) transfected with mGluR1 siRNA (SimGluR1) or scrambled siRNA (Scr) at P2 and treated with Veh or Pilo+Glut at P2 (n=3; 1-way ANOVA, P<0.0001; Tukey’s Multiple Comparison Test, *P=<0.05).
Of note, NSPCs maintained a similar impairment of calcium synchrony when transiently exposed to Pilo+Glut at the second passage (P2) and tested following 3 weeks of proliferative divisions (i.e. P5-memory) (Figs 3c,d). In addition, P5-memory NPSCs also demonstrated a small, but statistically significant decrease in Ca2+ synchrony when exposed at P2 with glut alone (Fig 3d and Supplementary Fig 2f). In accordance with the findings in P2 NPSCs, AIDA but not MK801 pre-treatment at P2 resulted in a rescue of Ca2+ de-synchrony in P5-memory cells, once again suggesting that the process is dependent on mGluR1 and not the NMDA receptor (Fig 3f). To more specifically establish the role of the mGluR1 in Ca2+ synchrony, P2 and P5 memory NPSCs transiently ablated of mGluR1 following transfection at P2 with siRNA were subject to Pilo+Glut exposure and analyzed for Ca2+ synchrony. As shown in Figures 3g and h, siRNA mediated knockdown of mGluR1 at P2 rescued the Pilo+Glut induced Ca2+ desynchrony in both P2 (Fig 3g) and P5-memory (Fig 3h) NPSCs. These data further support the notion that mGluR1 activation is only required transiently for long-term (i.e. to P5) alterations in NPSC function since knockdown of mGluR1 in two separate biological replicates was approximately 80% in P2 NPSCs but completely recovered in P5-memory NPSCs (Supplementary Fig. 1).
Muscarinic and glutamatergic stimulation in P2 NSPCs alters Cx43 phosphorylation and GJIC
Intercellular communication between coupled NSPCs in-vivo and in-vitro is mediated by gap junctions composed of connexin proteins (Elias and Kriegstein, 2008) with connexin 43 playing a particularly prominent role in NPSC differentiation, proliferation, and migration (Cheng et al., 2004; Elias et al., 2007; Samarasinghe et al., 2011). In addition, Cx43-containing gap junctions in NSPCs impact synchronized calcium transients between individually coupled cells as well as through more complex cellular networks (Malmersjo et al., 2013; Samarasinghe et al., 2011). These effects are critical to the coordinated progression of cells through the cell cycle and their appropriate exit from the cell cycle prior to differentiation (Samarasinghe et al., 2012; Weissman et al., 2004). Intercellular communication through Cx43-containing gap junctions is regulated by multiple signaling pathways that impact distinct phosphorylation sites on Cx43 (Samarasinghe et al., 2011). We therefore examined whether alterations in calcium transients observed following a combined activation of mAChR and mGluR1 in NSPCs was associated with dysregulation of Cx43 expression and/or phosphorylation.
As shown in Figure 4a, combined Pilo+Glut treatment of P2 NPSCs did not alter Cx43 expression (Figs 4a, b) but led to a reduction in phosphorylation at serine 368 (Figs 4c, d). Single treatments with Pilo or Glut alone did not elicit these changes in Cx43 phosphorylation (Figs 4c, d). Changes in other Cx43 phosphorylation sites including serines 279 and 282 were not observed (See Supplementary Figure 3a, b). In accordance with the decreased level of Cx43 phosphorylation, we also observed an increase in activity of the calcium-activated phosphatase, calcineurin/PP2B in response to combined Pilo+Glut treatment (Fig 4e). No significant changes in PP1/PP2A activity were noted (Supplementary Figure 4). No changes in expression were observed for connexin 36 (Cx36), another connexin protein expressed in NPSCs (Supplementary Figure 6a). Cx36 is not regulated by phosphorylation (Bruzzone and Dermietzel, 2006).
Fig 4. Decreased Cx43 phosphorylation in Pilo+Glut stimulated P2 NPSCs is associated with enhanced PP2B activity and increased GJIC.

Representative Western blots (a,c) and densitometric scans (b,d) of total Cx43 (a,b) and pCx43s368 in P2 NPSCs treated as indicated (d; n=3 independent experiments; 1-way ANOVA, P=0.0358; post hoc Tukey’s Multiple Comparison Test, *=P<0.05). (e) PP2B activity as assessed by a phosphate release assay in P2 NPSCs treated with vehicle or Pilo+Glut (n=3; student’s t-test; p=0.0409). Representative FRAP curves (f) from Calcein AM loaded P2 NPSCs treated as indicated with Box plots for t1/2 +/− SEM (g,h) shown of various treatment groups (n=5; g. Kruskal Wallis test p=0.0129; post hoc Dunn’s Multiple Comparison Test *=P<0.05, h. Kruskal Wallis test p=0.0057; post hoc Dunn’s Multiple Comparison Test *=P<0.05).
Since Cx43 phosphorylation sites affected by Pilo+Glut negatively regulate GJIC, hyperactivation of Cx43 gap junctions could contribute to the observed desynchronization of glutamate-induced calcium transients (Lampe and Lau, 2004). To directly test the impact of mAChR and mGluR1 activation on GJIC between coupled NSPCs, we used a quantitative fluorescence recovery after photobleaching assay (i.e. GAP-FRAP) in live cultures (Samarasinghe et al., 2011). As shown in Figures 4f and g, a combined Pilo+Glut treatment of P2 neurospheres, but not single treatments, led to a reduced t1/2 of calcein AM transfer between coupled cells, which equates to increased GJIC. The activation of mGluR1 is essential for this response as Pilo+Glut-enhanced GJIC in P2 neurospheres is blocked by prior treatment with AIDA (Fig 4h).
Transient muscarinic and glutamatergic stimulation elicits long-term changes in GJIC and Cx43 expression through increased Cx43 protein degradation
We also investigated whether the “memory” effect of dual Pilo+Glut exposure on the calcium response of P5 NSPCs is associated with dysregulation of GJIC through Cx43 gap junctions. As shown in Figures 5b and d, total Cx43 protein expression is reduced in P5-memory NPSCs that had received a prior exposure to Pilo+Glut or Glut alone. A decrease in total Cx43 levels was also noted in hippocampal lysates from chronic TLE rats (Figs 5a, c). Cx43 mRNA levels were not altered by these treatments (Supplementary Figure 5). Cx36 protein levels were also not affected in P5-memory samples (Supplementary Figure 6b). Cx43 is a highly dynamic protein with a half-life that can be as short as 1.5 hours (Laird, 2005). In fact, enhancement of Cx43 degradation is one mechanism responsible for stimulus-induced regulation of GJIC (Laird, 2005). In order to determine differential effects of Pilo+Glut co-treatment on Cx43 protein degradation in P5-memory NPSCs, we quantified total Cx43 protein levels following the inhibition of de-novo protein synthesis with cycloheximide. As shown in Figures 5e and g, Pilo+Glut treatment at P2 led to reduced stability of Cx43 protein in P5 memory NPSCs.
Fig 5. Pilo+Glut and Glut only P5-Memory NPSCs have lower total Cx43 protein levels and decreased GJIC.
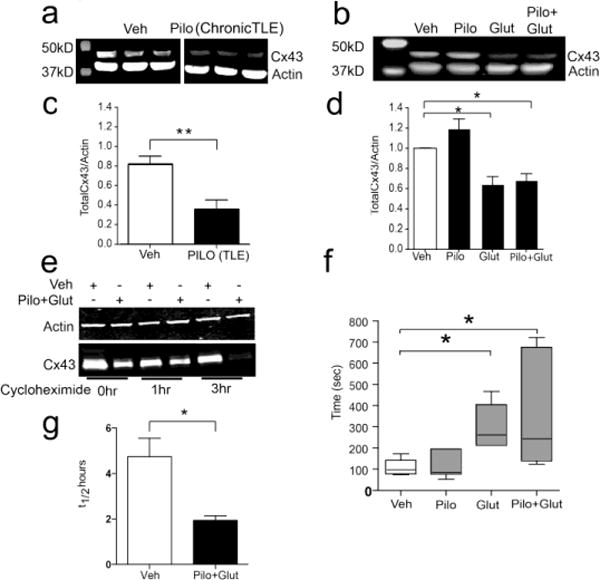
Representative Western blots (a,b) and densitometric scans (c,d) of total Cx43 in hippocampi of young adult rats 6 months following IP injection with vehicle or Pilo (c; n=3; student’s t-test; *=p<0.01) or P5-memory NPSCs treated as indicated during P2 (d; n=3 independent experiments; 1-way ANOVA, P=0.0015; post hoc Tukey’s Multiple Comparison Test, P<0.05). Representative Western blot of Cx43 protein half-life (e) or densitometric scans of Western blots (g) of total Cx43 from lysates of P5-memory NPSCs treated with Veh or Pilo+Glut at P2 and treated at P5 for 1, and 3hrs with the protein synthesis inhibitor cycloheximide (g; n=3, student’s t-test; *=p<0.05). P5-memory NPSCs treated at P2 as indicated were preloaded with Calcein AM and subjected to FRAP analysis (f). Box plots for t1/2 +/− SEM (n=6; Kruskal Wallis test p=0.0029; post hoc Dunn’s Multiple Comparison Test *=P<0.05).
In order to determine the quantitative effect of Pilo+Glut stimulation on GJIC, we employed GAP-FRAP in live P5-memory NPSCs. As shown in Figure 5f, and in accordance with the reduction in total Cx43 protein levels shown previously, the t1/2 for calcein transfer between adjacent NPSCs in a neurosphere is significantly higher for both the Glut alone and Pilo+Glut samples. This suggests a decreased rate of GJIC and may explain the Ca2+ de-synchrony observed in similar treatment groups of P5-memory NPSCs.
Muscarinic and glutamertergic activation of NPSCs derived from human IPSCs triggers enhanced production of immature GABAergic neurons
In order to more directly correlate the in-vitro and ex-vivo data obtained from animal tissues with human disease we utilized NPSCs derived from human induced pluripotent stem cells (IPSCs). As shown in Figures 6a–c, a 20-day differentiation of human NPSCs led to a decreased number of Tuj1+ cells (6b) and increased number of GAD67+ GABAergic type cells (6c) in dual Pilo+Glut treated samples, but not in cells treated with Veh, Pilo, or Glut alone. Furthermore, as in mouse NPSCs, this alteration in human NPSC differentiation requires mGluR1 activation as it is inhibited by pretreatment with AIDA (Figs 6a–c). These data are in accord with our findings in differentiated murine NPSC and suggests that neurogenesis can also be altered in human NPSCs by dual activation of mAchR and mGluR1.
Fig 6. Pilo+Glut treated human IPSC derived NPSCs demonstrate decreased neurogenesis and an enhanced immature GABAergic phenotype.

(a) Representative IIF images of differentiated NPSCs generated from human IPSCs, subject to treatment with Veh, Pilo+Glut, or AIDA+Pilo+Glut. Mean cell counts +SEM of Tuj-1 (b) and GAD67 (c) staining following IIF from differentiated human NPSCs treated as indicated (n=3; 1-way ANOVA, p=0.001 for Tuj-1; P=0.0001 for GAD67; post hoc Tukey’s Multiple Comparison Test, **= P<0.01; ***=P<0.005).
Discussion
The pathophysiological mechanisms linking an initial epileptic event during early life with delayed neurodevelopmental consequences including refractory adult seizures are multifactorial (Rakhade and Jensen, 2009). While a great deal of progress has been made in recent decades in our understanding of the molecular mechanisms of epileptogenesis, one relatively unexplored area is the impact of an epileptogenic stimulus on neural stem cells. Given the established role that alterations in stem cell fate can have on subsequent neurodevelopment and neuropathology and the observed changes in neurogenesis and cell differentiation observed in various models of epilepsy- a deeper understanding of the role of NPSCs in epileptogenesis may be of particular value in the treatment of this disease.
In the present study, we utilized dual Pilo+Glut treatment as an epileptogenic stimulus on NPSCs and observed both acute and chronic effects on stem cell differentiation fate that were directly correlated with treatment induced disruption of homeostatic mechanisms. Specifically, Pilo+Glut treatment, in an mGluR1 dependent manner, reduced Tuj-1+ neurogenesis and significantly increased the percentage of NPSCs that differentiated into immature GABAergic (SOX2+/GAD67+) cells. Pilo+Glut exposure reprogrammed NPSCs to attain this altered differentiation fate as it was still observed even if differentiation was delayed for three replicative divisions over a course of multiple weeks (i.e. P5-memory). These NPSC differentiation data are in accordance with ex-vivo data from chronic TLE rats where we also observed increased SOX2+/GAD67+ co-staining in the hillus of the DG and with data from differentiated NPSCs derived from human IPSCs subject to Pilo+Glut treatment. Further analysis revealed that dual Pilo+Glut exposure resulted in decreased synchrony of intercellular Ca2+ currents in P2 and P5-memory NPSCs. Interestingly, in P2 NPSCs this correlated with increased PP2B activity, decreased Cx43 phosphorylation, and increased GJIC, whereas in P5-memory NPSCs Ca2+ desynchrony was correlated with an increased rate of Cx43 protein degradation, overall decreases in total Cx43 protein expression, and decreased GJIC.
Prior studies that have interrogated the role of connexin 43 protein and GJIC in epilepsy have focused on the expression and function of this protein in the mature epileptic brain (Elisevich et al., 1997; Fonseca et al., 2002; Gigout et al., 2006; Naus et al., 1991). Uniquely, this study has instead examined the role of Cx43 and GJIC at the level of the stem/progenitor cell and its potential role during the process of epileptogenesis. A number of studies specifically examining the role of connexin 43 gap junctions/hemi channels in NPSCs have demonstrated a critical role for this protein during NPSC proliferation and differentiation (Cheng et al., 2004; Elias et al., 2007; Samarasinghe et al., 2011; Weissman et al., 2004). A provocative study by Elias et al., demonstrated a non-traditional role for Cx43 in neurodevelopment wherein the protein behaved as an essential anchor for radial glial cell migration (Elias et al., 2007). Interestingly, cortical dysplasia has been noted in surgical specimens from refractory TLE as well as in focal cortical dysplasia (FCD) type epileptic brains (Garbelli et al., 2011; Kuzniecky et al., 1991). The down-regulation of Cx43 protein expression in P5-memory NPSCs that we observed could further enhance the epileptic phenotype by impairing Cx43 mediated cellular migration. In addition to changes in cellular differentiation induced during epileptogenesis, the observed decrease in Cx43 expression suggests a further mechanistic link between an initial epileptogenic stimulus and subsequent CNS cellular disorganization observed in certain refractory epilepsies.
In addition to the potential influence of Cx43 dysregulation in subsequent NPSC migration, Cx43 dependent gap junction or hemichannel intercellular communication has a long-established role in neurodevelopmental processes (Bruzzone and Dermietzel, 2006). The importance of gap junction dependent Ca2+ wave propagation has been shown to be of particular importance to NPSC proliferation and differentiation (Bruzzone and Dermietzel, 2006; Ciccolini et al., 2003; Malmersjo et al., 2013; Samarasinghe et al., 2011; Weissman et al., 2004). We observed increased desynchrony of Ca2+ transients following a glutamate spike in Pilo+Glut exposed NPSCs. When these NPSCs were differentiated they demonstrated an increased immature GABAergic phenotype. In fact, spontaneous calcium transients have been shown to be essential for subsequent NPSC differentiation fate (Carey and Matsumoto, 1999; Gu and Spitzer, 1995). For example, induction of an increased frequency of Ca2+ transients in NPSCs was shown to increase GABAergic differentiation, although that study did not specifically examine synchrony (Ciccolini et al., 2003).
In P5-memory NPSCs, the small change in desynchrony noted in Glut only treated cells did not alter differentiation. Glut treatment alone at P2 also led to reduced Cx43 expression and GJIC in P5-memory NPSCs. In contrast, a more robust alteration in Ca2+ desynchrony triggered by a combined Pilo+Glut treatment (i.e. compared to Glut alone) was associated with the increased production of immature GABAergic cells. This suggests that a certain threshold level of change in Ca2+ signaling may be necessary to initiate a change in the fate specification of NPSCs that persists over multiple generations. mGluR1 activation alone is necessary but not sufficient to elicit this long-term change in differentiation potential and requires a “priming” event initiated by mAcR activation.
While the Pilo+Glut mediated phenotypic change in Ca2+ signaling (i.e. desynchrony) was preserved in P2 and P5-memory NPSCs, the mechanisms of action were markedly different. In acutely treated P2 cells, the activity of the Cx43 associated and Ca2+ dependent phosphatase PP2B was up-regulated and Cx43 phosphorylation was reduced (Laird, 2005; Lampe and Lau, 2004). In P5-memory cells, Cx43 degradation was enhanced and associated with loss of Cx43 protein levels and inhibition of GJIC. Therefore our findings suggest that epigenetic changes at early passage NPSCs (i.e. P2) impact the expression of genes that regulate Cx43 protein turnover that are maintained following multiple replicative divisions (i.e. P5-memory NPSCs). Nedd4, a Cx43 E3 ubiquitin ligase is an attractive candidate for future studies in our model given its established role in neurodevelopment (Kawabe et al., 2010; Liao et al., 2013).
In summary, we have demonstrated unique mechanisms in P2 and P5-memory NPSCs subject to an epileptogenic stimulus that impact on a gap junction dependent homeostatic state with subsequent effects on cell differentiation fate. The defining feature of our studies was that a single epileptogenic stimulus acting through diverse mechanisms was sufficient to alter NPSC differentiation acutely as well as chronically. The specific increases in a GABAergic phenotype and NKCC1 expression that we observed is in accord with findings of increased NKCC1 expression in epileptic human brain samples as well as with the finding that GABA antagonism is seizure-protective in the developing brain (Khalilov et al., 2005; Palma et al., 2006). Taken together, our data suggests a “multiple hit” hypothesis of NPSC dysfunction during epileptogenesis. Specifically, the present data support the notion that an initial epileptogenic stimulus is sufficient to immediately alter the differentiation pattern of affected NPSCs towards a proconvulsant, GABAergic phenotype. In addition, NPSCs that continue to proliferate will not only continue to be susceptible to an increased GABAergic pattern of differentiation, but also have additional decreases in Cx43 protein expression that may alter neural cell migration and further predispose the CNS towards refractory epilepsy. Finally, Cx43 protein, and Ca2+ signaling in particular, may be novel therapeutic targets for the treatment and prevention of refractory epilepsy.
Materials and Methods
Details of specific methods for each experimental protocol below are provided in the Supplemental Experimental Procedures.
Mouse NPSC Culture
Mouse NPSCs were prepared from E14.5 embryonic cortex according to the technical manual provided by StemCell Technologies, differentiation was also performed per the specifications outlined in the technical manual (StemCell Technologies, Vancouver, Canada). Cells were utilized at passage 2 and passage 5 as specified in the text.
Human iPSC Culture
Human iPSCs were derived from skin biopsy samples collected from the shoulder of normal individuals via 4 mm full thickness punch biopsies under local anesthesia. Subjects were enrolled at the University of Pittsburgh (PITT) and donated biopsies through their participation in studies approved by the PITT Institutional Review Board. iPSCs were derived and differentiated according to D’Aiuto (D’Aiuto et al., 2014).
Western Blot Analysis
Protein lysates obtained from NPSCs, differentiated NPSCs, and rat hippocampus were used in Western blot analysis using standard procedures. Lysates were resolved on standard or gradient gels as specified in the figure legends, and the Li-Cor system was utilized for imagining blots (Li-Cor, Lincoln, Nebraska). Specific antibodies can be found in Sl Materials. Images were quantified (densitometry) using NIH ImageJ software (ImageJ, http://rsbweb.nih.gov/ij/).
Gap-FRAP
NPSCs were loaded with 1ug/mL of Calcein AM (Invitrogen, Carlsbad, CA) 30 min prior to FRAP analysis, which was conducted on an Olympus IX81 confocal microscope equipped with Fluoview data collection software. FRAP recovery curves were fit to an exponential decay equation present in the GraphPad menu, and t1/2 was tabulated by the software from this fit.
Live Cell Intracellular Ca2+ Imaging
NPCs were loaded with 500nM Fura-2 AM for 30min prior to Ca2+ imaging, and alternatively illuminated with 340 and 380 nm light for Fura-2 using a Leica HC N PLAN BD 40X oil immersion objective (Leica, Buffalo Grove, IL). Data were collected and analyzed in Simple PCI software as the 340/380 ratio.
siRNA Treatment
Passage 2 NPSCs were transfected with either a control siRNA (Catalogue # sc-37007, Santa Cruz Biotechnology, Santa Cruz, CA) or an siRNA pool directed against mouse mGluR1 (Catalogue # sc-61027, Santa Cruz Biotechnology, Santa Cruz, C) using Lipofectamine LTX/Plus (Catalogue # 15338, Invitrogen Life Technologies, Grand Island, NY) employing conditions outlined by the manufacturer.
Animals
Adult male Sprague–Dawley rats (225–250 g) were purchased from Charles River (USA). All animal use followed University of Pittsburgh Institutional Animal Care and Use Committee approved protocols and was in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
In Vivo Pilocarpine Treatment
Studies were conducted applying the pilocarpine (Pilo) model of TLE in Sprague–Dawley rats (Curia)(Curia et al., 2008). Only rats developing SE were selected for subsequent analysis at 6 months end-point.
Phosphatase Activity Assay
Treated mouse NPSCs were prepared as per the technical manual provided by Enzo Life Sciences (Enzo Life Sciences, Farmingdale, New York). Briefly lysates were incubated with an RII phospho-serine peptide substrate and following equilibration, the reaction was terminated with BIOMOL GREEN TM reagent. After color development, the microplate was read at 620 nm. Calculations were conducted as per Enzo technical manual instructions, and phosphatase activity (both calcineurin and PP2A/1/5) was measured in nanomoles of phosphate released.
Cycloheximide Treatment
To assess the dynamics of intracellular connexin gap-junction protein degradation a 100μg/ml solution of cycloheximide (Sigma Aldrich, St. Louis, MO) was added to vehicle or Pilo+Glut treated neurospheres. Lysates from treated cells were subject to Western Blot. Normalized Cx43 levels (Cx43/Actin densitometry) were plotted vs time, a line of best fit was acquired using Excel software (Microsoft, Seattle, WA) and used to estimate the t1/2 (n=3 independent treatments, compared used an unpaired student’s t-test, with *= p<0.05).
Differentiation of NPSCs
Mouse NPSCs were prepared according to the technical manual provided by StemCell Technologies (StemCell Technologies, Vancouver, Canada). Cells were differentiated at 37°C for 10 days before collection.
Immunocytochemistry
Immunocytochemistry was performed according to standard procedures. Please refer to the detailed methods in SI for specific antibodies and concentrations. Differentiated NPSCs were visualized using an Olympus IX81 confocal microscope with Fluoview software (Olympus, Center Valley, PA). Hippocampal hilus images (4 specimens/animal) were acquired with an Olympus Fluoview 1000 laser scanning confocal microscope. The pinhole was adjusted to obtain a 1-μm-thick optical slice. The detection parameters were set in the control reaction and were kept constant across specimens. Correlation (Pearson’s index) analysis was performed in a semi-automated fashion with Metamorph software (Molecular Devices). Images were further analyzed on Photoshop (Adobe, San Jose, CA), and any manipulation of color levels was applied equally to all pseudocolors and all images within a panel.
Statistical Analysis
Unless otherwise specified within the text, statistical comparison was conducted by 1-way-analysis of variance (ANOVA) followed by post hoc Tukey’s multiple comparison’s test to determine within group differences. A p<0.05 was considered statistically significant.
Supplementary Material
Highlights.
Immature GABAergic cells are generated by NPSCs exposed to pilocarpine and glutamate
Immature GABAergic cells accumulate in the hippocampi of chronic epilepsy rats
Dual stimulation desynchronizes Ca++ bursts and alters Cx43 protein and function
Persistent changes in NPSC fate are triggered by pilocarpine and glutamate exposure
Acknowledgments
We thank Dr. Leonardo D’Aiuto, Department of Psychiatry, University of Pittsburgh, for the human iPSCs and we thank Dr. Mark Richardson, Department of Neurological Surgery, University of Pittsburgh Medical Center, for critical reading of the manuscript. This project was supported in part by NIH grant DK078394 (DBD). The authors acknowledge grant 1 UL1 RR024153 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bruzzone R, Dermietzel R. Structure and function of gap junctions in the developing brain. Cell Tissue Res. 2006;326:239–48. doi: 10.1007/s00441-006-0287-0. [DOI] [PubMed] [Google Scholar]
- Carey MB, Matsumoto SG. Spontaneous calcium transients are required for neuronal differentiation of murine neural crest. Dev Biol. 1999;215:298–313. doi: 10.1006/dbio.1999.9433. [DOI] [PubMed] [Google Scholar]
- Cheng A, et al. Gap junctional communication is required to maintain mouse cortical neural progenitor cells in a proliferative state. Dev Biol. 2004;272:203–16. doi: 10.1016/j.ydbio.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Ciccolini F, et al. Local and global spontaneous calcium events regulate neurite outgrowth and onset of GABAergic phenotype during neural precursor differentiation. J Neurosci. 2003;23:103–11. doi: 10.1523/JNEUROSCI.23-01-00103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curia G, et al. The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods. 2008;172:143–57. doi: 10.1016/j.jneumeth.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aiuto L, et al. Persistent Infection by HSV-1 Is Associated With Changes in Functional Architecture of iPSC-Derived Neurons and Brain Activation Patterns Underlying Working Memory Performance. Schizophr Bull. 2014 doi: 10.1093/schbul/sbu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer SC. Postnatal and adult neurogenesis in the development of human disease. Neuroscientist. 2008;14:446–58. doi: 10.1177/1073858408317008. [DOI] [PubMed] [Google Scholar]
- Di Maio R, et al. Pilocapine alters NMDA receptor expression and function in hippocampal neurons: NADPH oxidase and ERK1/2 mechanisms. Neurobiol Dis. 2011;42:482–95. doi: 10.1016/j.nbd.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Di Maio R, et al. Thiol oxidation and altered NR2B/NMDA receptor functions in in vitro and in vivo pilocarpine models: Implications for epileptogenesis. Neurobiol Dis. 2012;49C:87–98. doi: 10.1016/j.nbd.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Elias LA, Kriegstein AR. Gap junctions: multifaceted regulators of embryonic cortical development. Trends Neurosci. 2008;31:243–50. doi: 10.1016/j.tins.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias LA, et al. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–7. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- Elisevich K, et al. Hippocampal connexin 43 expression in human complex partial seizure disorder. Exp Neurol. 1997;145:154–64. doi: 10.1006/exnr.1997.6467. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Houser CR. Up-regulation of GAD65 and GAD67 in remaining hippocampal GABA neurons in a model of temporal lobe epilepsy. J Comp Neurol. 1999;412:488–505. [PubMed] [Google Scholar]
- Fonseca CG, et al. Upregulation in astrocytic connexin 43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res. 2002;929:105–16. doi: 10.1016/s0006-8993(01)03289-9. [DOI] [PubMed] [Google Scholar]
- Garbelli R, et al. Expression of connexin 43 in the human epileptic and drug-resistant cerebral cortex. Neurology. 2011;76:895–902. doi: 10.1212/WNL.0b013e31820f2da6. [DOI] [PubMed] [Google Scholar]
- Gigout S, et al. Effects of gap junction blockers on human neocortical synchronization. Neurobiol Dis. 2006;22:496–508. doi: 10.1016/j.nbd.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Gu X, Spitzer NC. Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+ transients. Nature. 1995;375:784–7. doi: 10.1038/375784a0. [DOI] [PubMed] [Google Scholar]
- Kawabe H, et al. Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron. 2010;65:358–72. doi: 10.1016/j.neuron.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalilov I, et al. Epileptogenic actions of GABA and fast oscillations in the developing hippocampus. Neuron. 2005;48:787–96. doi: 10.1016/j.neuron.2005.09.026. [DOI] [PubMed] [Google Scholar]
- Kuzniecky R, et al. Cortical dysplasia in temporal lobe epilepsy: magnetic resonance imaging correlations. Ann Neurol. 1991;29:293–8. doi: 10.1002/ana.410290311. [DOI] [PubMed] [Google Scholar]
- Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–82. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–86. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CK, et al. Lipopolysaccharide induces degradation of connexin43 in rat astrocytes via the ubiquitin-proteasome proteolytic pathway. PLoS One. 2013;8:e79350. doi: 10.1371/journal.pone.0079350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis SA, et al. Enumeration of neural stem and progenitor cells in the neural colony-forming cell assay. Stem Cells. 2008;26:988–96. doi: 10.1634/stemcells.2007-0867. [DOI] [PubMed] [Google Scholar]
- Malmersjo S, et al. Neural progenitors organize in small-world networks to promote cell proliferation. Proc Natl Acad Sci U S A. 2013;110:E1524–32. doi: 10.1073/pnas.1220179110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naus CC, et al. Gap junction gene expression in human seizure disorder. Exp Neurol. 1991;111:198–203. doi: 10.1016/0014-4886(91)90007-y. [DOI] [PubMed] [Google Scholar]
- Palma E, et al. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci U S A. 2006;103:8465–8. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, et al. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–38. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Jensen FE. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009;5:380–91. doi: 10.1038/nrneurol.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarasinghe RA, et al. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci U S A. 2011;108:16657–62. doi: 10.1073/pnas.1102821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarasinghe RA, et al. Cooperativity and complementarity: synergies in non-classical and classical glucocorticoid signaling. Cell Cycle. 2012;11:2819–27. doi: 10.4161/cc.21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankar R, et al. Granule cell neurogenesis after status epilepticus in the immature rat brain. Epilepsia. 2000;41(Suppl 6):S53–6. doi: 10.1111/j.1528-1157.2000.tb01557.x. [DOI] [PubMed] [Google Scholar]
- Smolders I, et al. NMDA receptor-mediated pilocarpine-induced seizures: characterization in freely moving rats by microdialysis. Br J Pharmacol. 1997;121:1171–9. doi: 10.1038/sj.bjp.0701231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, et al. Loss of cholecystokinin-containing terminals in temporal lobe epilepsy. Neurobiol Dis. 2014;62:44–55. doi: 10.1016/j.nbd.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thind KK, et al. Initial loss but later excess of GABAergic synapses with dentate granule cells in a rat model of temporal lobe epilepsy. J Comp Neurol. 2010;518:647–67. doi: 10.1002/cne.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman TA, et al. Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron. 2004;43:647–61. doi: 10.1016/j.neuron.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Young SZ, et al. NKCC1 knockdown decreases neuron production through GABA(A)-regulated neural progenitor proliferation and delays dendrite development. J Neurosci. 2012;32:13630–8. doi: 10.1523/JNEUROSCI.2864-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.