

Abstract
The use of viruses as transneuronal tracers has become an increasingly powerful technique for defining the synaptic organization of neural networks. Although a number of recombinant alpha herpesviruses are known to spread selectively in the retrograde direction through neural circuits only one strain, the H129 strain of herpes simplex virus type 1, is reported to selectively spread in the anterograde direction. However, it is unclear from the literature whether there is an absolute block or an attenuation of retrograde spread of H129. Here we demonstrate efficient anterograde spread, and temporally delayed retrograde spread, of H129 and three novel recombinants. In vitro studies revealed no differences in anterograde and retrograde spread of parental H129 and its recombinants through superior cervical ganglion neurons. In vivo injections of rat striatum revealed a clear bias of anterograde spread, although evidence of deficient retrograde transport was also present. Evidence of temporally delayed retrograde transneuronal spread of H129 in the retina was observed following injection of the lateral geniculate nucleus. The data also demonstrated that three novel recombinants efficiently express unique fluorescent reporters and have the capacity to infect the same neurons in dual infection paradigms. From these experiments we conclude that H129 and its recombinants efficiently infect neurons through anterograde transneuronal passage, but also are capable of temporally delayed retrograde transneuronal spread. In addition, the capacity to produce dual infection of projection targets following anterograde transneuronal passage provides an important addition to viral transneuronal tracing technology.
Keywords: Herpes Simplex Virus, Transneuronal Tracing, Anterograde, Retrograde, Recombinants, Fluorescent Reporters, Dual Infection
Introduction
Transneuronal tracing of neural circuitry with neuroinvasive viruses has become an increasingly powerful approach for defining the synaptic organization of neural pathways. The method is based upon the ability of viruses to invade neurons and produce infectious progeny that pass transneuronally at synapses to infect synaptically connected neurons. With increasing survival, the infection moves through neural networks, marking each neuron with distinctive viral or reporter proteins and thereby providing an effective means of defining the synaptic organization of functionally defined circuitry (Card and Enquist 2012; Ekstrand et al. 2008; Enquist et al. 2002; Loewy 1995; Kelly and Strick 2000; Card and Enquist 1994; Kuypers and Ugolini 1990; Kristensson et al. 1982; Turner and Jenkins 1997; Geerling et al. 2006).
The utility of viral infection for polysynaptic circuit definition has been improved significantly by the identification of viruses that are transported either anterogradely or retrogradely through circuitry, and through construction of recombinant viruses that express reporter proteins constitutively or conditionally (Boldogkoi et al. 2009; Boldogkoi et al. 2004; Enquist and Card 2003; Song et al. 2005; Zemanick et al. 1991). Because derivatives of neuroinvasive alpha herpesviruses possess both of these attributes, they have been widely applied for circuit analysis. Early studies demonstrated that a vaccine strain of pseudorabies virus (PRV-Bartha; (Bartha 1961)), a swine alpha herpesvirus with broad host range, was selectively transported retrogradely through neural circuits (Dolivo 1980). Subsequent analysis of the PRV-Bartha genome demonstrated that the retrograde-only phenotype was due to a deletion in the unique short region of the viral genome that also attenuated the virulence of the virus (Mettenleiter 1995; Pomeranz et al. 2005; Card and Enquist 1995; Pickard et al. 2002). The genomes of various PRV strains, including PRV-Bartha, have now been completely sequenced (Szpara et al. 2011). Collectively, these studies have established a foundation for the widespread use of PRV-Bartha for circuit analysis and stimulated the construction of PRV-Bartha recombinants that express a variety of reporter proteins (Enquist 1999). The availability of these reporter viruses opened the door for dual infection experiments that define neurons whose axons collateralize within complex circuits to innervate multiple neurons (Billig et al. 2000; Cano et al. 2004; Toth et al. 2008). Additionally, the recent exploitation of Cre-lox technology to construct alpha herpesviruses that replicate conditionally or carry conditional reporter genes now permits the identification of neural networks synaptically connected to phenotypically defined neurons (Card et al. 2011a; Card et al. 2011b; Lo and Anderson 2011; DeFalco et al. 2001; Campbell and Herbison 2007b, a; Yoon et al. 2005; Braz et al. 2009; Braz et al. 2002). While it is noteworthy that the host range of PRV includes all mammals and some birds, it does not infect higher primates, as does the human pathogen herpes simplex virus (HSV) (Pomeranz et al. 2005; Gustafson 1975). Moreover, HSV is widely used in rodent latency/reactivation models whereas PRV only establishes latency in its natural host, the adult pig (Wittmann and Rziha 1989; Enquist 1994; Roizman and Sears 1996).
The numerous PRV strains shown to be transported in the retrograde direction through neural circuits stand in marked contrast to the paucity of alpha herpesvirus mutants transported only in the anterograde direction through circuitry. In fact, only one strain of human HSV type 1 (HSV-1) has been reported to move selectively in the anterograde direction through neural circuits. This virus, HSV-1 H129, is an isolate from the brain of an individual who suffered from viral encephalitis (Dix et al. 1983), and was shown by Zemanick and colleagues to be transported preferentially in the anterograde direction following injection into motor cortex of cebus monkeys (Zemanick et al. 1991). In the same study, Zemanick and colleagues demonstrated that the anterograde phenotype of H129 spread contrasted with the selective retrograde spread of the McIntyre-B strain of HSV-1. A number of subsequent studies in rat confirmed the preferential anterograde transport of the H129 strain through neural circuits (Barnett et al. 1995; Garner and LaVail 1999; McGovern et al. 2012a; McGovern et al. 2012b; Rinaman and Schwartz 2004; Song et al. 2009; Vaughan and Bartness 2012; Sun et al. 1996; Dum et al. 2009; Kelly and Strick 2003). Additionally, two recent studies reported the construction of H129 recombinants that either replicate conditionally (Lo and Anderson 2011) or express an EGFP reporter constitutively (McGovern et al. 2012b). The recombinant constructed by Lo and Anderson is dependent upon the presence of cre recombinase (Cre) to replicate and its utility for circuit analysis was documented in transgenic mice expressing Cre in targeted populations of neurons. McGovern and colleagues engineered a recombinant that expresses an EGFP reporter under the control of the immediate early cytomegalovirus promoter and validated it for circuit analysis in airway circuits previously defined with parental H129.
Although the selective anterograde-only spread phenotype of HSV-1 H129 was asserted in the aforementioned literature, it remains unclear if retrograde transport is completely blocked or if it is reduced in efficiency as has been observed for PRV recombinants (Curanovic and Enquist 2009; Olsen et al. 2006; Zaidi et al. 2008). In fact, studies by Atherton and colleagues provided evidence for retrograde transport of H129 in the brain following inoculation of the anterior chamber of the eye (Archin et al. 2003; Archin and Atherton 2002b; Archin and Atherton 2002a). In the studies with PRV, mutations reduced particle egress from infected neuronal cell bodies and their dendrites, but did not completely block retrograde transneuronal passage of the virus. Recent complete sequencing of the H129 genome revealed 34 genes containing unique mutations compared to wild type strains of HSV-1 (Szpara et al. 2010). These mutations raise the possibility that H129 may also harbor a retrograde spread defect as part of a polygenic phenomenon, a possibility that is parsimonious with the data of Atherton and colleagues.
In preliminary studies we noted patterns of infection with H129 that suggested retrograde transneuronal spread of virus was delayed rather than completely blocked. Considered with the findings of Atherton and colleagues (Archin et al. 2003; Archin and Atherton 2002b; Archin and Atherton 2002a) we therefore conducted studies testing the hypothesis that HSV-1 H129 has a temporally delayed retrograde spread phenotype rather than a complete block. In addition, we characterized recombinants of H129 that express unique fluorescent reporter proteins for use in dual infection studies.
Materials and Methods
Construction of the H129 recombinants and the in vitro studies that characterized their invasiveness were conducted at Princeton University. All experiments conformed to the biosafety regulations of the Institutional Animal Care and Use Committee (IACUC) of the Princeton University Research board under protocol 1947-13.
Viruses and Vero Cells
The African green monkey kidney epithelial cell line Vero (ATCC cell line CCL-81) was used to propagate and titer HSV-1. Cells were grown in DMEM with 10% FBS and 1% penicillin/streptomycin (HyClone). HSV-1 H129, a low-passage clinical strain originally isolated from a patient with encephalitis, was obtained from Richard Dix (Dix et al. 1983). HSV-1 strain 17syn+ was obtained from Valerie Preston (McGeoch et al. 1988) and is referred to as wild type (WT) virus. To construct HSV-1 strain H129 mutant 414, we designed a plasmid containing a gene encoding the enhanced yellow fluorescent protein (EYFP) fluorescent protein whose transcription was driven by the cytomegalovirus (CMV) immediate-early promoter and terminated with the SV40 polyA sequence. The fluorophore expressing cassette was derived from those previously described (Taylor et al. 2012) with the modification that the nuclear localization sequence (NLS) was removed to allow diffuse fluorophore staining of cells. The plasmid contained the EYFP gene and an approximate 500 base pair (bp) sequence homologous to HSV-1 UL37 and UL38 genes, flanking both ends of the insertion fragment to target recombination into the intergenic region between those genes. To construct H129 recombinant 772 we used the pH129–EGFP plasmid containing the enhanced green fluorescent protein (EGFP) gene whose transcription was driven by the CMV immediate-early promoter and terminated with the SV40 polyA sequence. The plasmid was provided by Stuart Mazzone (McGovern et al. 2012b) and contains an 1800 bp sequence homologous to HSV-1 UL26/26.5 and UL27 genes to allow insertion in the intergenic region. To construct H129 recombinant 424 we used the approach of Nagel and colleagues (Nagel et al. 2008) to fuse the sequence of monomeric red fluorescent protein (mRFP) to the N terminus of the VP26 protein (encoded by the UL35 gene). The following primers were used to amplify a fragment of 1687 bp from the HSV1(17+) Lox-RFPVP26 BAC obtained from Beate Sodeik (Nygardas et al. 2013): 5′ATG TGG ATC AGC TGC TTT GT′3 and 5′GCT AGA CGC GCT CGA TTA T′3. The PCR fragment contains the mRFP-VP26 fusion gene and 478 bp upstream of the UL35 start codon and 130 bp downstream of the UL35 stop codon to target homologous recombination. There was no amino acid difference between strain 17syn+ and H129 in the 1687 bp fragment used to construct the gene encoding the mRFP-VP26 fusion protein. To produce recombinants 414 and 772, H129 nucleocapsid DNA was cotransfected with linearized plasmid and Lipofectamine 2000 following the manufacturer's protocol (Invitrogen). To make recombinant 424, the purified PCR product was cotransfected with nucleocapsid DNA. The properties of each of the recombinants are defined in Table 1. The H129 recombinants produced after cotransfection were plated on Vero cells and plaques expressing the appropriate fluorescence were visualized and isolated using an epifluorescence microscope. Putative recombinants were subjected to four rounds of purification. To determine the titer of stocks of each new recombinant, serial dilutions were plated in duplicate on Vero cells and grown for 3 to 4 days in media containing 1% methocel (Dow Chemical Company) before counting the number of plaques in each well. Titers are expressed in plaque forming units per milliliter (pfu/ml).
Table 1.
| Viral strain | Name | Reporter profile | Promoter | Insertion region |
|---|---|---|---|---|
| H129 | 414 | EYFP, diffusible | CMV | UL37/UL38 intergenic region |
| H129 | 424 | mRFP, nuclear | UL35 native | Fusion with the N terminus of UL35 |
| H129 | 772 | EGFP, diffusible | CMV | UL26/UL26.5-UL27 intergenic region |
Experiment 1: In Vitro Studies
Primary neuronal culture and trichamber system
The invasive phenotype of H129 and its recombinants was examined in vitro using the modified Campenot chamber model system developed and characterized by the Enquist laboratory (Ch'ng and Enquist 2005, 2006; Curanovic and Enquist 2009). This trichamber culture system contains three compartments separated by a Teflon tripartite ring coated with silicone grease. Axons of cultured sympathetic superior cervical ganglion neurons (SCG) penetrate the silicone grease to grow from a soma (S) compartment through a central methocel (M) chamber to gain access to the neurite (N) compartment. The silicone grease seal and methocel prevents diffusion between compartments, enabling analysis of viral invasiveness following inoculation of either the S or N chambers. SCG neurons cultured in this system grow axons, but not dendrites, and establish synaptic connections among neurons within the S chamber (cf., (Curanovic and Enquist 2009) for review). The transmitter status of synaptic connections between the cultured SCG neurons has been established as cholinergic and adrenergic (Potter et al. 1986) and alpha herpesvirus infection of neurons in this culture system increases the rate of action potential firing (McCarthy et al. 2009).
SCG were obtained from Sprague-Dawley rat embryos at day 17 of pregnancy (Hilltop Lab Animals, Inc., Scottdale, PA) and cultured as previously described (Ch'ng and Enquist 2005). Briefly SCG neurons were cultured in Neurobasal media (Invitrogen) supplemented with 1% of penicillin/streptomycin/glutamine (Invitrogen), B27 (Invitrogen) and 100 ng/ml of nerve growth factor 2.5S (Invitrogen) and allowed to mature for at least 18 days before infection with HSV-1. Neurons were cultured in trichamber Teflon CAMP320 isolator rings (Tyler Research) placed on top of 35 mm polystyrene dishes. The culture surface was previously coated with Poly-DL-ornithine (Sigma-Aldrich) at 500 μg/ml in Borate buffer followed by 10 μg/ml of mouse laminin (Invitrogen) in HBSS. To assess the capacity of these viruses to enter axons and be transported in the retrograde direction toward the cell bodies we added 1 × 106 pfu of each recombinant in 100 μl supplemented neuronal media to the axonal (N) compartment of the modified Campenot chambers. After 24 hours post infection (hpi), the contents of the S compartment were collected and titered for infectious virus on Vero cells. To quantitate primary and higher-order retrograde spread of infection in the S compartment, the lipophilic tracer DiI (Invitrogen) was added to the N compartment 3 hpi. It is important to emphasize that only a subset of neurons grow axons into the N compartment and that neurons that do not project into the N compartment establish synaptic contacts with neurons that do. It is also important to note that a large literature reporting upon the use of this culture system (e.g., (Koyuncu et al. 2013b)) has demonstrated that all neurons projecting into the N chamber are retrogradely labeled after application of DiI into that chamber. Neurons in the S compartment were imaged 24 hpi of the N compartment on a Nikon Ti-Eclipse inverted microscope as previously described (Curanovic et al. 2009). To determine the extent of first-order (primary) retrograde infection, we calculated the ratio between the number of cell bodies that were labeled with DiI and the number of cell bodies that were labeled with DiI and displayed appropriate fluorescence from 414, 772 or WT infection. To measure the extent of higher-order spread of infection from primary neurons, we calculated the ratio between cell bodies that were positive for HSV-1 infection but were not labeled with DiI and cell bodies that were both infected and labeled with DiI (primary infection). At least 600 cell bodies were counted in two chambers used for every condition.
To assess the anterograde spread of HSV-1, we used the same chamber system with SCG neurons. However, we plated 5 × 105 Vero cells on top of axons in the N compartment one day before infecting the S compartment. We then infected the S compartment with 1 × 106 pfu of each recombinant in 100 μl of supplemented neuronal media. After one hour, the inoculum was removed and replaced with conditioned neuronal media. Anterograde spread of infection occurs when progeny virus of infected SCG cell bodies move into axons, are transported to the N compartment, and released directly onto Vero cells where further infection and replication occurs. The contents of the S and N compartments were collected at 48 hpi and infectious virus was determined by plating on Vero cells. Non-neuronal spread of virus between compartments was eliminated by adding neuronal medium supplemented with 1% methylcellulose to the middle M compartment one hour prior to infection of either the N or S compartment (Ch'ng and Enquist 2005).
Experiments 2 – 4: Animal Studies
The in vivo studies were performed at the University of Pittsburgh. All experiments conformed to regulations stipulated in the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh IACUC, Recombinant DNA Committee, and the Division of Environmental Health and Safety (protocol # 12080947). Adult male Sprague-Dawley rats (weight: 225-350 grams) were used for all experiments. Animals were housed individually in a Biosafety Level 2+ facility certified for virus injections. The light/dark cycle was 12 hours light, 12 hours dark (light on at 7:00 am) and room temperature was held constant between 22 and 25°C. Access to food and water was provided ad libitum. A total of 38 rats were used for the experiments included in this report.
Experiment 2
To define the invasive profile of the H129 recombinants we first defined the replication and transport of virus through neural circuits following injection into the striatum. We selected the striatum for injection because of prior parametric studies conducted using PRV (Card et al., 1999) and the well characterized polysynaptic basal ganglia pathways defined in numerous investigations (e.g., (Gerfen 2004)). Animals (n=20) were deeply anesthetized with isoflurane gas throughout the experimental procedure. A beveled 1μl Hamilton Syringe was slowly lowered into the striatum through a craniotomy at coordinates anteroposterior (AP): 0.5 mm, mediolateral (ML): 2.5 mm, dorsoventral (DV): -5.0 mm in relation to Bregma according to the rat brain atlas of Swanson (Swanson 1998). The tissue was allowed to relax for 1 minute prior to pressure injection of 100 nanoliters (nl) of virus at a constant rate of 10 nl/minute using a nanopump (Model 310/Stoelting). Ten minutes after completion of the injection the syringe needle was slowly removed from the brain. This permitted the virus to bind to the tissue, thereby reducing backflow along the cannula tract. The burr hole was sealed with bone wax and the incision closed with surgical staples. Each animal received an injection of one of three recombinants; 414 (n = 6; 3.0 × 107 pfu/ml), 424 (n = 5; 2.0 × 108 pfu/ml), or 772 (n = 9; 5.0 × 108 pfu/ml). Three animals received an injection of a cocktail of the β-subunit of cholera toxin (CTβ; List Biochemical Laboratories, Campbell CA) combined with recombinant 424 (n=1) or 772 (n=2) according the method employed by O'Donnell and colleagues (O'Donnell et al. 1997). Briefly, 10 μl of virus was mixed with 10 μl of 0.25% CTβ and 100 nl of the mixture was injected using the procedures described above. CTβ is transported both anterogradely and retrogradely from the injection site but does not cross synapses (Luppi et al. 1990). Thus, including CTβ in the inoculum allowed a discrimination of first-order neurons from neurons infected by transneuronal passage of the virus, and also defined the projection fields of neurons at the injection site. Immediately after surgery, and every day post-inoculation until perfusion, animals received a subcutaneous injection of Ketofen (Henry Schein: Animal Health; 2 mg/kg). With the exception of one 414 infected animal that survived 4 days, all animals were anesthetized and perfused transcardially with an aldehyde based fixative (detailed below) 72 hours post-inoculation to standardize comparisons of viral spread between animals.
Experiment 3
To determine if retrograde transneuronal passage of H129 is completely blocked we analyzed viral transport within the retina following injection of H129 or the recombinants into the lateral geniculate nucleus (LGN). The LGN is among cell groups in the brain that receive a monosynaptic projection from retinal ganglion cells (RCGs). RCGs are the sole axonal efferent from the retina to the brain and receive synaptic input from neurons in deeper retinal layers (Lund et al. 1974; Sefton et al. 2004). Thus, infection of RCGs and synaptically connected neurons in the deeper layers of the retina provides a rigorous assay for retrograde transneuronal passage of virus. H129 and its recombinants were injected sterotaxically into the LGN using coordinates AP: -4.5 mm, ML: 4.2 mm, DV: -5.5 mm obtained from the Swanson brain atlas (Swanson 1998). In a subset of animals (n=5) we injected a cocktail of virus containing CTβ using the procedures previously described. The surgical approach employed for striatal injections was repeated for these experiments. Animals were anesthetized and perfused 4 (n=10) or 5 (n=1) days post-inoculation. These survival intervals were selected to optimize detection of retrograde transneuronal spread of infection to deep layers of the retina.
Experiment 4
Three dual injection paradigms were employed to determine the ability of two recombinants, 414 (EYFP reporter) or 772 (EGFP reporter) and 424 (mRFP reporter), to co-infect neurons and efficiently express unique fluorescent reporters. The EGFP and EYFP reporters of 772 and 414 produce cytoplasmic labels while the mRFP reporter of the H129 recombinant is part of a fusion protein that labels viral capsids. The first set of injections (n=2) were made into two adjacent areas in the cerebral cortex at coordinates AP: 0.5 mm, ML: 1.0 & 4.0 mm, DV: -0.8 & -1.1 mm. In the second paradigm (n=4) the recombinants were injected into homotypic areas of the left and right cerebral cortex at rostrocaudal level AP: 0.5 mm, ML: ±2.5 mm, DV: -1.0 mm. In the third experiment (n=1) recombinants were injected into the left and right striatum in the same approximate region injected in experiment 1. The animals were anesthetized and perfused either 3 (n=6) or 4 (n=1) days following virus injection.
Tissue preparation
At the post-inoculation survival intervals indicated above, animals were deeply anesthetized with Fatal Plus (Vortech Pharmaceuticals, Dearborn, Michigan; 390 mg/ml sodium pentobarbital) prior to transcardiac perfusion with 100 ml of 0.9% saline solution followed by 300 ml of 4% paraformaldehyde-lysine-periodate (PLP) fixative (McLean and Nakane 1974). The brains were removed and immersed in sequential changes of PLP fixative and sucrose solutions (20% and 30%) for one day each at 4°C. Cryoprotected brains were sectioned at 35 μm/section in the coronal (n=36) or sagittal (n=2) planes using a freezing microtome (SM2000R/Leica). Sections were collected serially into six bins of cryoprotectant (Watson et al. 1986) yielding a sampling frequency of 210 μm and stored at -20°C prior to processing for immunocytochemical localization of viral antigens or analysis of fluorescent reporter expression (described below). The retinae were from animals in experiments 3 were cryoprotected in buffered sucrose solutions and sectioned at 20 μm perpendicular to the long axis using a cryostat (HM 505E/Micron). Retina sections were mounted on gelatin coated slides and coverslipped with Vectastain Hard Set medium for analysis of reporter gene expression, either alone or in combination with immunocytochemical localization of CTβ. Retinae from animals infected with H129 recombinants were also processed for immunoperoxidase localization of viral antigens with a rabbit polyclonal anti-HSV-1 antiserum (Dako Cytomation).
Immunoperoxidase localizations
Free-floating sections from each case were incubated with the rabbit polyclonal anti-HSV-1 antiserum at a 1:20,000 dilution, a biotinylated donkey-anti-rabbit secondary antibody at 1:200 dilution (Jackson ImmunoResearch), and Vectastain Elite reagents (Vector Laboratories). The tissue was then immersed in 100 ml of a saturated diaminobenzidine solution for 10 minutes before addition of 35 μl of H2O2 to catalyze the peroxidase reaction. The reaction was terminated with repeated phosphate buffered saline rinses after 3.5 to 5 minutes. Reacted tissue was mounted and coverslipped on Superfrost Plus microscope slides (Fisher Scientific, Pittsburgh, PA). Tissue sections from some of the cases were counter-stained with Neutral Red or Cresyl Violet to aid cytoarchitectural analysis. Details of these procedures as applied in our laboratory have been published (Card and Enquist, 1999, 2012).
Immunofluorescence localizations
Immunofluorescence was used to localize viral antigens in combination with CTβ and/or glial fibrillary acidic protein (GFAP), an intermediate filament that is a specific marker of astrocytes (Reeves et al. 1989). Free-floating tissue sections were incubated with the Dako anti-HSV-1 polyclonal antiserum (1:2,000) combined with goat anti-CTβ (List Biochemicals; 1:5,000) or mouse anti- GFAP (Millipore; 1:1,000), either individually or in combination. Following dilution in the primary antisera for 24 hours at room temperature the tissue was washed in multiple changes of sodium phosphate buffer and incubated in a cocktail of species-specific secondary antibodies conjugated to Alexafluor 488 (Jackson ImmunoResearch Laboratories, 1:500) for detection of viral antigen, Cy3 (Jackson ImmunoResearch Laboratories, 1:250) for detection of CTβ, and/or Cy5 (Jackson ImmunoResearch Laboratories, 1:300) for detection of GFAP. The incubation in secondary antibodies was conducted at room temperature and terminated after 2 hours by room temperature buffer washes. The sections were then mounted on gelatin coated slides and coverslipped with Vectastain Hard Set Medium (Vector Laboratories).
Reporter protein localizations
Sections from each of the H129 recombinants were washed in repeated changes of sodium phosphate buffer at room temperature, mounted on gelatin coated slides, air dried, and coverslipped using Vectastain Hard Set medium (Vector Laboratories).
Analysis of cell morphology and progression of infection: Experiment 2
The stages of viral replication and the virulence of infection were determined for each recombinant virus. Sections prepared for immunoperoxidase and immunofluorescence localization of viral antigens were imaged using an Olympus BX51 photomicroscope equipped for bright field and epifluorescence analyses. Stages of infection were determined on the basis of the distribution of viral antigens within infected neurons. Differential concentration of viral antigens within the cell nucleus defined an “early” stage of infection. The presence of viral antigens within the nucleus, soma, and proximal dendrites cytoplasm defined an “intermediate” level of infection. Dense concentration of viral antigens within the nucleus and distribution throughout the somatodendritic compartment was classified as a “late” infection. Staging of infection using these objective criteria provided a qualitative means of assessing the progression of infection produced by each recombinant after injection of virus into striatum.
In prior studies examining the invasiveness of PRV, we demonstrated that the magnitude of reactive gliosis strongly correlates with both the virulence of the infecting virus as well as the temporal kinetics of viral infection (e.g., the most pronounced reactive gliosis was noted in relation to the longest infected neurons) (Card and Enquist 1995; Card et al. 1993). We therefore tested the hypothesis that the magnitude of reactive glial responses would provide a reliable index of the progression of infection of the H129 recombinants. We evaluated cytopathology displayed by infected neurons within cell groups infected by transneuronal passage of virus and also characterized the extent of reactive gliosis in the same cell groups. Markers of cytopathology included alteration of nuclear morphology (e.g., invaginations) and location within the cell soma and vesiculation of the soma cytoplasm. These markers were compared within equivalent components of infected circuitry from animals of the same postinoculation interval. Dual and triple labeling immunofluorescence was utilized to assess the extent of reactive astrogliosis and glial infection within infected circuitry. Sections from each case were imaged with an Olympus BX51 epifluorescence microscope equipped with filters specific for each of the fluorophores used in the analysis. Recruitment of cells of monocytic lineage to areas of infection, a marker of late stage infection, was also assessed via immunocytochemical localization of the ED1 antigen, a marker of immune cells of monocytic lineage. This was accomplished using a mouse monoclonal (Millipore) and donkey anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories) in immunoperoxidase localizations.
Quantitative measures were also used to assess the anterograde and retrograde progression of infection through basal ganglia circuitry for each recombinant. Numbers of neurons within second-order anterograde cell groups – the subthalamic nucleus (STN), the ventral lateral thalamic nucleus (VL), and the ventral medial thalamic nucleus (VM) – were counted in two equivalent coronal sections for cases in which the brain was sectioned in the coronal plane. Infected neurons were mapped at 40× magnification using a BX53F Olympus microscope equipped with a Ludl stage and StereoInvestigator image analysis software (version 8; MicroBrightfield, Inc.). Each cell group was systematically scanned using StereoInvestigator software and infected neurons were marked. The objective criterion for counting neurons was the presence of viral immunoreactivity within a clearly defined cell nucleus. Thus, the quantitative analysis included neurons in all stages of viral replication; e.g., early, intermediate, and late. In this fashion both the distribution and number of infected neurons within a cell group were recorded. The average number of infected neurons ± the standard error of the mean (SEM) was calculated for each recombinant virus in each region of interest. Student's t-tests (Bonferroni correction p=0.0167) were used to determine if differences in numbers of neurons within individual cell groups were statistically different among recombinant viruses. The stages of infection of neurons in superficial and deep cortical layers were also subjected to quantitative analysis. Two sections through somatosensory cortex (Swanson Atlas plates 27 & 28) were chosen for this analysis and cells were counted in 200 μm wide columns that were perpendicular to the pial surface and extended to the underlying white matter. The position of these columns in the lateral primary somatosensory cortex adjacent to the medial edge of secondary somatosensory cortex was standardized between cases. Neurons within these columns were classified as “early” in infection if viral antigen was confined to the cell nucleus and soma or “late” if viral antigen extended into the dendritic tree. Student's t-tests were used to compare the overall number of infected neurons in each layer and the proportions of late infected neurons within each layer number of late infected neurons divided by the total number of infected neurons). A significance value of P ≤ 0.05 was used for these comparisons and the data were presented as the mean ± the SEM.
Analysis of retrograde and anterograde transneuronal infection: Experiment 3
Retrograde transport of H129 and the recombinant viruses from the LGN was evaluated to assess the ability of H129 recombinants to infect by retrograde transneuronal transport. Sections of retina were initially examined using epifluorescence to determine areas that contained infected neurons. A minimum of 12 complete sections of each retina was examined for each case, and the extent of the spread of infection through retinal layers was determined throughout the entire extent of the section. Photographs of these regions taken with the epifluorescence microscope were then used to guide analysis with a Leica TCS SP5 II confocal microscope (Leica Microsystems, Buffalo Grove, IL) using a 63× oil objective. Photographs of individual optical planes and Z-stacks of infected regions of the retina were also obtained using confocal microscopy.
Anterograde transneuronal spread was quantified in four efferent targets of the LGN complex – the primary visual cortex, the zona incerta (ZI), the periaqueductal gray (PAG), and the superior colliculus (SC) (Figure 9; (Ribak and Peters 1975; Moore et al. 2000a)). For the PAG and ZI, the number of infected neurons within the entire nucleus in a single coronal plane corresponding to Swanson atlas plates 47 and 26, respectively was counted for each case. The number of infected neurons within a 200 μm wide column in the lateral third of the SC was counted in two coronal sections approximating level 38 of the Swanson atlas. The position of the column in the SC was standardized across cases and encompassed the full dorsoventral extent of superficial SC layers that are recipient of input from the intergeniculate and ventral subfields of LGN. Infected neurons within primary visual cortex, a target of the dorsal LGN, were also counted in a 200 μm wide column extending from the pial surface to the underlying white matter in one coronal section standardized between animals at level 40 of the Swanson atlas. Neurons within the primary visual cortex were also coded according to the lamina that they occupied.
Figure 9.
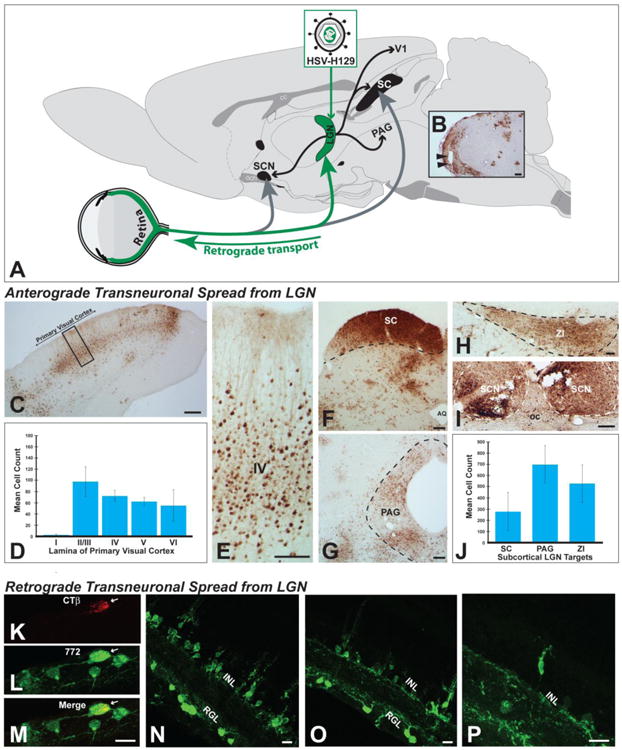
Anterograde and retrograde transneuronal infection of H129 and its recombinants in visual circuitry are illustrated. Figure A illustrates the experimental paradigm used for these assessments. Virus was injected into the lateral geniculate complex (LGN) and transneuronal infection was evaluated in the retina and in CNS cell groups innervated by LGN subdivisions. Injections centered in the LGN infected all subdivisions of the complex (dorsal lateral geniculate, ventrolateral geniculate, & intergeniculate leaflet; Fig. 9B). All central cell groups that receive projections from these LGN subfields exhibited infected neurons. These included the primary visual cortex (Figs. 9C & D) and the superior colliculus (SC; Fig. 9F), periaqueductal gray (Fig. 9G), zona incerta (Fig. 9I), and suprachiasmatic nuclei (Fig. 9I). Quantitative analysis revealed large numbers of neurons within each of these projection fields (Figs. 9D & J). Evidence for further anterograde transneuronal spread of virus from these cell groups was also observed. Retinal ganglion cells that project densely to all LGN subdivisions were also infected in each case. Figures K – M illustrate an infected RCG that retrogradely accumulated CTb and was infected by 772 after injection of a cocktail of these tracers into LGN. Figures N – P illustrate retrograde transneuronal spread of virus from retinal ganglion neurons that is largely confined to the inner nuclear layer of the retina. See text for more extensive discussion. Abbreviations: AQ = cerebral aqueduct, GCL = ganglion cell layer, INL = inner nuclear layer, oc = optic chiasm, PAG - periqueductal gray, SC = superior colliculus, SCN = suprachiasmatic nuclei, ZI = zona incerta. Marker bars for B, C, F & G = 200 μm, marker bar for D & I = 100 μm, marker bar for H = 50 μm, marker bars for K -P = 15 μm.
Analysis of reporter expression and dual infection: Experiment 4
Expression of the native fluorescence produced by reporter proteins unique to each recombinant was evaluated in all experimental animals. Reporter expression was photographed using epifluorescence followed by confocal microscopy as described above. Colocalization of reporters in neurons infected with recombinants 414, 424 and 772 in dual infection paradigms was assessed using the same approach.
Results
Each of the H129 recombinants used in this analysis exhibited multi-synaptic transneuronal spread that was heavily biased in the anterograde direction. Although anterograde transneuronal spread was the predominant mode of viral infection in each of the tested circuits we also documented temporally delayed retrograde transneuronal passage of the recombinants. All of the recombinants exhibited efficient reporter gene expression in both single and dual infection paradigms. In the following sections we, a) report in vitro data defining the replication kinetics and direction of virion transport of H129 and its recombinants within individual neurons, b) characterize cytopathology and the distribution of viral antigens resulting from infection and spread of H129 recombinants in vivo, c) define the invasive profiles and virulence of each recombinant following intracerebral injection of virus into the striatum or LGN, d) provide quantitative support for the conclusion that anterograde transneuronal spread occurs at a faster rate than retrograde transneuronal spread, and e) characterize the utility of reporter gene expression for labeling of neurons in single and dual infection paradigms.
Experiment 1: In Vitro Replication and Spread of H129 Recombinants
Replication Kinetics of H129 Recombinants in vitro
Three different regions of the H129 genome were targeted for insertion of fluorescent reporters (See Table 1). Recombinant 414 had the EYFP sequence inserted in the intergenic region between genes UL37 and UL38. As an alternative insertion locus, we tested insertion of the EGFP sequence in the intergenic region between genes UL26/UL26.5 and UL27 in recombinant 772. These two diffusible reporters are expressed from the same immediate early CMV promoter. Recombinant 424 expresses mRFP fused to the N terminus of the capsid protein VP26 under the control of its native promoter. This reporter produces a fluorophore that is differentially concentrated in the cell nucleus early in viral replication but spreads to the cell body as viral replication proceeds. In addition to being useful for in vivo tracing studies, recombinant 424 allows tracking of viral particle movement in live infected cells.
The replication kinetics of 414, 424 and 772 were measured in epithelial cells and compared to that of parental H129. Although all reporters showed strong fluorophore expression in infected cells (Figs. 1B-G), the single step growth curves revealed one important difference among them (Fig. 1A). Recombinant 414 grew less well and showed a one log-unit decrease in titer by 12-24 hours post infection. The growth defect also manifests as smaller plaques on Vero cells compared to plaques produced by H129, 772 or 424 (data not shown). This slightly reduced growth defect is in agreement with in vivo data showing that 414, which has an insertion in the intergenic space between UL37/UL38, is less virulent and produces a more restricted pattern of spread within equivalent survival intervals than either 772 or 424 (see in vivo data below). In contrast, recombinant 772 had a replication profile indistinguishable from that of parental H129, indicating that insertion of the reporter in the intergenic space of UL26/UL26.5-UL27 locus in H129 had no adverse effects (Fig 1A). Recombinant 424, had an apparent mild spread defect compared to H129, but this defect was not statistically significant. These data are consistent with prior reports involving replication of VP26-fluorophore fusions produced by other HSV-1 recombinants (Antinone et al. 2010; Nagel et al. 2008). The H129 recombinants 414 and 772 revealed strong diffuse fluorophore expression in infected epithelial or neuronal cells (Figs. 1B-E). Recombinants 414 and 772 provided strong labeling of neuronal cell bodies and also labeled axons (Figs. 1B & D and associated insets). Recombinant 424 showed a distinctive nuclear fluorescent profile consistent with the mRFP reporter being expressed as a fusion capsid protein (Figs. 1F & G), and was also useful for following particle transport through axons in live time imaging studies (data not shown).
Figure 1.
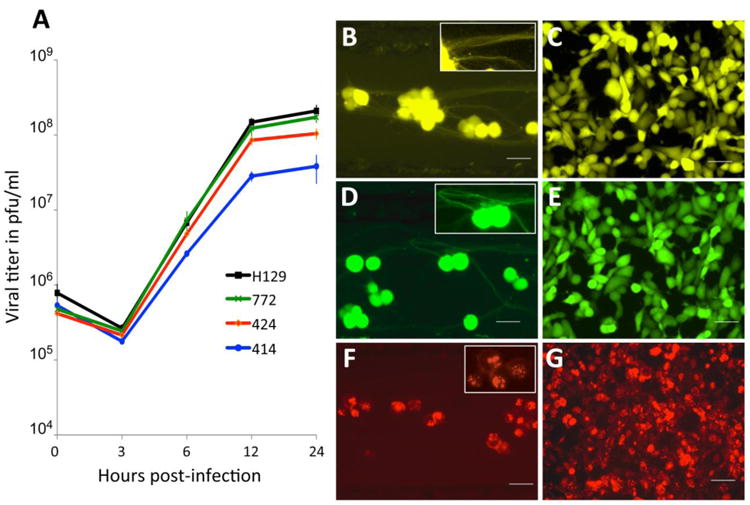
In vitro characterization of HSV-1 H129 recombinants. Single step growth curves for unmodified H129 and recombinants 414, 424 and 772 were measured. Vero cells were infected with each virus at an MOI of 10 and harvested at 0, 3, 6, 12 and 24 hpi. Viral titers expressed in pfu/ml were determined in Vero cells by limiting dilution and each point represents the average of triplicate assays with the standard deviation. The fluorescent profile of each recombinant infecting either SCG neurons (B, D and F) or Vero cells (C, E and G) is shown in B and C for 414, D and E for 772, and F and G for 424. The higher magnification insets illustrate fluorescent labeling in axons (B and D) or cell bodies (B, D, & F). Scale bar = 50 μm.
Anterograde spread of H129 in SCG neurons and epithelial cells
The anterograde spread capacity of H129 and the three new recombinants was measured in modified Campenot trichambers (Ch'ng and Enquist 2006). In this system, sympathetic SCG neurons were grown in the S compartment and axons penetrated into the N compartment (see methods for a more detailed description). Vero cells were plated on top of the axons in the N compartment (Fig. 2A). The cell bodies were infected and the infection was allowed to proceed for 48 hours prior to titration of infectious virus in both the S and N compartments (Fig. 2B). The viral titers in the S compartment were essentially identical for WT, H129, 424, and 772 recombinants. Titers of 414 were lower and are consistent with the aforementioned replication defect determined in Vero cells (Fig. 1A). The titers in the N compartment reflect spread to and replication of virus in Vero cells. H129, 424 and 772 have similar N compartment titers with no obvious defect in anterograde spread in vitro. The reduction in 414 titer is consistent with its replication defect in the S compartment and Vero cells. We conclude that H129 and recombinants 424 and 772 have no defects in a multi-step cycle that includes infection and replication in SCG cell bodies, sorting and anterograde transport of virus into axons, egress of virus from axons, and entry and spread of progeny virus in Vero cells.
Figure 2.
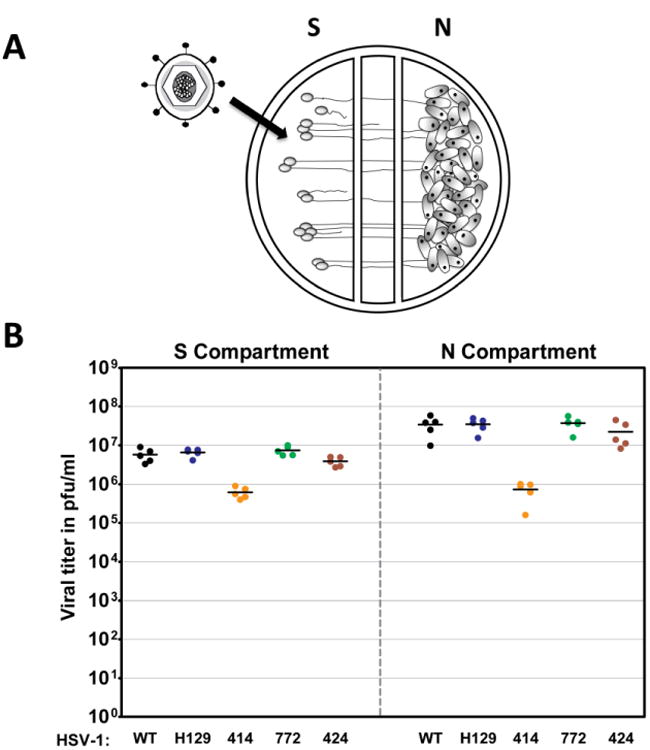
Anterograde spread of H129 and its recombinants in vitro. A schematized representation of the compartmentalized neuronal chamber system is shown in A. Sympathetic neurons seeded in the S compartment generate axons that grow into the N compartment of chambers. Infection of neurons in the S compartment leads to replication of virus in cell somas as well as anterograde transport and release of progeny virus in the N chamber. Infection of Vero cells seeded into the N compartment provides a quantitative measure of viral replication, spread and release from axon terminals. The content of the S and N compartments was collected 48 hpi with WT virus, H129, 414, 424 and 772 to determine the viral titer, expressed in pfu/ml, shown in B. Five chambers were used for each virus and the mean value for each set of chambers is denoted by a solid line.
Retrograde spread of H129 in SCG neurons
To determine if H129 is completely or partially defective in retrograde spread, we used the same chamber system as described above. In this analysis, we infected axons in the N compartment (no Vero cells added) and determined the replication of infectious virus in the SCG cell bodies in the S compartment (Fig. 3A). Parental H129 efficiently invaded axons and was transported retrogradely to cell bodies as well as WT. Similarly, recombinants 772 and 424 exhibited no statistically significant difference in the kinetics of replication when compared to H129 and WT (Fig. 3B). Recombinant 414 showed reduced titers in the S compartment most likely due to the replication defect noted above.
Figure 3.
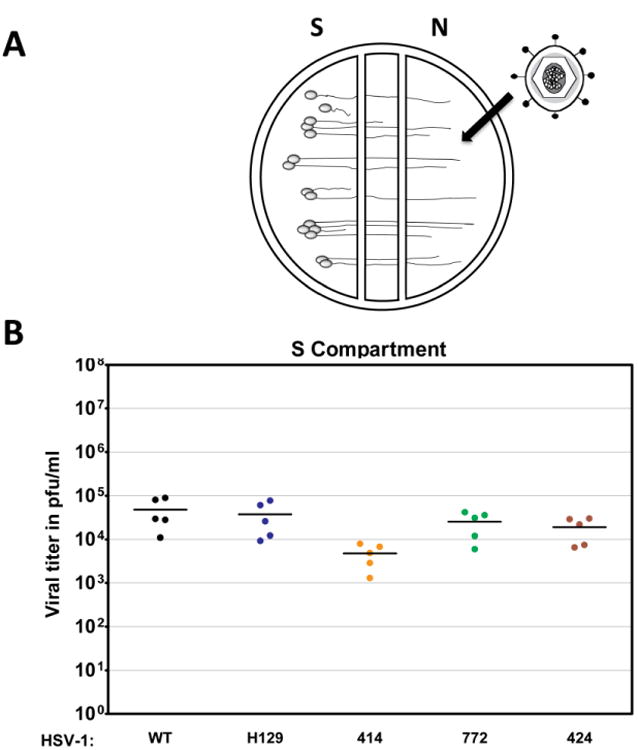
Retrograde infection of H129 and its recombinants in vitro. A schematized representation of the compartmentalized neuronal chamber system is shown in A. Sympathetic neurons seeded in the S compartment generate axons that grown into the N compartment of chambers. Virus was added to the N compartment to measure uptake and retrograde infection in the S compartment. The content of the S compartment of chambers was collected 24 hpi with WT virus, H129, 414, 424 and 772 to determine the viral titer expressed in PFU/ml as shown in B. Five chambers were used for each virus and the mean value for each set of chambers was denoted by a solid line.
We determined the extent of retrograde spread of infection in the S compartment from first-order infected neurons infected through their axonal projections into the N compartment to neurons that synapse upon first-order neurons but do not project axons to the N compartment. First-order neurons were labeled by application of DiI to the N compartment. Prior studies have demonstrated that lipophilic dyes such as DiI label neurons via lateral diffusion in the plasma membrane without transfer to unlabeled cells and without affecting cell viability or physiological properties of parent cell bodies in the S compartment (Fig. 4A; (Curanovic et al. 2009)). Infected neurons were counted by virtue of their reporter fluorescence and the ratio of primary infected neurons (DiI + reporter of infection) to synaptically linked neurons that do not project into the N chamber (no lipophilic dye labeling) (Fig. 4B). As shown in figure 4C, 95% and 94% of cell bodies directly connected to the N compartment were infected with either WT or 772 virus, indicating no significant difference in the rate or extent of primary retrograde infection from axons to cell bodies. Similarly, recombinant 414 infected 91% of primary neurons by 24 hours, a 3-4% reduction compared to WT and 772, indicating that it does not have a defect in primary neuron infection. Retrograde transneuronal infection of SCG neurons was measured by distinguishing primary infected cell bodies (virus reporter in dye labeled neurons) from those infected but not connected to the N compartment (virus reporter but no DiI labeling) (Fig. 4D). WT and 772 were indistinguishable in the extent of infection, infecting 40% and 39% of SCG neurons not projecting axons to the N compartment. Similarly, 414 infected 34% of the non-projecting axons. In evaluating these data it is important to note that prior studies have demonstrated that virus released from infected SCG neurons and axons remains tightly adherent to the cell membrane rather than being released to diffuse through the surrounding media (Curanovic et al. 2009; Koyuncu et al. 2013b). These data demonstrate similar rates of infection of first-order neurons and subsequent transneuronal spread of infection to synaptically linked SCG neurons following addition of WT or H129 recombinants to the N compartment (Figs.4C - D).
Figure 4.
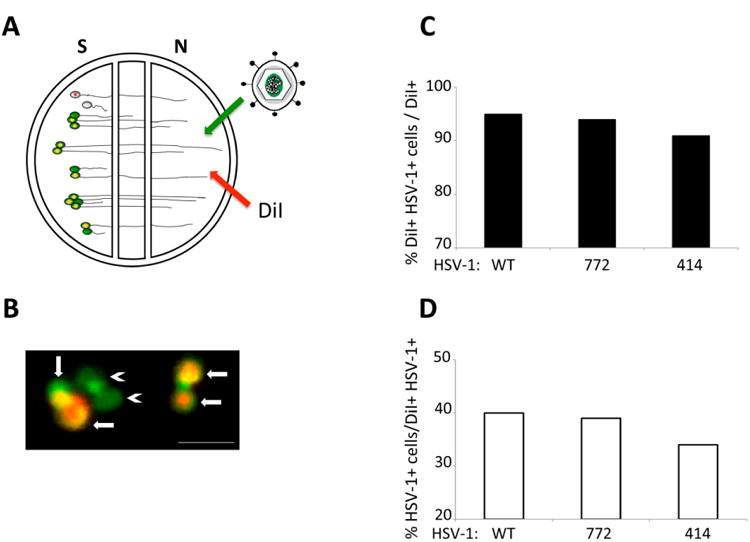
Primary and retrograde transneuronal spread of infection of H129. A schematized representation of the compartmentalized neuronal chamber system is shown in A. Sympathetic neurons seeded in the S compartment generate axons that grow into the N compartment of chambers. Virus was added to axons in the N compartment together with the lipophilic dye DiI. The dye migrates along axons to stain the parent cell bodies in the S compartment. Neurons that do not send an axon into the N compartment are not stained by dye. Cell bodies in the S compartment that were stained with dye (red fluorescence) and also infected with virus (green fluorescence) are depicted in B with arrows indicating primary infection. Cell bodies only stained in green and depicted with arrowheads represent higher-order infection resulting from retrograde transneuronal passage of virus. Scale bar = 50 μm. The amount of primary infection is shown in C as a percentage of the total number of cell bodies (DiI+) that are also infected with HSV-1 (DiI+ and HSV-1+). The amount of higher-order infection is shown in D as a percentage of primary infected cells (DiI+ and HSV-1+) over cells that are only infected with HSV-1 (HSV-1+). At least 600 cell bodies were counted in two chambers used for each condition.
Experiment 2: Replication and Spread through Basal Ganglia Circuits
First-Order Infection in the Striatum
The transport phenotype of the H129 recombinants was initially defined in basal ganglia circuitry following injection of virus into the striatum (Figs. 5A & B). Seventy-two hours after injection of 424 or 772 into the striatum, all cell groups that would be predicted on the basis of anterograde transneuronal spread were infected (Fig. 5B). In contrast, anterograde spread of 414 at survival intervals extending to 96 hours was far more restricted. The variability of 414 compared to 424 and 772 was also apparent in the spread of infection within the striatum. The most restricted spread resulted from injection of 414, which resulted in focal sites of viral immunoreactivity approximating 750 μm in widest diameter (Fig. 5C). Injection of 424 produced a substantially larger spread of infection around the cannula tract, with a widest diameter approximating 2,000 μm (Fig. 5D). The largest intrastriatal spread of infection resulted from injection of 772, which infected neurons in a zone approximating 3,000 μm in diameter (Fig. 5E). It is important to note that the distribution of viral antigens visible at 72 hours reflects both the direct infection of neurons around the cannula as well as replication and spread of virus from the earliest infected neurons. Thus, the subsequent anterograde spread of infection to striatal projection targets will reflect a temporal gradient in which the earliest transneuronal infection is derived from neurons immediately around the injection cannula, followed by temporally delayed spread of infection from striatal neurons infected by spread of infection through intrastriatal circuitry. Faster replication kinetics through local circuit connections within the striatum likely accounted for the more extensive intrastriatal spread of 424 and 772. Given these issues and the 72-hour survival intervals used in this study we cannot determine the precise zone of first-order infection based solely on the distribution of viral antigens within the striatum.
Figure 5.
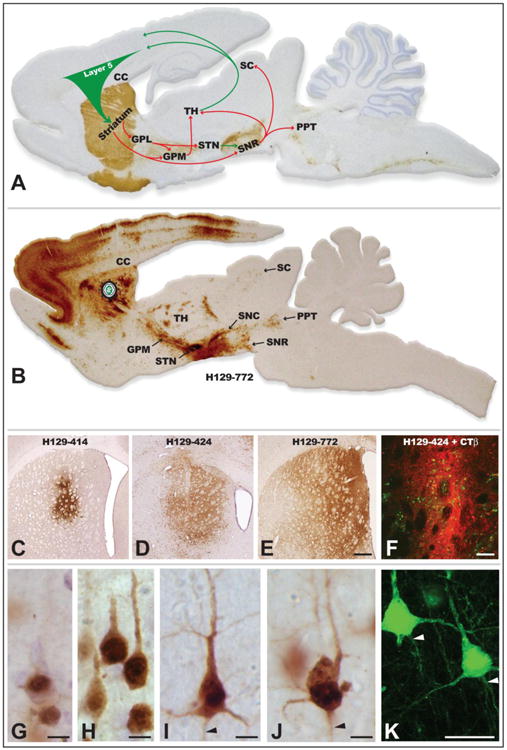
The organization of basal ganglia circuitry used to characterize the invasiveness of H129 recombinants is illustrated in A. Tyrosine hydroxylase was localized in the sagittal section using immunoperoxidase procedures (brown reaction product) to highlight the nigrostriatal dopaminergic projection that is a major component of the circuit and the section was counterstained with cresyl violet. Arrows indicate the direction of projection pathways that would produce anterograde transneuronal passage of virus after injection of virus into striatum. Green arrows indicate excitatory projections and red arrows inhibitory projections. Figure B illustrates the distribution of viral infection 72 hours after injection of H129 recombinant 772 into the striatum. The sagittal section shows a plane of section similar to that illustrated in A and infected cells were localized using an anti-HSV antiserum and immunoperoxidase procedures. Images C – F illustrate the distribution of infected neurons in the striatum 72 hours after injection of H129 recombinants into the striatum, either alone (C – E) or in combination with the classical tracer CTβ. Note the variability of the intrastriatal spread of infection produced by recombinants 414, 424 and 772. The primary site of injection is marked by the dense deposit of CTβ (red fluorescence), which distinguishes the zone of first order infection from transneuronal spread of virus (green fluorescence localizes infected cells). Stages of viral infection illustrative of the temporal kinetics of viral infection are illustrated in images G – K. Image K illustrates viral antigen localized with immunofluorescence and imaged with confocal microscopy. Infected cortical pyramidal neurons early in viral replication demonstrate differential concentration of viral antigens within the cell nucleus (G). At intermediate stages of infection (H) the dense staining of the nuclei of infected pyramidal neurons is complemented by the appearance of viral antigens in cytoplasm of cell somas and primary dendrites. With advancing replication (I – K) viral antigen fills the entire somatodendritic compartment and is also evident within the initial segments of axons (arrowheads). Cytopathology is also evident in the longest infected neurons, and is characterized by swelling of the cell body, displacement and invagination of the cell nucleus within the cell soma, and occasional beading of dendrites (J). Abbreviations: cc = corpus callosum, GPL = lateral subdivision of globus pallidus, GPM = medial subdivision of globus pallidus, PPT = pedunculopontine tegmental nucleus, SC = superior colliculus, SNC = substantia nigra, pars compacta, SNR = substantia nigra, pars reticulata, STN = subthalamic nucleus, TH = thalamus. Marker bar in E = 500 μm and images C – E are of the same magnification, marker bar in F = 100 μm, marker bars in G – J = 10 μm, marker bar in K = 25 μm.
In order to more precisely define the zone of first-order infection within the striatum we injected cocktails of virus and the classical tracer CTβ. In a prior investigation using this approach with PRV-Bartha, we demonstrated that the dense deposit of CTβ is larger than the effective zone of virus uptake (O'Donnell et al. 1997). Thus, we posited that dense CTβ deposition would provide an effective means of defining the region of first-order infection of the H129 recombinants. Injection of the recombinant virus-CTβ cocktails produced dense deposits of CTβ that were of similar size between cases. Since CTβ is also transported anterogradely into axons we also observed labeled fibers extending from the dense deposits into the surrounding striatum (Fig. 5F), consistent with the documented local circuit connections within striatum (e.g., (Bolam et al. 1986; Bolam and Izzo 1988; Wilson and Groves 1980). Infected neurons were present within the area of dense deposition, as well as in the surrounding striatum, with the number and density of infected neurons outside the dense deposit varying among recombinants (Fig. 5F). Since 414 produced the most restricted transport phenotype among the three recombinants we reasoned that it would provide the best perspective on the primary zone of infection. Analysis of cases injected with the 424/772-CTβ cocktail revealed a dense concentration of infected neurons within the confines of the dense CTβ deposit and scattered groups of infected neurons in the surrounding area (Fig. 5F). The distribution of the CTβ deposit around the cannula tract in the cases injected with 424 and 772 approximated the zone of infected neurons surrounding the cannula in cases injected with 414. On the basis of these findings we estimated the area of first-order infection occupies a radius of approximately 650 μm around the injection cannula.
Cellular Labeling, Cytopathology, and the Progression of Infection
In each of our experiments we initially localized infected neurons by means of immunoperoxidase methodology using a rabbit polyclonal antiserum reactive with HSV-1 structural proteins. Data from those studies revealed distinct stages of infection in which immunoreactivity was differentially concentrated in different compartments of infected neurons. At early stages of infection viral antigens were largely confined to the cell nucleus and, to a limited extent, the cell soma (Fig. 5G). With advancing replication viral antigens were concentrated in dense, punctate aggregates within the cell nucleus, throughout the cell soma, and within proximal dendrites (Fig. 5H). With further replication, viral immunoreactivity filled the dendritic tree, labeling even the most distal dendrites, and was also present in the axon hillock and initial segments of axons (Figs. 5I-K). Cytopathic changes in cell morphology provided further indications of the temporal kinetics of viral replication within infected neurons. In particular, we noted changes in nuclear morphology and pathological alteration of the dendritic tree. Nuclei of neurons advanced in replication characteristically developed invaginations and were displaced within cell bodies, often found at the polar aspects of the cell soma (Fig. 5J). Neurons exhibiting such changes also typically exhibited swelling of the soma (Fig. 5J). Thus, insight into the progression of infection through a circuit can be inferred by the distribution of viral antigens within neurons and the presence or absence of cytopathology.
As noted above, viral antigen was present within the axon hillock and initial segment of infected neurons (Figs. 5I-K). We therefore determined if the terminal fields of axons of infected neurons also contained viral antigen by examining projection fields of the striatum (GPL, GPM, and SNR) in cases injected with the CTβ:424/772 cocktail. CTβ transport from the injection sites robustly labeled terminal fields of striatal neurons in each of these areas (e.g., Fig. 6A). However, analysis of material dual stained for CTβ and viral antigen using epifluorescence and confocal microscopy only rarely revealed viral antigen within CTβ labeled afferents (arrowheads and insets in Figures 6a – c). Nevertheless, we commonly observed HSV immunopositive varicose axons within these projection fields that did not contain CTβ (yellow arrows in Figs 6a, a″, b, b″ and c, c″) and interpret these profiles as local circuit axons of interneurons or recurrent collaterals of neurons infected by anterograde spread of infection.
Figure 6.
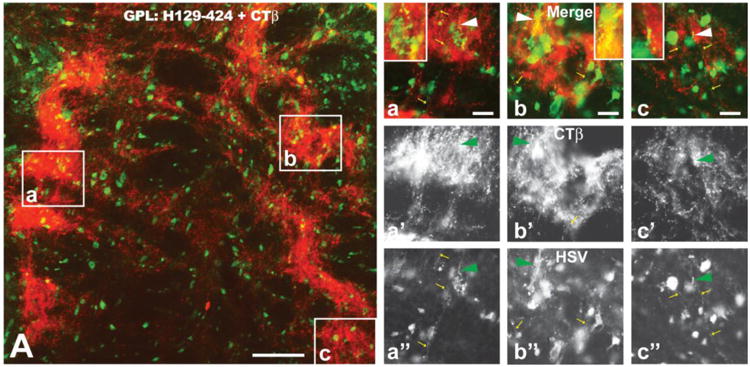
The distribution of viral antigens (green fluorescence) in relation to CTβ - labeled (red fluorescence) striatal axons in the lateral subdivision of the globus pallidus (GPL) is illustrated 72 hours after injection of a cocktail of CTβ and H129 recombinant 424 into the dorsomedial quadrant of the striatum. The GPL is shown in sagittal section with the left border of image A representing the rostral pole of GPL. Note the dense termination of striatal afferents within subregions of GPL. Infected neurons coextensive with striatal afferents were likely labeled by first-order anterograde transneuronal spread of virus. Infected neurons outside of fields containing labeled striatal afferents were likely labeled by anterograde spread of the recombinant within striatum and subsequent anterograde spread to GPL. The boxed areas in image A are shown at higher magnification in a, b and c. Images a′, b′ & c′ and a″, b″ & c″ show only the red (CTβ) or green (viral antigen) channels in a, b & c. Note that CTβ labeled afferents rarely contain viral antigens (arrowhead a – c″ and insets of a - c) but that axons of local circuit neurons or recurrent collateral of infected GPL neurons (small yellow arrows in a – c and a″ – c″) are immunopositive for viral antigens. See text for a more detailed explanation. Marker bar in figure A = 100 μm; the marker bars in a, b and c = 20 μm and the corresponding images of the red and green channels are of the same magnification.
Reactive Gliosis and the Progression of Infection
Reactive gliosis revealed insights into the progression of infection that may play an important role in biasing transport of virus through synaptic connections. As noted previously, reactive glial responses provide insight into the progression of PRV spread through a circuit, with the most robust reactive gliosis associated with neurons that have been replicating virus the longest period of time and/or that exhibit cytopathology (Card et al. 1993; Rinaman et al. 1993). Particularly important in this regard is the demonstration that reactive astrogliosis is the earliest component of this temporally organized response and that reactive astroglia ultimately lose their intermediate filament -GFAP - immunocytochemical phenotype at long post inoculation intervals (Rinaman et al. 1993). Localization of viral antigens in combination with detection of GFAP revealed the presence of reactive astrocytes throughout infected circuitry. Furthermore, the magnitude of reactive astrogliosis and the intensity of GFAP immunoreactivity reflected the extent of infection within cell groups contributing to anterograde spread of infection. For example, at the site of virus injection in the striatum we identified three distinct zones with differing degrees of reactive gliosis or GFAP immunoreactivity. Within the area of first-order infection (as determined by the dense deposition of CTβ), GFAP labeling was highly reduced or absent (Fig. 7B & 7b″′). This finding is consistent with prior light and electron microscopic analyses with PRV demonstrating that GFAP immunoreactivity in reactive astrocytes in areas of advanced infection is markedly reduced (Card et al. 1993; Rinaman et al. 1993). Examination of cell groups within the circuitry infected by anterograde transneuronal spread of infection also demonstrated altered levels of GFAP immunoreactivity in relation to infected neurons. For example, GFAP immunopositive reactive astrocytes were prevalent among neurons infected by transneuronal spread of virus to GPL (Fig. 7C). In contrast, the dense concentration of infected neurons in the subthalamic nucleus, which occurs subsequent to GPL infection (Fig. 5A), were surrounded by GFAP+ reactive astroglia, but exhibited only lightly stained GFAP+ astrocytes among the infected neurons (Fig. 7D, 7d & 7d′). Dual localizations also revealed viral antigen in a subset of reactive glia (Fig. 7E). Infected cells exhibiting oligodendrocyte morphology were also observed among axons in myelinated fiber tracts (Fig. 7F). This finding was particularly apparent within portions of the corpus callosum between infected homotypic regions of the cerebral hemispheres. In two cases virus was injected directly into corpus callosum rather than into the subjacent striatum. These injections produced local infection of oligodendrocytes within the white matter, but there was no evidence of infected cortical neurons projecting through that portion of the corpus callosum. We therefore conclude that H129 is not directly taken up by fibers of passage and that oligodendrocytes do not pass virus to the axons that they myelinate; rather the only means of spread is through direct infection of neurons and subsequent axonal transport of progeny virus.
Figure 7.
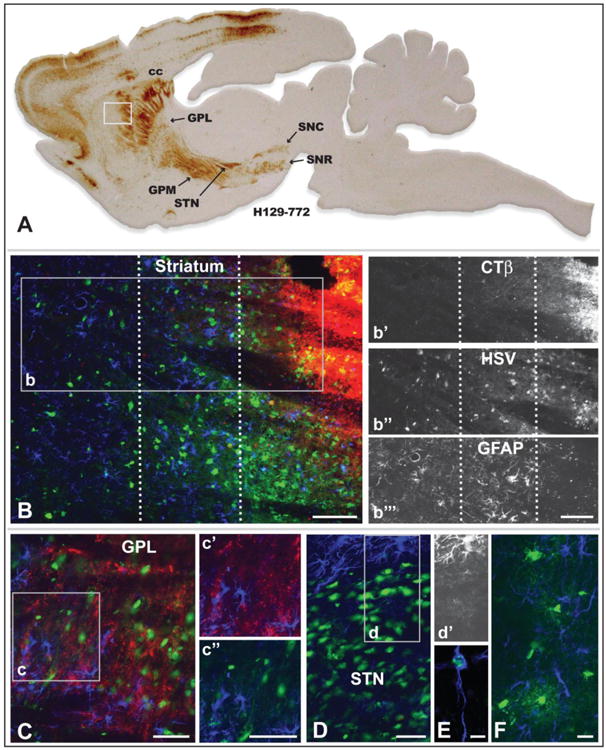
Reactive gliosis and non-neuronal infection in relation to infected neurons 72 hours after injection of a cocktail of recombinant 772 and CTβ into striatum is illustrated. Figure A is of a parasagittal plane through the neuraxis revealing the distribution of viral antigens localized with immunoperoxidase procedures. The white box in A illustrates the location of image B, which illustrates the distribution of CTβ (red fluorescence), viral antigens (green fluorescence), and GFAP-labeled astrocytes (blue fluorescence) localized by immunofluorescence in an adjacent section. The dotted lines in B define 3 zones distinguished by the differential distribution of CTβ, primary versus secondary spread of infection, and differing degrees of reactive astrogliosis. Individual channels for CTβ, HSV and GFAP in area b are shown in b′, b″, & b″′, respectively. Note that the primary site of first-order infection marked by the dense deposit of CTβ is distinguished by a dense aggregation of infected neurons and the down-regulation of the GFAP phenotype in reactive astrocytes (Fig. b″′). In the immediately adjacent region GFAP immunoreactivity reveals densely packed reactive astroglia exhibiting hyperplastic cell bodies and densely stained thick processes (Fig. b″′). Reactive astrogliosis is less marked in the zone most distant from the site of injection and is most prominent in relation to infected neurons (Fig. b″′). Figure C demonstrates that reactive astrogliosis is present throughout the primary site of infection in the lateral subdivision of globus pallidus. GFAP+ astroglia in the region of the inset are shown either in relation of CTβ labeled striatal axons or infected neurons. In contrast to GPL, the dense aggregation of infected neurons in the subthalamic nucleus (STN; image D) exhibits a GFAP phenotype similar to the site of recombinant injection in the striatum; e.g., the GFAP phenotype in reactive astroglia among infected STN neurons is down regulated while reactive astroglia in the area immediately surrounding the STN are densely immunopositive for GFAP. This is clearly apparent in image d′, which selectively illustrates the channel for GFAP immunoreactivity. A subset of reactive astroglia also become infected and replicate virus (E), as do oligodendrocytes within the corpus callosum (F) and other myelinated fiber pathways involved in the anterograde spread of infection. See text for a more detailed discussion. Marker bar in B and insets = 100 um, marker bars for C and areas of inset = 50 μm, marker bar for D = 50 μm with D and d′ the same magnification, marker bar for E = 7.5 μm, marker bar for F = 20 μm.
Anterograde Spread of Infection through the Basal Ganglia
As noted above, we observed anterograde spread of virus through the well-characterized circuitry of the basal ganglia (Figs. 5A & B, 6A). Infected cell groups included both GPL and GPM, the subthalamic nucleus, substantia nigra pars compacta and pars reticulata, the pedunculopontine tegmental nucleus, the superior colliculus, and transneuronal infection of cortical lamina through neurons of the motor thalamus (Figs. 5A & 6A). The distribution of viral immunoreactivity within neurons in each area varied such that we observed neurons in all stages of infection (e.g., early, intermediate, and late stages). This finding is consistent with neurons being infected by two waves of infection; directly from the injection site and in a temporally delayed manner that results from circuit related spread of infection within the striatum. Further support of this interpretation was evident in animals in which H129 was co-injected with CTβ. In these cases the projection fields of axons arising from the site of injection were densely labeled with CTβ and coextensive with large numbers of neurons advanced in infection (Figs. 7C). In contrast, the distribution of viral antigens within the majority of infected neurons outside of the CTβ labeled projection fields indicated that the neurons were earlier in the course of infection.
The distribution of viral antigens within infected neurons provided a reliable index of the temporal kinetics of infection within neurons. Analyzing these features within the context of polysynaptic connections among basal ganglia cell groups was informative in evaluating the progression of infection. For example, analysis of the laminar distribution of infected cortical neurons after injection of 772 into striatum revealed neurons in in both superficial and deep cortical lamina. In this regard, it is important to note that prior studies employing H129 for circuit analysis have demonstrated that the virus is transported in the retrograde direction from the injection site to infect neurons (Rinaman and Schwartz 2004; Zemanick et al. 1991). On this basis, one would expect two populations of neurons within the cortex after injection of virus into striatum; neurons concentrated in lamina V infected by retrograde transport of virus from the injection site (McGeorge and Faull 1989; Cowan and Wilson 1994) and neurons in both superficial and deep cortical layers infected by anterograde transneuronal spread of virus through thalamocortical and cortico-cortical projections (Herkenham 1979; Desbois and Villanueva 2001; Mitchell and Cauller 2001; Yamamoto et al. 1990; Aldes 1988). Further one would predict that neurons retrogradely infected from striatum would be more advanced in infection than those infected by anterograde polysynaptic spread of infection. In fact, this is what was observed (Figs. 8A-H, L & M). Neurons in the thalamus that give rise to projections to the cortex (VM & VL) were advanced in infection (Figs. 8A – D) and their projection fields in the cortex exhibited an abundance of neurons in early stages of infection (Figs 8E & F). Similarly, neurons in lamina V exhibited both advanced and early stages of infection (Figs. 8E & G). Quantitative analysis confirmed these assertions. Larger numbers of neurons were present in lamina V compared to superficial lamina (Fig. 8L) and the percentage of neurons in late stages of infection was significantly higher in lamina V (Fig. 8M). Additionally, a higher proportion of neurons early in infection were present in the superficial lamina compared to lamina V (0.61 ± 0.08 in lamina II/III compared to 0.50 ± 0.06 in lamina V; P = 0.005). The differential concentration of corticostriatal projection neurons in lamina V was confirmed in cases in which the striatum was injected with a cocktail of CTβ and 424 or 772. In these cases infected neurons labeled by retrograde transport of CTβ were concentrated within lamina V (Fig. 8H).
Figure 8.
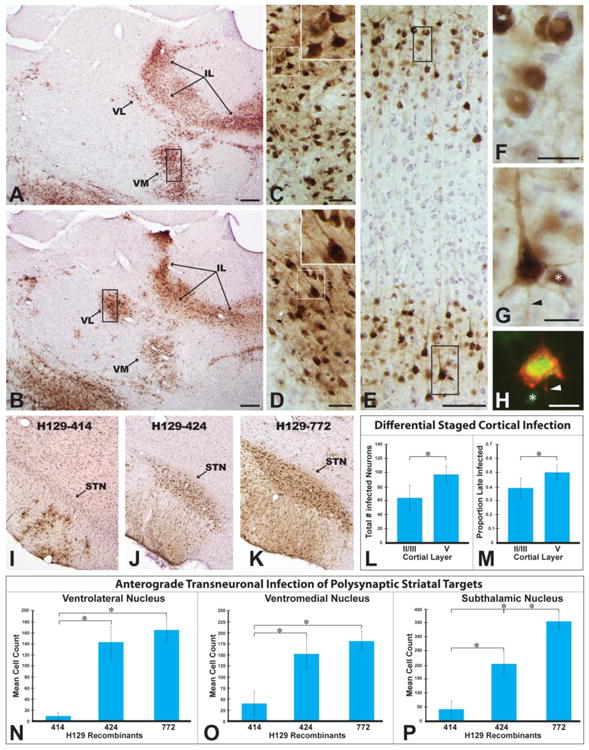
The progression of infection through basal ganglia circuitry following injection of H129 recombinants into striatum is illustrated. Immunoperoxidase localization of viral antigens in images A & B reveals the extent of anterograde transneuronal infection of ventromedial (VM) and ventrolateral (VL) thalamic cell groups at two rostrocaudal levels of the neuraxis 72 hours following injection of recombinant 772 into dorsomedial striatum. High magnification images of the boxed area of each VL in C and VM in D, and the associated insets, demonstrate that neurons are advanced in infection based upon the extensive dense intracellular distribution of viral antigens. The differential distribution of infected neurons in somatosensory cortex that results from anterograde transneuronal infection through thalamocortical projections is illustrated in E. Neurons early in infection are present in both superficial and deep cortical lamina (E, F & G). Lamina V also contains neurons advanced in infection (E & H), consistent with retrograde transport of recombinants from the striatum. Confirmation that neurons advanced in infection project to striatum was demonstrated by colocalization of CTβ (red fluorescence) and viral antigens (green fluorescence) in lamina V pyramidal neurons (H). Asterisks in G & H mark infected neurons of interneuron morphology and arrowheads mark labeled axons. Figure L demonstrates a significant difference in the total numbers of infected neurons within superficial and deep lamina of a standardized subfield of somatosensory cortex. Figure M demonstrates that lamina V in the same region of somatosensory cortex sampled in Figure L contains a higher percentage of late infected neurons than superficial cortical lamina in the same region. Figures I - K illustrate variable rates of infection of the subthalamic nucleus (STN) 72 hours after injection of recombinant 414 (I), 424 (J), or 772(K) into striatum. The graphs in figures N-P present quantitative data on the number of infected neurons within VL (L), VM (M), and STN (N) 72 hours after injection of the three recombinants into striatum. Additional abbreviation: IL = intralaminar nuclei. Marker bars for A & B = 200 μm, marker bars for C & D = 50 μm, marker bar for E = 100 μm, marker bar for F – H = 20 μm.
Additional quantitative analysis of the number of infected neurons that contribute to anterograde transneuronal spread of virus confirmed the differences in temporal kinetics of viral transport among H129 recombinants noted in the qualitative analysis. HSV-1 immunopositive neurons displaying a clearly discernible cell nucleus were counted with image analysis software in two equivalent coronal planes of section through VM, VL, and STN. The data demonstrated statistically significant differences in the number of neurons infected in each of these cell groups in animals infected with 414 compared to animals infected with 424 or 772 (Figs. 8N-P). In every case, the number of infected neurons in each of these cell groups was significantly less in animals infected with 414 compared to animals infected with either 424 or 772 (Figs 8N-P). Fewer numbers of infected neurons were also present in each of these cell groups in animals infected with 424, but the number was only statistically significant in the STN (Fig. 8P).
Retrograde Transport of H129 Recombinants in Basal Ganglia
As noted above, previous studies have reported that H129 infects neurons by retrograde transport from the site of injection, but that progeny virus does not pass retrogradely across synapses (Rinaman and Schwartz 2004; Zemanick et al. 1991). Consistent with this observation we observed infected neurons in all areas known to project to striatum. Notable among these regions were the intralaminar thalamic nuclei (central median, paracentral and central lateral), the parafasicular thalamic nucleus, the basolateral amygdala, and extensive territories in the cerebral cortex. Confirmation that neurons in these regions projected monosynaptically to the site of virus injection in the striatum was verified by retrograde labeling of infected neurons with CTβ in animals injected with the CTβ:virus cocktail. In some of these regions, e.g., neocortex, the extensive transneuronal passage of virus raised the possibility that neurons may have been infected by retrograde transneuronal spread of virus, albeit at a substantially lower temporal progression than anterograde transneuronal passage. For example, infected neocortical lamina V pyramidal neurons with extensive intracellular distribution of viral antigens were often associated with immediately adjacent small neurons of local circuit morphology at early stages of infection (Fig. 8G & H). Interneurons of this morphology synapse upon pyramidal neurons but are also often contacted by recurrent collaterals of pyramidal neurons. Thus, interneuron infection within this circuitry could result from either anterograde or retrograde transneuronal spread of infection.
The pattern of infection within cortical columns also suggested that the H129 recombinants may have spread via retrograde transneuronal passage through local circuit connections. For example, neurons in superficial cortical lamina II & III project to lamina V that, in turn, projects into striatum. As noted above, quantitative analysis of the stage of viral replication (early versus late) in columns within somatosensory cortex revealed that the number of infected neurons in layer V was significantly higher than in superficial layers (layers II/III = 64.86 ± 18.4; layer V = 95.93 ± 13.22; P = 0.011) and layer V contained a higher proportion of late infected neurons compared to layers II/III (layers II/III = 0.39 ± 0.075; layer V = 0.504 ± 0.056; P = 0.006). However it is also possible that infected neurons in layers II/III were infected by anterograde transneuronal passage of virus through VL and VM thalamocortical afferents that terminate within superficial layers.
Experiment 3: Replication and Spread through Retinal Circuitry
The possibility that H129 was infecting by retrograde transneuronal passage of virus is consistent with reports from Atherton's group (Archin and Atherton 2002a; Archin et al. 2003; Archin and Atherton 2002b). We therefore sought to test this hypothesis in visual circuitry. The rat visual system provides a rigorous means of detecting retrograde transneuronal infection since RCGs are the sole source of retinal projections to the brain and do not have recurrent collaterals into the deeper layers of the retina (Lund et al. 1974; Sefton et al. 2004). Furthermore, polysynaptic connections link photoreceptors with RCGs. Therefore, infection of neurons in the inner layers of the retina following retrograde transport of virus from retinorecipient regions of the neuraxis (e.g., the lateral geniculate nucleus) would occur only through viral replication and retrograde transneuronal spread from RCGs. We therefore injected parental H129 and its recombinants into the lateral geniculate nucleus of the thalamus (LGN) and surveyed the contralateral retina for infected neurons 4 or 5 days post-inoculation (Fig. 9A).
The injection sites for animals in this experiment involved one or more of the three LGN subdivisions (dorsal, ventral, intergeniculate leaflet; Fig. 9B). All of these subdivisions receive a dense retinal input from RGCs and all injections led to retrograde infection of RCGs. Additionally, replication of virus within LGN subfields led to anterograde transneuronal infection of neurons in regions that receive projections from the respective LGN subdivisions (Figs. 9C – J). For example, infected neurons were apparent in lamina IV of primary visual cortex, a projection target of dorsal LGN, and infection had spread to other lamina of the same region as well as to neurons in extrastriate cortical regions (Figs 9C – E). Additionally, infected neurons were present in the superior colliculus (Fig. 9F), periaqueductal gray (PAG; Fig. 9G), zona incerta (Fig. 9H), and suprachiasmatic nuclei (Fig.9I), all of which are projection targets of the vLGN or IGL (Moore et al. 2000b; Harrington 1997). Each of these regions contained large numbers of infected neurons that were advanced in replication, based upon the distribution of viral antigens and the presence of cytopathology. This was particularly apparent in the suprachiasmatic nucleus, which receives dense synaptic input from the intergeniculate leaflet of LGN (Card and Moore 1984; Card and Moore 1989; Moore and Card 1994), and exhibited cytopathology in neurons throughout the cell group bilaterally. Furthermore, neurons that are synaptic targets of SCN neurons (e.g., the subparaventricular zone and hypothalamic paraventricular nucleus, (Watts and Swanson 1987)) also were infected, consistent with further replication and anterograde transneuronal spread. The dense neuronal packing density and extent of cytopathology within the SCN precluded quantitative analysis. However, we did conduct a quantitative analysis of the magnitude of infection in four other projection targets of LGN neurons 96 hours following injection of recombinant 772 into the LGN. Each of these cases had injection sites that encompassed all three subdivisions of the LGN and large numbers of neurons were evident within the areas receiving projections from both the dorsal (primary visual cortex) and ventral (SC, PAG, ZI) subdivisions of LGN (Figs. 9D & J). This analysis consistently demonstrated large numbers of neurons within the LGN projection targets, late stage infection, cytopathology within the infected cell groups, and evidence for further anterograde transneuronal spread.
Clear evidence of retrograde infection of retinal ganglion neurons and retrograde transneuronal infection of synaptically connected neurons in deep layers of the retina was observed in all cases of virus injection into the LGN. Inspection of sections cut perpendicular to the plane of the retina revealed virus infected RGCs and transneuronal spread of infection to inner layers of the retina in a columnar fashion (Figs. 9E-H). A minimum of 12 complete sections was examined in each experimental case, and the extent of viral infection within cortical lamina was scored. We observed infection or retinal ganglion neurons in every section from each case (Figs. 9K – P). We also observed retrograde spread of infection into the deeper layers of retina in every section (Figs 9N – P). However, retrograde transneuronal passage of virus from RCGs was entirely confined to the inner nuclear layer, where we identified neurons principally exhibiting amacrine cell morphology (Figs 9N – P). Within columns of infected cells we also observed infection of cells exhibiting Müller cell morphology, which made accurate counts of numbers of infected neurons difficult to determine. Nevertheless, we never observed infected neurons in the photoreceptor layer, and infected bipolar neurons were rarely observed. In all cases, retrograde transneuronal infection was largely confined to cells exhibiting amacrine morphology in the inner nuclear layer. Since neurons in the inner nuclear layer project to but do not receive a projection from RGCs and do not project outside of the retina, the only means of infection in these cells from LGN injections is through multi-synaptic retrograde transport through RGCs. Furthermore, the retrograde transneuronal passage of virus through all retinal layers stood in marked contrast to the anterograde transneuronal passage of virus to projection targets of the LGN in the same experimental animals.
Experiment 4: Reporter Gene Expression & Dual Infection of Circuitry
The H129 recombinants all expressed the appropriate fluorescent protein reporters of infection. Dense cytoplasmic labeling of infected neurons with EYFP and EGFP resulted from infection with 414 and 772, respectively (Figs. 10A &B). Fluorophores expressed by these recombinants filled the soma and dendrites of infected neurons, with the density of labeling correlating with the stage of viral infection; e.g., the most extensive labeling occurred in neurons advanced in viral replication. The punctate capsid labeling with mRFP in 424 infected neurons reflected the replication kinetics of the recombinant virus in infected neurons (Fig. 10C). Labeling was first observed within the cell nucleus, producing increasingly dense aggregates of mRFP fluorescence as new capsids were assembled during replication. With advancing replication mRFP labeling was apparent in the cell cytoplasm as capsids migrated out of the nuclei of infected cells for envelopment and sorting into axons.
Figure 10.
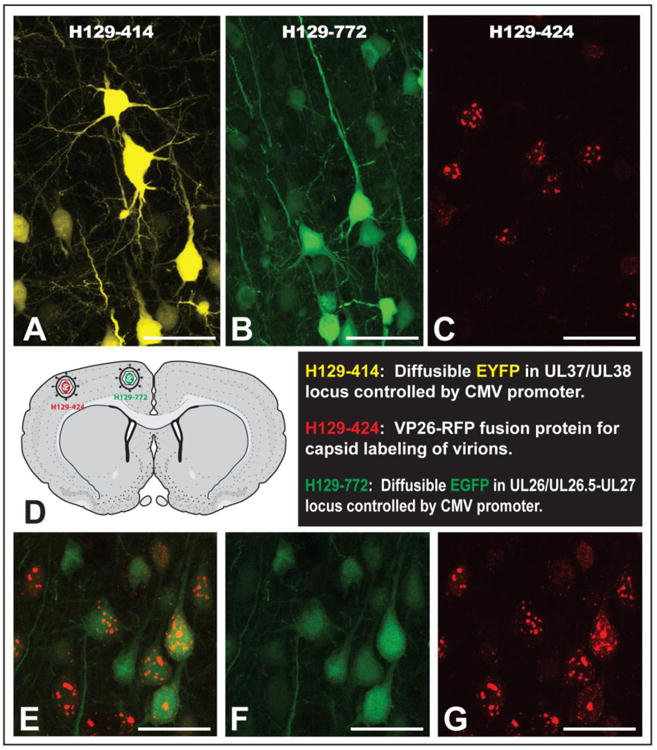
Efficiency of reporter gene expression and the utility of H129 recombinants for dual tracing studies are illustrated. The 414 and 772 reporters generate diffuse cytoplasmic fluorophores that densely stain the somatodendritic compartment of infected neurons (A & B). The 424 recombinant expresses mRFP as part of a fusion protein with the UL35 (VP26) gene under the control of its native promoter. VP26 is a capsid protein so mRFP appears as punctate labeling within infected neurons (C). Furthermore, the distribution of punctate mRFP labeling is reflective of the stage of viral replication such that early labeling is characterized by differential concentration of mRFP in cell nuclei and the subsequent appearance of mRFP in the cell soma, dendrites and axons occurs with advancing viral replication. Figure D illustrates one of three dual infection paradigms included in the analysis, each of which involved recombinant 424 in combination with either 414 or 772. In each paradigm anterograde transneuronal spread of recombinants resulted in the dual infection of neurons that were easily identified due to the differential concentration of reporters in the nucleus versus the cell cytoplasm (E – F). Marker bars A – G = 50 μm.
An important goal of our studies was to determine if neurons could be co-infected with HSV-1 H129 recombinants expressing unique fluorescent reporters. Toward this end we injected recombinant 424 in combination with 414 (n = 6) or 772 (n = 1) in spatially separated areas within the telencephalon. Three paradigms were used: a) injections into adjacent cortical areas of the same hemisphere (n=2), b) cortical injections into homotypic areas of opposite hemispheres (n=4), or c) injections into homologous regions of the left and right striatum (n=1). In each experimental paradigm we detected infected neurons expressing mRFP and EFYP or EGFP, demonstrating the capacity for co-infection and efficient reporter expression. Similar to single infection experiments, EYFP/EGFP expression was diffuse throughout the somatodendritic compartment and absent in the nucleus, while mRFP expression was punctate and heavily concentrated in infected cell nuclei. The differential concentration of reporter gene expression within the nuclear versus cytoplasmic compartments provided an unambiguous means of resolving both reporters in the same neurons (Figs. 6E-G). Single confocal optical planes and projection stacks of multiple optical planes acquired with confocal microscopy definitively demonstrate that individual neurons expressed both fluorophores (Figs. 6E-F).
Discussion
Over the last 25 years HSV-1 strain H129 has been used as a transneuronal tracer to define neural connections through anterograde transneuronal spread of infection. The anterograde phenotype has been confirmed by a number of investigators in different neural circuits and it is clear that the virus is efficiently replicated and transported anterogradely through polysynaptic pathways (Barnett et al. 1995; Lo and Anderson 2011; McGovern et al. 2012a; McGovern et al. 2012b; Rinaman and Schwartz 2004; Zemanick et al. 1991; Sun et al. 1996; Song et al. 2009; Vaughan and Bartness 2012; Dum et al. 2009; Kelly and Strick 2003). Nevertheless, the complex synaptology characteristic of many systems and the absence of detailed temporal analyses have interfered with the ability to definitively determine if there is a quantitative or absolute block to retrograde transneuronal spread. In this regard, evidence for retrograde transneuronal transport in the brain has been provided after inoculation of the anterior chamber of the eye (Archin and Atherton 2002a; Archin et al. 2003; Archin and Atherton 2002b). In this report we provide in vitro and in vivo evidence that H129 and its recombinants have the ability to spread retrogradely through neural circuits, albeit at a rate in vivo that is delayed relative to anterograde spread. In addition, we characterize reporter gene expression and invasiveness of three new H129 recombinants, whose availability will expand the utility of H129 as a circuit-tracing tool.
The H129 strain of HSV-1 was isolated from the brain of a patient suffering from acute necrotizing encephalitis in 1977 and was included in an analysis of neurovirulence of 23 strains of HSV-1 conducted by Dix and colleagues in 1983 (Dix et al. 1983). In that study it was reported that the HSV-1 strains segregated into three groups differing in virulence, leading the authors to conclude that “a strain-dependent variation in neural spread exists that may influence the ability of the virus to cause acute neurological disease.” Evidence in support of this conclusion was subsequently provided by Zemanick, Strick and Dix in a study analyzing the replication and spread of H129 and the McIntyre-B strains of HSV-1 (Zemanick et al. 1991). In that study evidence of differential transport of the two strains of virus was demonstrated, with H129 spreading preferentially in the anterograde direction and McIntyre-B spreading retrogradely from the primary motor cortex of cebus monkeys. However, it is important to note that neurons infected with H129 were also observed in areas that project to the motor cortex in that study (e.g., ventrolateral thalamus), and that the reciprocity of connections between these regions and the primary motor cortex prevented a definitive determination of the possibility that neurons were also infected by temporally delayed retrograde transneuronal passage.
Further evidence of biased anterograde transneuronal spread of H129 was provided by Rinaman and Schwartz, who characterized the spread of H129 infection through vagal circuitry of rats (Rinaman and Schwartz 2004). Inoculation of the stomach wall with H129 in that study produced retrograde transport and replication of virus in sympathetic postganglionic neurons but did not produce retrograde transneuronal infection of sympathetic preganglionic neurons in the spinal cord. Similarly, replication of H129 was observed in caudal brainstem dorsal motor vagal (DMV) neurons that innervate the stomach. However, in an important control in which anterograde transport of H129 into the dorsal motor vagal complex was prevented by deafferentation of vagal sensory afferents, no evidence of retrograde transneuronal spread H129 from infected DMV neurons was observed within 120 hours of inoculation of the stomach wall. Thus, it was clear that within the post inoculation intervals examined, H129 did not infect neural circuitry by retrograde transneuronal passage. Importantly, within the same time interval in intact animals, H129 produced extensive anterograde transneuronal infection.
The rapid and efficient anterograde transport of H129 through neural circuitry is also consistent with early studies conducted by LaVail and colleagues. In these investigations, the rates of spread of three strains of HSV-1 (H129, McIntyre-B, and F) were examined in either the trigeminal system following corneal inoculation (LaVail et al. 1997) or in central visual pathways following intravitreal injection (Gardner and LavVail 1999). Viral invasion of these circuits was characterized by detecting viral antigens in different segments of the circuits using Western blots of tissue extracts, through immunocytochemical localization of infected neurons, and in the trigeminal studies with transmission electron microscopy. In both systems H129 spread anterogradely through circuitry significantly faster than the F strain while the McIntyre-B strain was defective in anterograde spread. Importantly, in vitro analysis of the replication kinetics of these three strains in Vero cells revealed no statistically significant differences. Our data are consistent with these observations in that parental H129 and recombinants 424 and 772 showed no differences in replication kinetics in single step growth curves in vitro. Additionally, the same viruses were as efficient as WT virus in replication and anterograde spread through SCG neurons to infect Vero cells in modified Campenot chambers. Finally, in vivo each of these viruses rapidly spread anterogradely through the polysynaptic neural networks of the basal ganglia and thalamocortical projections within 72 hours of injection virus into striatum. Thus, even though we observed replication of H129 and these recombinants in neurons with first order projections to the site of injection in striatum, transneuronal transport of H129 and recombinants 424 and 772 was biased in the anterograde direction.
While our data are consistent with the many investigations demonstrating that H129 infection spreads efficiently through neural circuits in the anterograde direction, we also provide clear evidence that H129 can infect circuits through retrograde transneuronal spread. Injection of parental H129, 414, 424 and 772 into the LGN produced retrograde infection of retinal ganglion cells and invasion of neurons in deeper layers of retina that are presynaptic to RCGs 4 and 5 days post injection. Given the fact that RCGs do not have recurrent collaterals into the retina and are the only retinal cells to project to the brain, these data provide unequivocal proof of retrograde transneuronal passage of H129 within neural circuitry. These data are also in accord with the observations of Atherton and colleagues (Archin and Atherton 2002a; Archin et al. 2003; Archin and Atherton 2002b) who analyzed the spread of H129 through central circuits following inoculation of the anterior chamber of the eye. In their experiments evidence was provided for sequential retrograde transport of H129 through central circuits to infect retinorecipient neurons in the suprachiasmatic nucleus of the hypothalamus and, ultimately, RCGs and their presynaptic partners in deeper layers of the retina. The circuitry that they demonstrated to be infected in their study is the same autonomic circuitry that Pickard and colleagues demonstrated was infected by retrograde transneuronal passage of the Bartha strain of PRV from the eye (Pickard et al. 2002). We therefore conclude that the transport phenotype of H129, while predominantly anterograde, has a retrograde component.
The multi-synaptic retrograde transport property of H129 presents both challenges and opportunities for the fields of neurobiology and virology. We do confirm here that H129 is transported primarily in the anterograde direction and therefore remains the most effective means of defining multisynaptic pathways through anterograde transneuronal transport. However, special care must be taken to distinguish retrograde from anterograde transport. As demonstrated in the current investigation, combined injection of H129 with a retrograde or bidirectional inert tracer (e.g., CTβ) provides a clear means of distinguishing first order retrograde transport from neurons that may have been infected by retrograde transneuronal passage. In addition to the complication of deficient retrograde transport, it is vital to recognize that a retrogradely infected neuron will also spread virus anterogradely though its axon collaterals. For example, retrogradely labeled pyramidal cells from striatal injections will replicate H129 and subsequently send virions back down its axon to the striatum as well as its collateral targets to cortical interneurons, adjacent pyramidal cells, contralateral cortices and subcortical targets. Because of this possibility, one must be cautious in interpreting the route of infection in circuitry at longer post inoculation intervals.
Despite the caveats demonstrated here, many opportunities are available for researchers employing H129 and its derivatives as anterograde tracers of neuronal circuits. In particular the availability of recombinants expressing unique reporters permits dual infection studies via anterograde transneuronal passage. Here we successfully co-infected neurons with two recombinant strains of H129 demonstrating that this same technique may be applied in other experimental designs. For example further investigation of the basal ganglia with these recombinants could include studying the degree of convergence or divergence of striatal outputs onto the globus pallidus, or how di-synaptic striatal and mono-synaptic cortical outputs converge/diverge onto the subthalamic nucleus. Since H129 and its derivatives replicated well in all tested circuits, the present recombinants should be amenable to dual infection paradigms in neural systems of the investigators choosing.
When using multiple recombinants of H129, sound experimental design is critical for successful dual infections. Cells can replicate only a finite number (<7-10) copies of viral genomes (Kobiler et al. 2010). Accordingly, an infected neuron's ability to replicate more than one strain will be dependent upon the viral genomes of both viruses reaching the neuron in close temporal proximity. If one recombinant is present when the cell's replicative capacity saturates, then it will be extremely unlikely that the cell will replicate a second recombinant (Banfield et al. 2003). Therefore, in order to infect a neuron with multiple recombinants, parametrics must be conducted to optimize the chances that viruses will reach the target neuron in close temporal proximity. For example if one were to study the di-synaptic striatal and mono-synaptic cortical input to the subthalamic nucleus, one would have to inject the striatum at least 12-24 hrs prior to injecting the cortex. If both regions were injected during the same surgery, direct cortical input to the STN would arrive long before indirect striatal inputs. Neurons in the STN would then become saturated by the first recombinant and would be unable to transcribe viral DNA of the second recombinant. Without this taken into consideration, investigators are likely to underreport neurons receiving convergent inputs. The possibility of false negatives also emphasizes the importance of only interpreting positive data in studies of this nature.
Interestingly, we observed no defect in the temporal kinetics of H129 spread in either the anterograde or retrograde direction in our in vitro studies, but noted a clear temporal delay in the retrograde transneuronal spread of the virus in vivo. Although the mechanisms underlying these differences in the kinetics of anterograde versus retrograde transport remain to be defined, they are likely related to the more complex environment of the intact nervous system. It is also important to emphasize that the temporal kinetics of viral spread through circuitry in either the anterograde or retrograde direction will likely vary among different circuits due to differences in circuit architecture (e.g., numbers of neurons, lengths of axons, complexity of the branching architecture of the dendritic tree, and topographical differences in the termination of projection specific classes of afferents upon the somatodendritic compartment). The long post inoculation survival intervals in our analysis were used to optimize the possibility of observing retrograde transneuronal spread. Therefore, while our data demonstrate faster kinetics of anterograde spread compared to retrograde spread, they do not provide insight into the timing of the first order retrograde infection. This will certainly vary with the circuit under study, so defining the temporal spread of virus from injection sites through analysis of different post inoculation intervals should be incorporated into each novel use of H129 for circuit analysis.
The complete genome sequence of H129 has been determined and compared it to the wild type HSV-1 sequence (Szpara et al. 2010). That analysis demonstrated that more than 50% of the H129 genes contain missense or silent mutations, but no deletions or other null mutations were identified. It is likely that the retrograde spread defect involves several gene products. Because the retrograde spread defect of H129 was only detected in vivo and not in vitro, we have no rapid and facile method to assay the contribution of the many sequence changes in H129 to the phenotype. There are many differences between the in vitro chamber system and in vivo models; chambers contain a monoculture of sympathetic SCG neurons unlike the rodent CNS and PNS where several different cell types coexist, including immune effector cells (Koyuncu et al. 2013a). One key difference between the systems is that SCG cultures develop robust axonal processes that extend from the S to the N compartments, but the neurons do not grow dendritic arborizations in culture as they do in vivo (Lein and Higgins 1989; Tomishima and Enquist 2001; Fierbach et al. 2007). It may be that the H129 retrograde spread defect could be related to somatodendritic sorting and egress or with differences in the axonal versus dendritic partition of viral progeny.
Acknowledgments
This research was supported by NIH grants P40 RR018604 and PEW 2010-000225-002 postdoctoral fellowship to EAE. We thank Dr. Nicholas Brecha for critical evaluation of the retinal data, Vivian Allahyari for technical assistance with the animal studies, Beate Sodeik for providing the HSV-1 BAC used to produce H129-424, Jessica Brooks for assistance in construction of H129-772, and Stuart Mazzone for providing the plasmid used to construct the H129-772 recombinant. Santiago Rompani provided essential preliminary insights into H129 transport that were instrumental in the inclusion and design of the visual system experiments.
References
- Aldes LD. Thalamic connectivity of rat somatomotor cortex. Brain Research Bulletin. 1988;20:333–348. doi: 10.1016/0361-9230(88)90063-9. [DOI] [PubMed] [Google Scholar]
- Antinone SE, Zaichick SV, Smith GA. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. Journal of Virology. 2010;84(24):13019–13030. doi: 10.1128/JVI.01296-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin NC, Atherton SS. Infliltration of T-lymphocytes in the brain after anterior chamber inoculation of a neurovirulent and neuroinvasive strain of HSV-1. Journal of Neuroimmunology. 2002a;130:117–127. doi: 10.1016/s0165-5728(02)00213-8. [DOI] [PubMed] [Google Scholar]
- Archin NC, van den Boom L, Perelygina L, Hilliard JM, Atherton SS. Delayed spread and reduction in virus titer after anterior chamber inoculation of a recombinant of HSV-1 expressing IL-16. Investigative Ophthalmology & Visual Science. 2003;44(7):3066–3076. doi: 10.1167/iovs.02-1071. [DOI] [PubMed] [Google Scholar]
- Archin NM, Atherton SS. Rapid spread of a neurovirulent strain of HSV-1 through the CNS of BALB/c mice following anterior chamber inoculation. Journal of Neurovirology. 2002b;8(2):122–135. doi: 10.1080/13550280290049570. [DOI] [PubMed] [Google Scholar]
- Banfield BW, Kaufman JD, Randall JA, Pickard GE. Development of pseudorabies virus strains expressing red fluorescent proteins: new tools for multisynaptic labeling applications. Journal of Virology. 2003;77(18):10106–10112. doi: 10.1128/JVI.77.18.10106-10112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett EM, Evans GD, Sun N, Perlman S, Cassel MD. Anterograde tracing of trigeminal afferent pathways from the murine tooth pulp to cortex using herpes simplex virus type 1. Journal of Neuroscience. 1995;15:2972–2984. doi: 10.1523/JNEUROSCI.15-04-02972.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartha A. Experimental reduction of virulence of Aujeszky's disease virus. Magy Allatorv Lapja. 1961;16:42–45. [Google Scholar]
- Billig I, Foris JM, Enquist LW, Card JP, Yates BJ. Definition of neuronal circuitry controlling the activity of phrenic and abdominal motoneurons in the ferret using recombinant strains of pseudorabies virus. Journal of Neuroscience. 2000;20(19):7446–7454. doi: 10.1523/JNEUROSCI.20-19-07446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Ingham CA, Izzo PN, Levey AI, Rye DB, Smith AD, Wainer BH. Substance P-containing terminals in synaptic contact with cholinergic neurons in the neostriatum and basal forebrain: a double immunocytochemical study in the rat. Brain Research. 1986;397:279–289. doi: 10.1016/0006-8993(86)90629-3. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Izzo PN. The postsynaptic targets of substance P-immunoreactive terminals in the rat neostriatum with particular reference to identified spiny striatonigral neurons. Experimental Brain Research. 1988;70:361–377. doi: 10.1007/BF00248361. [DOI] [PubMed] [Google Scholar]
- Boldogkoi Z, Balint K, Awatramani GB, Balya D, Busskamp V, Viney TJ, Lagali PS, Duebel J, Pasti E, Tombacz D, Toth JS, Takacs IF, Scherf BG. Genetically timed, activity-sensor and rainbow transsynaptic viral tools. Nature Methods. 2009;6(2):127–130. doi: 10.1038/nmeth.1292. [DOI] [PubMed] [Google Scholar]
- Boldogkoi Z, Sik A, Denes A, Reichart A, Toldi J, Gerendai I, Kovacs KJ, Palkovits M. Novel tracing paradigms - genetically engineered herpesviruses as tools for mapping functional circuits within the CNS: present status and future prospects. Progress in Neurobiology. 2004;72:417–445. doi: 10.1016/j.pneurobio.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Braz JM, Enquist LW, Basbaum AI. Inputs to serotonergic neurons revealed by conditional viral transneuronal tracing. Journal of Comparative Neurology. 2009;514:145–160. doi: 10.1002/cne.22003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz JM, Rico B, Basbaum AI. Transneuronal tracing of diverse CNS circuits by Cre-mediated induction of wheat germ agglutinin in transgenic mice. Proceedings of the National Academy of Science USA. 2002;99(23):15148–15153. doi: 10.1073/pnas.222546999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RE, Herbison AE. Defining the gonadotrophin-releasing hormone neuronal network: Transgenic approaches to understanding neurocircuitry. Journal of Neuroendocrinology. 2007a;19(7):561–573. doi: 10.1111/j.1365-2826.2007.01561.x. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Herbison AE. Definition of brainstem afferents to gonadotropin-releasing hormone neurons in the mouse using conditional viral tract tracing. Endocrinology. 2007b;148(12):5884–5890. doi: 10.1210/en.2007-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano G, Card JP, Sved AF. Dual viral transneuronal tracing of central autonomic circuits involved in the innervation of the two kidneys in the rat. Journal of Comparative Neurology. 2004;471:462–481. doi: 10.1002/cne.20040. [DOI] [PubMed] [Google Scholar]
- Card JP, Enquist LW. Use of pseudorabies virus for definition of synaptically linked populations of neurons. In: Adolph KW, editor. Molecular Virology Techniques, Part A. Vol. 4. Academic Press; San Diego: 1994. pp. 363–382. [Google Scholar]
- Card JP, Enquist LW. Neurovirulence of pseudorabies virus. Critical Reviews in Neurobiology. 1995;9(2 & 3):137–162. [PubMed] [Google Scholar]
- Card JP, Enquist LW. Use and visualization of neuroanatomical viral transneuronal tracers. In: Badoer E, editor. Visualization Techniques: From Immunohistochemistry to Magnetic Resonance Imaging. Vol. 70. Neuromethods Humana Press; London: 2012. pp. 225–268. [Google Scholar]
- Card JP, Kobiler O, Ludmir EB, Desai V, Sved AF, Enquist LW. A dual infection pseudorabies virus conditional reporter approach to identify projections to collateralized neurons in complex neural circuits. PLoS One. 2011a;6:1–12. doi: 10.1371/journal.pone.0021141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card JP, Kobiler O, McCambridge J, Ebdlahad S, Shan Z, Raizada MK, Sved AF, Enquist LW. Microdissection of neural networks by conditional reporter expression from a Brainbow herpesvirus. Proceedings of the National Academy of Sciences, USA. 2011b;108(8):3377–3382. doi: 10.1073/pnas.1015033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card JP, Moore RY. The suprachiasmatic nucleus of the golden hamster: Immunohistochemical analysis of cell and fiber distribution. Neuroscience. 1984;13(2):415–431. doi: 10.1016/0306-4522(84)90240-9. [DOI] [PubMed] [Google Scholar]
- Card JP, Moore RY. Organization of lateral geniculate-hypothalamic connection in the rat. Journal of Comparative Neurology. 1989;284:135–147. doi: 10.1002/cne.902840110. [DOI] [PubMed] [Google Scholar]
- Card JP, Rinaman L, Lynn RB, Lee BH, Meade RP, Miselis RR, Enquist LW. Pseudorabies virus infection of the rat central nervous system: Ultrastructural characterization of viral replication, transport, and pathogenesis. Journal of Neuroscience. 1993;13(6):2515–2539. doi: 10.1523/JNEUROSCI.13-06-02515.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'ng TH, Enquist LW. Neuron-to-cell spread of pseudorabies virus in a compartmentalized neuronal culture system. Journal of Virology. 2005;79(17):10875–10889. doi: 10.1128/JVI.79.17.10875-10889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'ng TH, Enquist LW. An in vitro system to study trans-neuronal spread of pseudorabies virus infection. Veterinary Microbiology. 2006;113:193–197. doi: 10.1016/j.vetmic.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Cowan RL, Wilson CJ. Spontaneous firing patterns and axonal projections of single corticostriatal neurons in the rat medial agranular cortex. Journal of Neurophysiology. 1994;71:17–32. doi: 10.1152/jn.1994.71.1.17. [DOI] [PubMed] [Google Scholar]
- Curanovic D, Ch'ng TH, Szpara ML, Enquist LW. Compartmented neuron cultures for directional infection by alpha herpesviruses. Current Protocols in Cell Biology. 2009;43:26.24.21–26.24.23. doi: 10.1002/0471143030.cb2604s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curanovic D, Enquist LW. Directional transneuronal spread of alpha-herpesvirus infection. Future Virology. 2009;4(6):591–603. doi: 10.2217/fvl.09.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFalco J, Tomishima MJ, Liu H, Zhao C, Cai X, Marth JD, Enquist LW, Friedman JM. Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science. 2001;291:2608–2613. doi: 10.1126/science.1056602. [DOI] [PubMed] [Google Scholar]
- Desbois C, Villanueva L. The organization of lateral ventromedial thalamic connections in the rat: A link for the distribution of nociceptive signals to widespread cortical regions. Neuroscience. 2001;102:885–898. doi: 10.1016/s0306-4522(00)00537-6. [DOI] [PubMed] [Google Scholar]
- Dix RD, McKendall RR, Baringer JR. Comparative neurovirulence of herpes simplex virus type 1 strain after peripheral or intracerebral inoculation of BALB/c mice. Infection and Immunity. 1983;40(1):103–112. doi: 10.1128/iai.40.1.103-112.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolivo M. A neurobiological approach to neurotropic viruses. Trends in Neuroscience. 1980;3:149–152. [Google Scholar]
- Dum RP, Levinthal DJ, Strick PL. The spinothalamic system targets motor and sensory areas in the cerebral cortex of monkeys. Journal of Neuroscience. 2009;29(45):14223–14235. doi: 10.1523/JNEUROSCI.3398-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Enquist LW, Pomerantz RJ. The alpha-herpesviruses: molecular pathfinders in nervous system circuits. Trends in Molecular Medicine. 2008;14(3):134–140. doi: 10.1016/j.molmed.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist LW. Infection of the mammalian nervous system by pseudorabies virus (PRV) Seminars in Virology. 1994;5:221–231. [Google Scholar]
- Enquist LW. Life beyond eradication: veterinary viruses in basic science. Archives of Virology. 1999;15:87–109. doi: 10.1007/978-3-7091-6425-9_7. [DOI] [PubMed] [Google Scholar]
- Enquist LW, Card JP. Recent advances in the use of neurotropic viruses for circuit analysis. Current Opinion in Neurobiology. 2003;13:603–606. doi: 10.1016/j.conb.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Enquist LW, Tomishima MJ, Gross MJ, Smith GA. Directional spread of alpha-herpesvirus in the nervous system. Veterinary Microbiology. 2002;86:5–16. doi: 10.1016/s0378-1135(01)00486-2. [DOI] [PubMed] [Google Scholar]
- Fierbach B, Bisher M, Goodhouse J, Enquist LW. In vitro analysis of transneuronal spread of an alphaherpesvirus infection in peripheral nervous system neurons. Journal of Virology. 2007;81(13):6646–6857. doi: 10.1128/JVI.00069-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner JA, LavVail JH. Differential anterograde transport of HSV type 1 viral strains in the murine optic pathway. Journal of NeuroVirology. 1999;2:140–150. doi: 10.3109/13550289909021996. [DOI] [PubMed] [Google Scholar]
- Garner JA, LaVail JH. Differential anterograde transport of HSV type 1 viral strains in the murine optic pathway. Journal of Neurovirology. 1999;5(2):140–150. doi: 10.3109/13550289909021996. [DOI] [PubMed] [Google Scholar]
- Geerling JC, Mettenleiter TC, Loewy AD. Neuroanatomical Tract-Tracing 3: Molecules, Neurons, Systems. Springer; New York: 2006. Viral tracers for the analysis of neural circuits; pp. 263–303. [Google Scholar]
- Gerfen CR. Basal Ganglia. In: Paxinos G, editor. The Rat Nervous System. Elsevier Academic Press; Amsterdam: 2004. pp. 455–508. [Google Scholar]
- Gustafson DP. Pseudorabies. In: Dunne HW, Leman AD, editors. Diseases of Swine. The Iowa State University Press; Ames: 1975. pp. 391–410. [Google Scholar]
- Harrington ME. The ventral lateral geniculate nucleus and the intergeniculate leaflet: Interrelated structures in the visual and circadian systems. Neuroscience and Biobehavioral Reviews. 1997;21(5):705–727. doi: 10.1016/s0149-7634(96)00019-x. [DOI] [PubMed] [Google Scholar]
- Herkenham M. The afferent and efferent connections of the ventromedial thalamic nuclues of the rat. Journal of Comparative Neurology. 1979;183:487–517. doi: 10.1002/cne.901830304. [DOI] [PubMed] [Google Scholar]
- Kelly RM, Strick PL. Rabies as a transneuronal tracer of circuits in the central nervous system. Journal of Neuroscience Methods. 2000;103:63–71. doi: 10.1016/s0165-0270(00)00296-x. [DOI] [PubMed] [Google Scholar]
- Kelly RM, Strick PL. Cerebellar loops with motor cortex and prefrontal cortex of a nonhuman primate. Journal of Neuroscience. 2003;23(23):8432–8444. doi: 10.1523/JNEUROSCI.23-23-08432.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobiler O, Lipman Y, Therkelsen K, Daubechies I, Enquist LW. Herpesviruses carrying a Brainbow cassette revel replication and expression of limited numbers of incoming genomes. Nature Communications. 2010;1:146. doi: 10.1038/ncomms1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell Host & Microbe. 2013a;13(4):379–393. doi: 10.1016/j.chom.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuncu OO, Perlman DH, Enquist LW. Efficient retrograde transport of pseudorabies virus within neurons requires local protein synthesis in axons. Cell Host & Microbe. 2013b;13(1):54–66. doi: 10.1016/j.chom.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensson K, Nennesmo I, Persson L, Lycke E. Neuron to neuron transmission of herpes simplex virus. Transport from the skin to brainstem nuclei. Journal of Neurological Sciences. 1982;54:149–156. doi: 10.1016/0022-510x(82)90227-1. [DOI] [PubMed] [Google Scholar]
- Kuypers HGJM, Ugolini G. Viruses as transneuronal tracers. TINS. 1990;13:71–75. doi: 10.1016/0166-2236(90)90071-h. [DOI] [PubMed] [Google Scholar]
- LaVail JH, Topp KS, Giblin PA, Garner JA. Factors that contribute to the transneuronal spread of herpes simplex virus. Journal of Neuroscience Research. 1997;49:485–496. [PubMed] [Google Scholar]
- Lein PJ, Higgins D. Laminin and a basement membrane extract have different effects on axonal and dendritic outgrowth from embryonic rat sympathetic neurons in vitro. Developmental Biology. 1989;136(2):330–345. doi: 10.1016/0012-1606(89)90260-1. [DOI] [PubMed] [Google Scholar]
- Lo L, Anderson DJ. A Cre-dependent, anterograde transsynaptic viral tracer for mapping output pathways of genetically marked neurons. Neuron. 2011;72:938–950. doi: 10.1016/j.neuron.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewy AD. Pseudorabies virus: A transneuronal tracer for neuroanatomical studies. In: Kaplitt MG, Loewy AD, editors. Viral Vectors Gene Therapy and Neuroscience Applications. Academic Press; San Diego: 1995. pp. 349–366. [Google Scholar]
- Lund RD, Lund JS, Wise RP. The organization of the retinal projection to the dorsal lateral geniculate in pigmented and albino rats. Journal of Comparative Neurology. 1974;158(4):383–403. doi: 10.1002/cne.901580403. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Fort P, Jouvet M. Iontophoretic application of unconjugated cholera toxin B subunit combined with immunohistochemistry of neurochemical substances: a method for transmitter identification of retrogradely labeled neurons. Brain Research. 1990;534:209–224. doi: 10.1016/0006-8993(90)90131-t. [DOI] [PubMed] [Google Scholar]
- McCarthy KM, Tank DW, Enquist LW. Pseudorabies virus infection alters neuronal activity and connectivity in vitro. PLoS Pathogens. 2009;5(10):e1000–1640. doi: 10.1371/journal.ppat.1000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. Journal of General Virology. 1988;69(1531-1574):1531–1574. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- McGeorge AJ, Faull RLM. The organization of the projection from the cerebral cortex to the striatum in the rat. Neuroscience. 1989;29:503–537. doi: 10.1016/0306-4522(89)90128-0. [DOI] [PubMed] [Google Scholar]
- McGovern AE, Davis-Poynter N, Farrell MJ, Mazzone SB. Transneuronal tracing of airways-related sensory circuitry using herpes simplex virus 1, strain H129. Neuroscience. 2012a;207:148–166. doi: 10.1016/j.neuroscience.2012.01.029. [DOI] [PubMed] [Google Scholar]
- McGovern AE, Davis-Poynter N, Rakoczy J, Phipps S, Simmons DG, Mazzone SB. Anterograde neuronal circuit tracing using a genetically modified herpes simplex virus expressing EGFP. Journal of Neuroscience Methods. 2012b;209:158–167. doi: 10.1016/j.jneumeth.2012.05.035. [DOI] [PubMed] [Google Scholar]
- McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixative for immunoelectron microscopy. Journal of Histochemistry and Cytochemistry. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- Mettenleiter TC. Molecular properties of alphaherpesviruses used in transneuronal pathway tracing. In: Kaplitt MG, Loewy AD, editors. Viral Vectors Gene Therapy and Neuroscience Applications. Academic Press; San Diego: 1995. pp. 367–393. [Google Scholar]
- Mitchell DB, Cauller LJ. Corticocortical and thalamocortical projections to layer I of the frontal neocortex in rats. Brain Research. 2001;921:68–77. doi: 10.1016/s0006-8993(01)03084-0. [DOI] [PubMed] [Google Scholar]
- Moore RY, Card JP. Intergeniculate leaflet: An anatomically and functionally distinct subdivision of the lateral geniculate complex. Journal of Comparative Neurology. 1994;344:403–430. doi: 10.1002/cne.903440306. [DOI] [PubMed] [Google Scholar]
- Moore RY, Weis R, Moga M. Efferent projections of the intergeniculate leaflet and the ventral lateral geniculate nucleus of the rat. Journal of Comparative Neurology. 2000a;420:398–418. doi: 10.1002/(sici)1096-9861(20000508)420:3<398::aid-cne9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Moore RY, Weis R, Moga MM. Efferent projections of the intergeniculate leaflet and the ventral lateral geniculate nucleus in the rat. Journal of Comparative Neurology. 2000b;420:398–418. doi: 10.1002/(sici)1096-9861(20000508)420:3<398::aid-cne9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Nagel CH, Dohner K, Fathollahy M, Strive T, Borst EM, Messerle M, Sodeik B. Nuclear egress and envelopment of herpes simplex virus capsids analyzed with dual-color fjourescence HSV1(17+) Journal of Virology. 2008;82(6):3109–3124. doi: 10.1128/JVI.02124-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygardas M, Paavilainen H, Muther N, Nagel CH, Roytta M, Sodeik B. A herpes simplex virus-derived replicative vector expressing LIF limits experimental demyelinating disease and modulates autoimmunity. PLos One. 2013;8(5):e64200. doi: 10.1371/journal.pone.0064200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell P, Lavin A, Enquist LW, Grace AA, Card JP. Interconnected parallel circuits between rat nucleus accumbens and thalamus revealed by retrograde transynaptic transport of pseudorabies virus. Journal of Neuroscience. 1997;17(6):2143–2167. doi: 10.1523/JNEUROSCI.17-06-02143.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen LM, Ch'ng TH, Card JP, Enquist LW. Role of pseudorabies virus Us3 kinase during neuronal infection. Journal of Virology. 2006;80(1):6387–6398. doi: 10.1128/JVI.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard GE, Smeraski CA, Tomlinson CC, Banfield BW, Kaufman J, Wilcox CL, Enquist LW, Sollars PJ. Intravitreal injection of the attenuated pseudorabies virus PRV Bartha results in infection of the hamster suprachiasmatic nucleus only by retrograde transsynaptic transport via autonomic circuits. Journal of Neuroscience. 2002;22:2701–2710. doi: 10.1523/JNEUROSCI.22-07-02701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeranz LE, Reynolds AE, Hengartner CJ. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiology and Molecular Biology Reviews. 2005;69:462–500. doi: 10.1128/MMBR.69.3.462-500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter DD, Landis SC, Matsumoto SG, Furshpan EJ. Synaptic functions in rat sympathetic neurons in microcultures. II. Adrenergic/cholinergic dual status and plasticity. Journal of Neuroscience. 1986;6(4):1080–1098. doi: 10.1523/JNEUROSCI.06-04-01080.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves SA, Helman LJ, Allison A, Israel MA. Molecular cloning and primary structure of human glial fibrillar acidic protein. Proceedings of the National Academy of Sciences, USA. 1989;86(13):5178–5182. doi: 10.1073/pnas.86.13.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribak CE, Peters A. An autoradiographic study of projections from the lateral geniculate body of the rat. Brain Research. 1975;92:341–368. doi: 10.1016/0006-8993(75)90322-4. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Card JP, Enquist LW. Spatiotemporal responses of astrocytes, ramified microglia, and brain macrophages to central neuronal infection with pseudorabies virus. Journal of Neuroscience. 1993;13(2):685–702. doi: 10.1523/JNEUROSCI.13-02-00685.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L, Schwartz GJ. Anterograde transneuronal viral tracing of central viscerosensory pathways in rats. Journal of Neuroscience. 2004;24:2782–2786. doi: 10.1523/JNEUROSCI.5329-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Sears E. Herpes simplex viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fundamental Virology. Lippincott-Raven; Philadelphia: 1996. pp. 1043–1107. [Google Scholar]
- Sefton AJ, Dreher B, Harvey A. Visual System. In: Paxinos G, editor. The Rat Nervous System. 3rd. Elsevier Academic Press; San Diego: 2004. pp. 1083–1165. [Google Scholar]
- Song CK, Enquist LW, Bartness TJ. New developments in tracing neural circuits with herpesviruses. Virus Research. 2005;111:235–249. doi: 10.1016/j.virusres.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Song CK, Schwartz GJ, Bartness TJ. Anterograde transneuronal viral tract tracing reveals central sensory circuits from white adipose tissue. American Journal of Physiology. 2009;3:501–511. doi: 10.1152/ajpregu.90786.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Cassell MD, Perlman S. Anterograde, transneuronal transport of herpes simplex virus type 1 strain H129 in the murine visual system. Journal of Virology. 1996;70:5405–5413. doi: 10.1128/jvi.70.8.5405-5413.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW. Brain Maps: Structure of the Rat Brain. second. Elsevier; Amsterdam: 1998. [Google Scholar]
- Szpara ML, Parson L, Enquist LW. Sequence variability in clinical and lab isolates of Herpes Simplex 1 reveals new mutations. Journal of Virology. 2010;84(10):5303–5313. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. A wide extent of inter-strain diversity in virulent and vaccine strains of Alphaherpesviruses. PLoS Pathogens. 2011;7(10):e1002282. doi: 10.1371/journal.ppat.1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MP, Kobiler O, Enquist LW. Alphaherpesvirus axon-to-cell spread involves limited virion transmission. Proceedings of the National Academy of Sciences, USA. 2012;109:17046–17051. doi: 10.1073/pnas.1212926109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomishima MJ, Enquist LW. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. Journal of Cell Biology. 2001;154(4):741–752. doi: 10.1083/jcb.200011146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth IE, Wiesel O, Toth DE, Boldogkoi Z, Halasz B, Gerendai I. Transneuronal retrograde viral labeling in the brain stem and hypothalamus is more intense from the left than from the right adrenal gland. Microscopy Research Techniques. 2008;71:503–509. doi: 10.1002/jemt.20578. [DOI] [PubMed] [Google Scholar]
- Turner SL, Jenkins FJ. The roles of herpes simplex virus in neuroscience. Journal of NeuroVirology. 1997;3:110–125. doi: 10.3109/13550289709015801. [DOI] [PubMed] [Google Scholar]
- Vaughan CH, Bartness TJ. Anterograde transneuronal viral tract tracing reveals central sensory circuits from brown fat and sensory denervation alters its thermogenic responses. American Journal of Physiology. 2012;302:R1049–R1058. doi: 10.1152/ajpregu.00640.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RE, Wiegand ST, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–159. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- Watts AG, Swanson LW. Efferent projections of the suprachiasmatic nucleus: II. Studies using retrograde transport and simultaneous peptide immunohistochemistry in the rat. Journal of Comparative Neurology. 1987;258:230–252. doi: 10.1002/cne.902580205. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Groves PM. Fine structure and synaptic connections of the rat neostriatum: A study employing intracellular injection of horseradish peroxidase. Journal of Comparative Neurology. 1980;194:599–615. doi: 10.1002/cne.901940308. [DOI] [PubMed] [Google Scholar]
- Wittmann G, Rziha HJ. Aujeszky's disease (pseudorabies) in pigs. In: Wittmann G, editor. Herpesvirus Diseases of Cattle, Horses and Pigs. Kluwer; Boston: 1989. pp. 230–325. [Google Scholar]
- Yamamoto T, Kishimoto Y, Yoshikawa H, Oka H. Cortical laminar distribution of rat thalamic ventrolateral fibers demonstrated by the PHA-L anterograde labeling method. Neuroscience Research. 1990;9:145–154. doi: 10.1016/0168-0102(90)90030-i. [DOI] [PubMed] [Google Scholar]
- Yoon H, Enquist LW, Dulac C. Olfactory inputs to hypothalamic neurons controlling reproduction and fertility. Cell. 2005;123(669-682) doi: 10.1016/j.cell.2005.08.039. [DOI] [PubMed] [Google Scholar]
- Zaidi FN, Todd K, Enquist LW, Whitehead MC. Types of taste circuits synaptically linked to a few geniculate ganglion neurons. Journal of Comparative Neurology. 2008;511:753–772. doi: 10.1002/cne.21869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemanick MC, Strick PL, Dix RD. Direction of transneuronal transport of herpes simplex virus 1 in the primate motor system is strain-dependent. PNAS. 1991;88:8048–8051. doi: 10.1073/pnas.88.18.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]