

Abstract
Mast cell degranulation triggers hypersensitivity reactions at the body–environment interface. Adenosine modulates degranulation, but enhancement and inhibition have both been reported. Which of four adenosine receptors (ARs) mediate modulation, and how, remains uncertain. Also uncertain is whether adenosine reaches mast cell ARs by autocrine ATP release and ecto-enzymatic conversion. Uncertainties partly reflect species and cell heterogeneity, circumvented here by focusing on homogeneous human LAD2 cells. Quantitative PCR detected expression of A2A, A2B, and A3, but not A1, ARs. Nonselective activation of ARs with increasing NECA monotonically enhanced immunologically or C3a-stimulated degranulation. NECA alone stimulated degranulation slightly. Selective AR antagonists did not affect C3a-stimulated degranulation. NECA's enhancement of C3a-triggered degranulation was partially inhibited by separate application of each selective antagonist, and abolished by simultaneous addition of antagonists to the three ARs. Only the A2A antagonist separately inhibited NECA's enhancement of immunologically stimulated degranulation, which was abolished by simultaneous addition of the three selective antagonists. Immunological or C3a activation did not stimulate ATP release. NECA also enhanced immunologically triggered degranulation of mouse bone marrow derived mast cells (BMMCs), which was partially reduced only by simultaneous addition of the three antagonists or by the nonselective antagonist CGS15943. BMMCs also expressed A2A, A2B, and A3 ARs. but not A1AR detectably. We conclude that (a) A1AR is unnecessary for LAD2 degranulation or AR enhancement; (b) A2A, A2B, and A3 ARs all contribute to pharmacologic AR enhancement of LAD2 and BMMC degranulation; and (c) LAD2 cells depend on microenvironmental adenosine to trigger AR modulation.
Keywords: FcεRI, C3a, A2A, A2B, A3, ATP release
Introduction
Mast cells are inflammatory cells that arise from bone marrow and circulate in the blood to body tissues where they mature, expressing tissue-specific phenotypes [1]. These cells are critical in triggering hypersensitivity reactions of organs at the interface with the external environment, including the lung, upper airways, skin, gastrointestinal tract, and eye [1, 2]. Multiple stimuli can activate mast cells to degranulate, releasing preformed histamine, TNF-α, proteases, and heparin [3, 4]. Lipid mediators are later released over periods of minutes, with subsequent release of additional cytokines [5]. Immunologic activation is mediated by multivalent antigens crosslinking immunoglobulin IgE bound to high-affinity receptors (FcεRI) on the cell surface [3], triggering a principal signaling cascade [6]. In addition, degranulation can be stimulated by complementary signaling cascades initiated through agonist binding to cytokine and G protein-coupled receptors (GPCRs), including C3a complement component receptor [1, 6]. Both immunologic and C3a stimulation play major roles in activating mast cells in allergic asthma [7].
Adenosine has been known to be a major modulator of mast cell function for more than 35 years [8] and is thought to play a role in the pathogenesis of asthma and chronic obstructive pulmonary disease by its actions on mast cells [9, 10]. Which of the four subtypes of P1 adenosine receptors (ARs) mediates adenosine's modulation of degranulation has been unclear, in part because agonists' actions strongly depend on multiple factors. First, the species of origin is important since opposite effects have been reported in mouse and human mast cells [11], and the responses of even mouse and rat differ [12]. Second, the organ of origin plays a role. For example, low concentrations of adenosine enhance FcεRI-mediated degranulation of lung, but not skin, mast cells [13]. Third, heterogeneity of a mast cell population can be a confounding issue, even within a single organ [10, 14]. As an example, most mast cells in the lung are of the connective-tissue subtype lacking C3a receptors, but mucosal subtype mast cells expressing C3a receptors do infiltrate the lung [15]. Fourth, opposite actions have been reported over different concentration ranges of adenosine [10, 13] and at different times after agonist addition [10, 16]. Fifth, time has been found of importance in some studies, in terms of both whether the AR agonist is applied before, during or after applying the degranulating stimulus and whether the effect is assessed shortly or late after applying the purine (the "Discussion" section). Given these confounding variables, identification of the AR receptor(s) regulating degranulation has proven challenging. For example, both A1 [10, 11] and A3 ARs [11, 17] have been postulated to be responsible for adenosine-enhanced degranulation. In contrast, A2BARs have recently been suggested to inhibit mast cell degranulation [10, 16, 18]. Those data conflict with earlier data suggesting that activating A2BAR stimulates degranulation of canine BR mastocytoma cells [19]. There has also been disagreement whether A2AARs do [20] or do not [16] inhibit degranulation.
Another uncertainty concerning AR regulation of degranulation is whether human mast cells release ATP to be ecto-enzymatically converted to adenosine, or whether the adenosine must be delivered by other cells. Rat basophilic leukemia cells, a model of mast cells, have been reported to release ATP during antigen-stimulation of degranulation [21]. Rat pleural and peritoneal mast cells and mouse bone marrow-derived mast cells have also been found to release ATP-dependent adenosine in response to degranulating stimuli [22].
In the present study, we have circumvented the issue of cell heterogeneity by studying the widely-used LAD2 mature human mast cell line [23]. In contrast to HMC-1 human mast cell leukemia cells, LAD2 cells consistently express the surface FcεRI receptors characteristic of mature human mast cells in vivo, and also respond to C3a and C5a. In expressing chymase, this cell line corresponds to mucosal mast cells. We have also compared the responses of the human LAD2 cells and mouse bone marrow-derived mast cells (BMMCs) to several AR antagonists. In addition, we have directly tested whether the LAD2 human mast cells release ATP by real-time measurements based on the luciferin–luciferase reaction.
Materials and methods
Cellular model
LAD2 bone marrow-derived human mast cells [23] were a gift from Drs. Dean Metcalfe and Arnold Kirshenbaum (Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD). Unless otherwise noted, materials for cell culture were purchased from Gibco (Invitrogen, Grand Island, NY). Cells were grown in StemPro-34 medium supplemented with StemPro-34 nutrient, 2 mM L-glutamine, 100 ng/ml rhSCF (Peprotech, Rocky Hill, NJ) and 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C, with 5 % CO2 and 95 % air in a water-jacketed incubator. Cells were sub-cultured weekly with fresh medium 1:2, and maintained at 0.25 to 0.5 million cells/ml.
The mouse bone marrow-derived mast cells (BMMCs) were generated as previously described [24]. Briefly, bone marrow cells were obtained from femurs and tibias of WT BALB/c mice. BMMCs were prepared by culturing bone marrow cells in mast cell medium [MCM; RPMI-1640, 15 % fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, 2.9 mg/ml glutamine, 50 mM 2-mercaptoethanol, 1 mM sodium pyruvate, 1× non-essential amino acids, 10 mM HEPES] containing 10 ng/ml interleukin (IL)-3 (Peprotech) and 12.5 ng/ml rmSCF (Peprotech) for 6–8 weeks with media replenished twice weekly. BMMCs were used when >95 % of cells expressed high levels of FcεRI and c-Kit (CD117), as determined by flow cytometry.
Chemicals and solutions
The degranulation assay solution comprised (in mM): 124.5 NaCl, 5.9 KCl, 11.5 glucose, 1.2 MgCl2, 1.5 CaCl2, 10 HEPES, 32 mannitol, and 0.1 % bovine serum albumin (BSA) at ∼310 mosmol/kg H2O and pH 7.4. Tyrode's buffer contains (in mM): 130 NaCl, 10 HEPES, 1 MgCl2, 5 KCl, 1.4 CaCl2, 5.6 glucose and 1 mg/ml BSA, at ∼310 mosmol/kg H2O and pH 7.4. All chemicals were obtained from Sigma-Aldrich (St Louis, MO) except for MRS 1754, SCH 442416, and CGS 15943 (Tocris Bioscience, Ellisville, MO). As previously [25], the ATP assay solution was prepared in accordance with the manufacturer's instructions by diluting one vial of ATP assay mix (FL-AAM, Sigma-Aldrich) with 5 ml of deionized water together with 5 ml ATP assay mix dilution buffer (FL-AAB).
Measurement of degranulation
Cells were seeded in 48-well culture plates with 0.2 million cells per well. In experiments involving degranulation mediated by cross-linking FcεRI receptors, mast cells were preincubated overnight with 1 μg/ml Human Myeloma IgE (Calbiochem, EMD Chemicals, Gibbstown, NJ). LAD2 mast cells were activated either by the indicated concentrations of C3a peptide (63–77; AnaSpec, Fremont, CA) or by 5 μg/ml anti-human IgE (Sigma-Aldrich), with or without drugs, for a further 30 min at 37 °C. Unless otherwise stated, AR agonists were added at the same time as the C3a or anti-IgE, and AR antagonists were introduced 30 min before stimulating degranulation. The numbers of experiments and wells are symbolized by N and n, respectively.
To induce degranulation in BMMCs, cells were rested overnight in MCM containing IL-3 (10 ng/ml; Peprotech) and IL-4 (10 ng/ml; Peprotech). Thereafter, BMMCs were incubated with 1 μg/ml anti-DNP IgE (clone SPE-7; Sigma-Aldrich) for 2 h at 37 °C, washed, preincubated for 30 min with or without drugs at the specified concentrations, and subsequently stimulated with various concentrations of dinitrophenol-conjugated human serum albumin (DNP-HSA; Sigma-Aldrich) for 1 h in Tyrode's buffer.
To assay degranulation, 20 μl of supernatant was collected and the cells were then lysed in 0.1 % of the nonionic surfactant Triton X-100. Aliquots (20 μl) of cell lysates or supernatants were incubated with 20 or 40 μl, with similar results, of 1 mM p-nitrophenyl N-acetyl-β-D-glucosaminide in a 96-well plate at 37 °C for 1.5 h. Reaction was stopped by adding 210 μl of 0.1 M Na2CO3/NaHCO3 buffer and the absorbance was measured at 405 nm. Percentage degranulation was monitored as percentage β-hexosaminidase release calculated as the amount of β-hexosaminidase in the supernatant (measured as optical density, ODsuper) divided by the total amount of β-hexosaminidase in the supernatant and in the lysate (ODlysate):
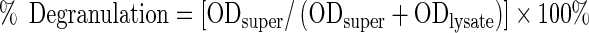 |
1 |
The percentage inhibition was calculated from Eq. 2:
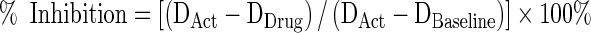 |
2 |
Where DAct is the percentage degranulation of drug-untreated cells after activation, DDrug is the percentage degranulation of drug-treated cells after activation, and DBaseline is the baseline percentage degranulation of drug-untreated cells without activation. Where drug-induced enhancement was noted, % Enhancement was calculated analogously to % Inhibition:
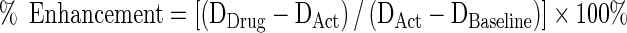 |
3 |
Detection of LAD2-cell AR gene expression by quantitative PCR (qPCR)
Total mRNA in LAD2 cells was isolated following the protocol of the RNeasy Mini Kit (Qiagen, Valencia, CA). Reverse transcription was completed using Superscript III First Strand Synthesis Supermix kit (Invitrogen, Carlsbad, CA). The cDNA was then amplified by PCR with gene-specific primers for ARs and GAPDH on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) according to the manufacturer's protocol using SYBR Green PCR MasterMix. Amplification parameters were as follows: one cycle of 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. The primers (Table 1) were synthesized by Eurofins MWG Operon (Huntsville, AL). The expression levels of indicated genes were calculated by the 2−ΔΔCT method normalized to the A2A subtype receptor, with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the endogenous control. Each gene expression assay was run in quadruplicate.
Table 1.
Gene-specific primers for qPCR of LAD2 cells
| Target | Forward primer | Reverse primer | Product (bp) |
|---|---|---|---|
| A1 AR | CTACCTAAT CCGCAAGCAGC | GTCATCAGGCCTCTCTTCTGG | 367 |
| A2A AR | AACCTGCAGAACGTCACCA | GTCACCAAGCCATTGTACCG | 245 |
| A2B AR | GTGCCACCAACAACTGCACAGAAC | CTGACCATTCCCACTCTTGACATC | 518 |
| A3 AR | CACCACCTTCTATTTCATTGTCTCT | GGTACTCTGAGGTCAGTTTCATGTT | 338 |
| GAPDH | ATTCCATGGCACCGTCAAGGCT | TCAGGTCCACCACTGACACGTT | 572 |
Detection of BMMC AR gene expression by qPCR
Total BMMC mRNA was isolated and cDNA generated as with the LAD2 cells. However, cDNA was then amplified by PCR with gene-specific Taqman, rather than SYBR Green, probes for ARs and β-actin in 96-well plates using a 7300 Real-Time PCR System (Applied Biosystems) and default thermocycler program. FAM-labeled MGB Taqman probes (Applied Biosystems) utilized in the assays are listed in Table 2. The expression levels of indicated genes were calculated by the 2−ΔΔCT method with β-actin as the endogenous control. As for the LAD2 cells, each gene expression assay was performed in quadruplicate.
Table 2.
Gene-specific primers for qPCR of BMMCs
| Target | Assay ID of FAM-labeled MGB probe | Product (bp) |
|---|---|---|
| A1 AR | Mm01308023_m1 | 58 |
| A2A AR | Mm00802075_m1 | 85 |
| A2B AR | Mm00839292_m1 | 61 |
| A3 AR | Mm01296602_m1 | 65 |
| β-Actin | Mm00607939_s1 | 115 |
ATP measurement
ATP release was measured by the bioluminescent luciferin-luciferase reaction. Light emission was recorded with a microplate luminometer (Synergy 2; BioTEK, Winooski, VT), as previously described [25]. In brief, cells were plated onto 96-well microplates (Corning Costar, Corning, NY) at 0.1 million per well. To minimize ATP release in the course of pre-incubations, culture media were removed and replaced by 100 μl of isotonic solution for either 1 or 4–5 h, as discussed in the "Results" section. Thereafter, 75 μl of isotonic solution was replaced by an equal volume of test solution to establish the final osmolalities or activate degranulation. After addition of test solutions, 10 μl of the ATP assay solution was injected into each well through the internal dispenser system within 1 min. Measurements were begun immediately after dispensing the solutions, and taken at 2-min intervals for each well for 2 h, with an integration time of 0.2 s/measurement. ATP levels were calculated at each time point from a standard curve transforming arbitrary light units into ATP concentrations. Separate standard curves were utilized in experiments involving either changes in ionic strength or test substances at concentration high enough to interfere with the ATP assay.
Statistical analysis
Unless otherwise stated, all data were presented as means ± standard errors of the means (SE). The paired or unpaired Student's t test was used for comparing two means, as appropriate. Unless otherwise stated, one-way ANOVA with Dunnett's Multiple Comparison Test was used for comparison of multiple sets of data, and two-way ANOVA with Bonferroni Multiple Comparison Test was applied to compare concentration-response relationships. Statistical analyses were performed with SigmaStat (Aspire Software International, Ashburn, VA). Results were considered significant when the probability (P) of the null hypothesis was <0.05.
Results
Adenosine receptors of LAD2 cells
LAD2 cells expressed A2A, A2B, and A3 ARs, but A1AR was undetected (N = 3, Fig. 1). Quantified by qPCR, the relative expression of A2A, A2B, and A3 ARs normalized to A2AAR was 1.00, 0.24 ± 0.03, and 0.32 ± 0.02, respectively.
Fig. 1.

Relative gene expression of AR subtypes in LAD2 mast cells, quantified by qPCR (N = 3). The AR expressions were normalized to the A2A receptor subtype, displaying a ranking of A2A > A3 > A2B, with A1 undetected
Effects of nonselectively activating LAD2 ARs
Nonselective activation of all ARs with the poorly metabolized agonist NECA monotonically enhanced both C3a- and FcεRI-initiated degranulation with increasing concentration (Fig. 2). The maximal enhancements were of comparable magnitude, whether initiated by C3a or immunologically. NECA's enhancement of C3a-triggered degranulation was observed at all three C3a concentrations applied, 0.2, 0.5, and 1.0 μM (Fig. 2a, b). In the presence of 0.2 μM C3a, 5–20 μM NECA enhanced degranulation by 23–27 % (Fig. 2a). NECA displayed comparable efficacy in enhancing FcεRI-mediated degranulation. In the presence of 5 μg/ml anti-human IgE antibody, NECA increased degranulation by 27–31 % (Fig. 2a).
Fig. 2.

Nonselective NECA activation of ARs enhanced degranulation triggered immunologically or by C3a. a Degranulation was initiated either by adding 0.2 μM C3a (n = 12, lower NECA concentration scale) or by adding 5 μg/ml anti-human IgE antibody (n = 6, upper NECA concentration scale) after overnight pre-sensitization with 1 μg/ml human myeloma IgE. NECA enhanced C3a-triggered degranulation monotonically over the concentration range applied from 50 nM to 20 μM. Similarly, NECA enhanced FcεR1-mediated degranulation monotonically, but displayed saturation at approximately 2 μM. b In contrast to panel (a), the C3a concentration was varied in both experimental and control wells, whereas the experimental wells were exposed to a fixed NECA concentration of 1 μM (n = 18–24). NECA significantly enhanced the C3a-induced degranulation at the three C3a concentrations studied (P < 0.0001, two-way ANOVA)
Despite the similar efficacies, NECA displayed much higher potency in enhancing FcεRI-mediated degranulation (Fig. 2a). NECA increased FcεR1-triggered LAD2 degranulation monotonically over the range from 10 nM to 2 μM, but higher NECA concentrations of 4 and 22 μM had no further effect (Fig. 2a). In contrast, NECA continued to enhance C3a-mediated LAD2 degranulation monotonically over the full concentration range applied, from 50 nM to 20 μM (Fig. 2a). The enhanced degranulations at NECA concentrations of 50 nM and 1 μM were very much larger for FcεR1- than for C3a-triggered LAD2 degranulation (P < 0.0001 by ANOVA with Bonferroni post-test). Estimated from fits to the Michaelis–Menten equation, the half maximal effective NECA concentrations (EC50s) differed by an order of magnitude. The estimated EC50s were 29 ± 8 nM (n = 12) and 0.5 ± 0.2 μM (n = 6) for enhancing the immunologically and C3a-stimulated degranulations, respectively.
Adding 1 μM NECA alone also increased LAD2 cell degranulation significantly by 2.3 ± 0.9 % (∼20 % above baseline, n = 18, N = 3, P < 0.025 by unpaired t test). The magnitude of that stimulation was sevenfold to eightfold smaller than that triggered by 0.2 μM C3a (Fig. 2b). As noted in the foregoing paragraph, maximal NECA-enhanced degranulation stimulated either by 0.2 μM C3a or 5 μg/ml anti-human IgE antibody were of similar magnitude (Fig. 2a). The results suggested that nonselective activation of ARs can itself stimulate degranulation of LAD2 cells, but with far lower efficacy than that of FcεR1 or C3a (the "Discussion" section).
Effects of selective agonists on LAD2 degranulation
The foregoing observations documented that nonselective activation of ARs enhances degranulation triggered by C3a or immunologically. Recent reports [10, 11] have suggested that activation of A1 and A3 ARs can be responsible for such enhancement (Introduction). In the absence of detectable A1AR expression, we expected that application of the highly selective A3AR agonist Cl-IB-MECA [26] would replicate the enhancement produced by NECA (Fig. 2b).
Contrary to prediction, application of 3, 30 or 100 nM of the selective A3 agonist Cl-IB-MECA together with the C3a had no significant effect on C3a-triggered degranulation (one-way ANOVA with Bonferroni's multiple comparison test, n = 35–36, N = 6). In parallel measurements conducted with aliquots of the same cells (n = 35–36, N = 6), 0.5 μM of the nonselective agonist NECA enhanced C3a-triggered degranulation by 41 ± 9 % (P < 0.00001). Application of the selective A2AAR agonist CGS21680 (100 nM) also had no significant effect on C3a-triggered degranulation, applied either at the same time as the C3a (n = 24, N = 4) or 30 min earlier (n = 24, N = 4) in separate experiments. These results suggested that multiple ARs might be interacting, directly or indirectly, to generate the enhancement produced by NECA. In the absence of a selective agonist for A2BAR, this possibility was pursued by nonselectively activating the three expressed ARs, following preincubation with selective AR antagonists.
Effects of selective AR antagonists on LAD2 cells
Cells were incubated for 30 min with selective AR antagonists, applied alone or together, before triggering degranulation with 0.2 μM C3a or 5 μg/ml antihuman IgE antibody (Fig. 3). In the absence of NECA, 100-nM concentrations of the A2AAR antagonist SCH442416 [27, 28], the A2BAR antagonist MRS1754 [29] and the A3AR antagonist MRS1191 [30] exerted no significant effect on C3a-stimulated LAD2 degranulation (Fig. 3a). However, C3a-triggered degranulation enhanced with 1 μM NECA was significantly inhibited by separate addition of the A2AAR, A2BAR, and A3AR antagonists (Fig. 3b). Simultaneous addition of the selective antagonists to the A2A, A2B and A3 ARs abolished the NECA-induced enhancement of C3a-triggered degranulation (Fig. 3b).
Fig. 3.

Effects of selective AR antagonists on LAD2 cell degranulation (*P < 0.05). All drug-treated groups were pre-incubated for 30 min with 100-nM concentrations of the selective A2A antagonist SCH442416, the selective A2B antagonist MRS1754, and the selective A3 antagonist MRS1191, separately or in combination. Drug-treated cells were stimulated by either 0.2 μM C3a or 5 μg/ml anti-human IgE antibody, following preincubation with antagonists and 1 μM NECA. Values displayed within the bars indicate the number of wells (n) studied, and asterisks denote significant differences from NECA-enhanced degranulation in the absence of antagonists. a Direct effects of the antagonists on C3a-triggered degranulation in the absence of NECA (n = 6, N = 1). b Effects of the antagonists on NECA-enhanced degranulation triggered by adding C3a (n = 48–78, N = 8–13). c Effects of the antagonists on NECA-enhanced degranulation triggered by crosslinking FcεRI (n = 72–78, N = 12–13). Data were analyzed by Kruskal–Wallis one-way analysis of variance by ranks, with Dunn's test of pairwise multiple comparisions
Application of the A2AAR, A2BAR, and A3AR antagonists together also abolished NECA's enhancement of FcεR1-mediated degranulation (Fig. 3c). However, when added singly, only the A2AAR antagonist SCH442416 significantly but incompletely inhibited NECA-enhanced, FcεR1-triggered degranulation (Fig. 3c).
The results obtained with selective AR antagonists suggested that activation of the three expressed ARs interactively enhance both C3a- and FcεR1-stimulated LAD2 degranulation.
ATP release by LAD2 cells
The preceding results indicated that ARs modulate LAD2 degranulation. We wondered whether agonist might be physiologically delivered to these receptors by autocrine and paracrine release of ATP and its ectoenzymatic conversion to adenosine, as we have found in other cells [31, 32]. Initially, we followed our practice of minimizing baseline extracellular ATP concentration during the pre-incubation periods by replacing culture media with isotonic solutions for 1 h before beginning experiments. Using this approach in initial measurements, the baseline extracellular ATP concentration was relatively high (36 ± 4 nM, n = 24, N = 3). In previous measurements of 4,472 wells containing native cells and cell lines from ocular ciliary epithelium and trabecular meshwork, the baseline extracellular ATP concentration ranged from 6.1 ± 0.3 nM (n = 978) to 15.7 ± 0.7 nM (n = 670) in isotonic solution [25, 31–33], with a mean of ∼9 nM. Since a high isotonic baseline level might potentially obscure physiologically significant, degranulation-linked ATP release, we prolonged the recovery time to 4–5 h after transferring cells to the wells. With this approach, the baseline level in the bathing solution was reduced by ∼86 % to 5.2 ± 0.3 nM (n = 32, N = 4), a value more in keeping with our measurements in other cells.
As expected from other studies, hypotonicity (50 %) significantly increased ATP release by ∼160 % (Fig. 4). In contrast, neither cross-linking FcεRI nor adding C3a affected extracellular ATP concentration. The latter result, together with the data obtained using selective AR antagonists, suggests that purinergic modulation of degranulation in vivo would necessarily depend upon adenosine delivery from other cells (the "Discussion" section).
Fig. 4.

Effects of hypotonicity ("Hypo"), C3a, and cross-linking FcεRI ("IgE") on ATP concentration of external solution bathing LAD2 cells. "Iso" refers to measurements of isotonic bath solutions measured in parallel with the hypotonic and C3a experimental solutions. "IgE(−)" refers to the isotonic control solution without anti-IgE, studied in parallel with the IgE-treated experimental preparations ("IgE"). The values within the bars were the numbers of wells. For the Iso, Hypo, and C3a groups, N = 4, and for the IgE and IgE(−) groups, N = 2
Adenosine receptors of BMMCs
Nonselective activation of ARs has been reported to exert different actions on preincubated rat peritoneal mast cells and cultured human mast cells derived from the buffy coat of peripheral blood [11]. In a limited number of experiments, we have compared the LAD2 results with the effects of NECA and selective AR antagonists on FcεRI-mediated degranulation of another murine preparation, mouse BMMCs. As in the case of LAD2 cells, BMMCs expressed A2A, A2B, and A3 ARs, but A1AR was undetected (Fig. 5). Quantified by qPCR, the relative expression of A2A, A2B, and A3 ARs normalized to A2AAR was 1.00, 2.49 ± 0.25, and 0.64 ± 0.06, respectively.
Fig. 5.

Relative gene expression of AR subtypes in BMMCs, quantified by qPCR (n = 5). The AR expressions were normalized to the A2A receptor subtype, displaying a ranking of A2B > A2A > A3, with A1 undetected
Effects of NECA and selective AR antagonists on FcεRI-mediated degranulation of BMMCs
Consistent with published data [34], immunologic stimulation increased baseline degranulation by ∼200 % [compare IgE with non-treated control (NC), Fig. 6], and 1 μM NECA enhanced the FcεRI-mediated degranulation by another 200 % (compare IgE/NECA with IgE, Fig. 6).
Fig. 6.

Effects of selective AR antagonists on NECA-enhanced BMMC degranulation. As in Fig. 3, drug-treated wells were pre-incubated with 100-nM concentrations of selective A2A, A2B, and A3 AR antagonists. In contrast to the LAD2-cell degranulation, the BMMC degranulation was only partially reduced by simultaneous application of the antagonists and was not significantly altered by pre-incubation separately with the A2A, A2B, or A3 AR antagonists
The enhancement increased monotonically with increasing NECA concentrations from 10 nM to 1 μM (filled circles, Fig. 7). In contrast to the results obtained with LAD2 cells, separate addition of the selective A2A, A2B, and A3 AR antagonists had relatively little effect on NECA's enhancement of BMMC degranulation. That enhancement was partially reduced only by the simultaneous application of the A2A, A2B, and A3 AR antagonists (Fig. 6). However, the NECA-induced enhancement could be markedly inhibited by increasing concentrations of the nonselective AR antagonist CGS15943 (Fig. 7). Thus, NECA's action on BMMC degranulation was mediated by ARs. The CGS15943 appeared more efficacious in blocking ARs in LAD2 cells, as well. In contrast to simultaneously blocking expressed ARs with selective antagonists, applying the nonselective antagonist significantly inhibited C3a-triggered degranulation by 18 ± 4 % even in the absence of NECA (one-way ANOVA with Holm–Sidak test, P < 0.01, N = 3). Evidently, this inhibition was not a nonspecific effect since addition of the CGS15943 did not alter baseline LAD2 degranulation. The different responses of LAD2 and BMMCs presumably reflect different pharmacologic profiles (the "Discussion" section).
Fig. 7.

Effects of the nonselective AR antagonist CGS15943 on NECA-enhanced, FcεRI-mediated mouse BMMC degranulation. Degranulation was measured without NECA and at increasing NECA concentrations from 10 nM to 1.0 μM. NECA-enhanced degranulation was measured in the absence of the antagonist and in the presence of 100 nM, 500 nM, and 1 μM CGS15943 (n = 6–54). At 1 μM, the nonselective AR antagonist produced a concentration-dependent inhibition of both the NECA enhancement (P < 0.0001, two-way ANOVA with Bonferroni multiple comparison test) and the FcεRI-mediated degranulation in the absence of NECA (P < 0.01, one-way ANOVA with Dunnett's multiple comparison test)
Discussion
Summary of major results
The major results of the present study of a homogenous preparation of human mast cells were that (a) in the absence of pharmacologic activation, ARs played a minor role in modulating mast cell degranulation; (b) A1AR was unnecessary either for LAD2 cell degranulation or NECA-mediated enhancement; (c) nonselective activation of the three expressed ARs with NECA directly stimulated LAD2-cell degranulation; (d) increasing concentrations of NECA monotonically enhanced degranulation, triggered immunologically or by C3a; (e) inhibition of each of the three expressed ARs reduced the enhancement of C3a-activated LAD2 cell degranulation; (f) immunologic or C3a activation of degranulation did not stimulate ATP release from LAD2 cells; and (g) selective inhibition of only one of the three ARs had little effect on NECA's enhancement of FcεRI-mediated BMMC degranulation, whereas simultaneous application of all three antagonists or application of the nonselective antagonist CGS15943 significantly inhibited that enhancement.
Role of A1AR in modulating mast cell degranulation
In a recent study of cultured mast cells derived from the buffy coat of human peripheral blood, Yip et al. [10] found that only A1 agonists could replicate adenosine or NECA in enhancing immunologically triggered degranulation. The current work demonstrates that detectable A1AR expression is not required for NECA to enhance degranulation stimulated either immunologically or by C3a in human LAD2 cells. NECA also enhanced immunologically stimulated degranulation by BMMCs. A1ARs have not been detected in BMMCs under baseline conditions [35], although Möller et al. [34] did find that A1AR could be upregulated in these cells 6 h after crosslinking IgE bound to FcεRI. Gene expressionn of A1 ARs could not be detected in our BMMCs. Agonist activation of A1ARs can certainly play a role in modulating mast cell degranulation, but our data demonstrate that detectable expression of A1AR is not a necessary requirement for AR enhancement of that degranulation.
Effect of nonselective AR activation on baseline LAD2 mast cell degranulation
Among the many uncertainties concerning adenosine's role is whether activation of one or more AR receptors can directly stimulate mast cell degranulation. Multiple studies of human and rat mast cells did not detect such stimulation [8, 10, 11, 36, 37]. In contrast, several publications have reported evidence of direct AR stimulation of human, rat and mouse mast-cell degranulation [17, 34, 38]. The current results are in agreement with the latter reports. The NECA-stimulated degranulation of LAD2 cells was ∼20 %, consistent with published estimates ranging from ∼5 % [17] to 33 % [34]. Where compared in the same study, FcεRI- or C3a-mediated activation was threefold [34], fourfold [17], and sevenfold to eightfold (current study) more efficacious than NECA- or adenosine-triggered activation in stimulating mast cell degranulation. The results suggest that pharmacologic AR activation can indeed directly stimulate mast cell degranulation, but its significance in vivo likely depends on: AR expression levels, type and organ of origin of the mast cell, microenvironment and whether the more efficacious immunologic and C3a stimuli are also at play. For example, allergen challenge increases the ATP levels in bronchoalveolar lavage (BAL) fluid both in human asthmatcs and mice with experimentally induced asthma [39]. Likewise, adenosine levels are elevated in BAL fluid of asthmatics and are acutely increased in lung lavage fluid by allergen challenge of sensitized rabbits [40].
Effect of nonselective AR activation on LAD2 degranulation triggered immunologically or by C3a
In addition to stimulating LAD2 degranulation directly, nonselective AR activation enhanced the degranulation triggered immunologically or by C3a. The enhancement of C3a-triggered degranulation was directly dependent on the applied NECA concentration over the range 50 nM to 20 μM (Fig. 2a). NECA's potencies in enhancing the degranulations initiated by cross-linking FcεRI and activating C3a receptors differed quantitatively (Fig. 2a), consistent with the non-identical principal and complementary signaling cascades involved [6].
The monotonic AR enhancement observed with LAD2 cells agrees with the first peer-reviewed report of nonselective AR agonist action on immunologically triggered mast cell degranulation [8]. Marquardt et al. [8] also observed AR enhancement of rat pleural and peritoneal mast cell degranulation triggered by other stimuli, including concanavalin A, cpd 48/80 and the Ca2+ ionophore A23187. However, additional patterns have been observed, as well. Both monotonically inhibitory [20] and biphasic [14] trajectories have been reported, even from the same laboratory. For example, Gomez et al. [13] noted a biphasic response in which 1 μM adenosine enhanced FcεRI-mediated degranulation of enzymatically dispersed human lung mast cells by ∼25 %, whereas 1 mM adenosine inhibited that degranulation by ∼75 %. The same group reported a monotonically inhibitory modulation of enzymatically dispersed human skin mast cells [13]. The modulatory pattern of nonselective AR activation has been found dependent not only on the species and organ origin of the mast cells, but also on the time relationship between application of the AR agonists and the initiating degranulating stimulus. For example, Hughes et al. [41] found that nonselective AR agonists produced a monotonic enhancement of immunologically mediated degranulation when the agonist was added 5 min after exposure of mechanically dispersed human lung mast cells to anti-IgE, but produced a monotonic inhibition when added 15 min beforehand. The same 100-micromolar concentration of NECA enhanced degranulation by ∼25 % when added after anti-IgE and inhibited degranulation by ∼50 % when added before anti-IgE. The time sequence was also observed to be of importance in studying the effects of nonselective AR activation of human mast cells derived from the buffy coat of peripheral blood [11], but oppositely to that reported by Hughes et al. [41]. Yip et al. [11] found that 10 nM adenosine or NECA enhanced degranulation only if the nonselective agonists were added 10 min before, and not concurrently with, the anti-IgE.
The preceding seemingly inconsistent patterns raise the possibility that multiple interactive signaling cascades might mediate the modulatory actions of nonselective AR agonists exerted on mast cell degranulation. This possibility is consistent with results obtained in whole animals. Aerosol NECA administration has been found to cause degranulation-dependent enhancement of airway hyperresponsiveness in mice within 10 min [17]. However, the same group later observed that 20–24 h after NECA application, the AR activation had an opposite, inhibitory effect; after the more prolonged time, NECA inhibited antigen-triggered histamine release and fall in body temperature, which the authors regarded as an index of mast cell degranulation [16]. Nevertheless, prolonged elevation of adenosine concentration in adenosine deaminase-deficient mice has been observed to produce chronic enhancement of lung mast cell degranulation [42]. Human asthmatics are also exposed to chronically high adenosine concentrations in the lung [43], so that the degranulation-enhancing action of AR activation may be particularly relevant clinically.
Contributions of A2A, A2B, and A3 ARs to the enhancement of C3a-activated LAD2 cell degranulation
Contrary to the effects of nonselective AR activation, separate application of the selective agonists for A2A, CGS21680, and for A3, Cl-IB-MECA, had no effect on C3a-triggered LAD2-cell degranulation. As noted above, A1AR was not expressed and no selective agonist of A2BAR is yet available. It is conceivable that C3a might have already stimulated one or another of the expressed ARs, but separate application of selective antagonists to each of the three expressed ARs had no effect on C3a-mediated degranulation (Fig. 3a). More informative than applying selective agonists was to nonselectively activate the ARs with or without selective antagonists. Separate inhibitions of A2A, A2B, and A3 ARs significantly reduced NECA-enhanced, C3a-stimulated degranulation of LAD2 cells, and simultaneous inhibition of the three expressed ARs abolished the NECA enhancement, altogether (Fig. 3b). The single contributions of each AR were less prominent in enhancing FcεRI-mediated LAD2 degranulation. Only the A2A antagonist SCH442416 significantly inhibited the NECA-enhanced, immunologically stimulated LAD2 degranulation, but simultaneous inhibition of the three expressed ARs nearly completely inhibited that enhancement (Fig. 3c). Our measurements of mouse BMMC degranulation complemented these data. Only simultaneous application of the same antagonists used for the LAD2 cells slightly, but significantly, inhibited immunologically stimulated NECA-enhanced BMMC degranulation (Fig. 6). The large inhibition produced by a nonselective AR antagonist (1 μM CGS15943, Fig. 7) suggests that at least half of the immunologically triggered BMMC degranulation was mediated by ARs.
These data raise the possibility that A2A, A2B, and A3 ARs may have all contributed to the observed NECA enhancement of LAD2, and possibly BMMC, cell degranulation. Interestingly, additive effects by A1, A2A and A3 ARs on phosphoinositide breakdown have also been observed in the rat basophilic leukemia RBL-2H3 cell line [44]. If so, what could be the signaling cascades mediating this modulation? A1 and A2 ARs were first distinguished by their effects on adenylyl cyclase; A1 and A3 ARs inhibit the enzyme by coupling to Gi/o, whereas A2A and A2B stimulate adenylyl cyclase through coupling to Gs [45, 46]. However, additional pathways must be involved since A1 and A2A ARs exert opposing effects on adenylyl cyclase, and yet affect MAPKs similarly [47]. In part this reflects the fact that ARs can couple to more than one G protein, such as Gq, and A2A ARs can couple different G proteins in different regions [45]. In addition, ARs can oligomerize with the same or other ARs, with other GPCRs (such as dopamine) and with other proteins (such as calmodulin) [48], in principle providing the basis for triggering a wide range of signaling cascades.
Summary
Nonselective activation of A2A, A2B, and A3 adenosine receptors directly stimulates degranulation of human LAD2 mast cells. The three receptors all contribute to a much larger enhancement of FcεRI- or C3a-mediated degranulation of LAD2 cells. Stimulating degranulation either immunologically or with C3a does not trigger LAD2 release of ATP, so that delivery of adenosine to ARs in vivo depends on availability of ATP or adenosine from other cells in the microenvironment. In contrast to the monotonically enhancing actions of ARs on degranulation observed with LAD2 cells, many mast cell preparations display monotonically inhibitory or biphasic patterns of modulation. The multiple patterns of AR modulation observed in many other preparations likely reflect AR activation of multiple interactive signaling cascades, in part mediated by coupling to multiple G proteins and oligomerization with other receptors and proteins [48].
Acknowledgments
Supported by NIH Research Grant EY13624 and Core Grant EY01583 (M.M.C.); University of Pennsylvania Research Foundation and NIDDK Intramural Research Program, National Institutes of Health. We thank Drs. Arnold Kirshenbaum and Dean Metcalfe (NIAID, NIH, Bethesda, MD, USA) for providing LAD2 cells, and Dr. Hydar Ali (University of Pennsylvania) for helpful discussions.
References
- 1.Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2006;117(2 Suppl Mini-Primer):S450–S456. doi: 10.1016/j.jaci.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 2.Irani AM. Ocular mast cells and mediators. Immunol Allergy Clin North Am. 2008;28(1):25–42. doi: 10.1016/j.iac.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Turner H, Kinet JP. Signalling through the high-affinity IgE receptor Fc epsilonRI. Nature. 1999;402(6760 Suppl):B24–B30. doi: 10.1038/35037021. [DOI] [PubMed] [Google Scholar]
- 4.Duffy SM, Cruse G, Brightling CE, Bradding P. Adenosine closes the K + channel KCa3.1 in human lung mast cells and inhibits their migration via the adenosine A2A receptor. Eur J Immunol. 2007;37(6):1653–1662. doi: 10.1002/eji.200637024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broide DH. Molecular and cellular mechanisms of allergic disease. J Allergy Clin Immunol. 2001;108(2 Suppl):S65–S71. doi: 10.1067/mai.2001.116436. [DOI] [PubMed] [Google Scholar]
- 6.Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat Rev Immunol. 2006;6(3):218–230. doi: 10.1038/nri1782. [DOI] [PubMed] [Google Scholar]
- 7.Venkatesha RT, Berla Thangam E, Zaidi AK, Ali H. Distinct regulation of C3a-induced MCP-1/CCL2 and RANTES/CCL5 production in human mast cells by extracellular signal regulated kinase and PI3 kinase. Mol Immunol. 2005;42(5):581–587. doi: 10.1016/j.molimm.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 8.Marquardt DL, Parker CW, Sullivan TJ. Potentiation of mast cell mediator release by adenosine. J Immunol. 1978;120(3):871–878. [PubMed] [Google Scholar]
- 9.van den Berge M, Hylkema MN, Versluis M, Postma DS. Role of adenosine receptors in the treatment of asthma and chronic obstructive pulmonary disease: recent developments. Drugs R D. 2007;8(1):13–23. doi: 10.2165/00126839-200708010-00002. [DOI] [PubMed] [Google Scholar]
- 10.Yip KH, Lau HY, Wise H. Reciprocal modulation of anti-IgE induced histamine release from human mast cells by A(1) and A(2B) adenosine receptors. Br J Pharmacol. 2011;164(2b):807–819. doi: 10.1111/j.1476-5381.2011.01446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yip KH, Wong LL, Lau HY. Adenosine: roles of different receptor subtypes in mediating histamine release from human and rodent mast cells. Inflamm Res. 2009;58(Suppl 1):17–19. doi: 10.1007/s00011-009-0647-9. [DOI] [PubMed] [Google Scholar]
- 12.Broide DH, Metcalfe DD, Wasserman SI. Functional and biochemical characterization of rat bone marrow derived mast cells. J Immunol. 1988;141(12):4298–4305. [PubMed] [Google Scholar]
- 13.Gomez G, Zhao W, Schwartz LB (2011) Disparity in FcepsilonRI-induced degranulation of primary human lung and skin mast cells exposed to adenosine. J Clin Immunol 31 (3):479–487 [DOI] [PMC free article] [PubMed]
- 14.Peachell PT, Lichtenstein LM, Schleimer RP. Differential regulation of human basophil and lung mast cell function by adenosine. J Pharmacol Exp Ther. 1991;256(2):717–726. [PubMed] [Google Scholar]
- 15.Zaidi AK, Amrani Y, Panettieri RA, Ali H. Response to C3a, mast cells, and asthma. Faseb J. 2006;20(2):199. doi: 10.1096/fj.06-0204ufm. [DOI] [PubMed] [Google Scholar]
- 16.Hua X, Chason KD, Jania C, Acosta T, Ledent C, Tilley S. Gs-coupled adenosine receptors differentially limit antigen-induced mast cell activation. J Pharmacol Exp Ther. 2013;344(2):426–435. doi: 10.1124/jpet.112.198978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hua X, Chason KD, Fredholm BB, Deshpande DA, Penn RB, Tilley SL (2008) Adenosine induces airway hyperresponsiveness through activation of A3 receptors on mast cells. J Allergy Clin Immunol 122 (1):107–113, 113 e101-107 [DOI] [PMC free article] [PubMed]
- 18.Hua X, Kovarova M, Chason KD, Nguyen M, Koller BH, Tilley SL. Enhanced mast cell activation in mice deficient in the A2b adenosine receptor. J Exp Med. 2007;204(1):117–128. doi: 10.1084/jem.20061372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Auchampach JA, Jin X, Wan TC, Caughey GH, Linden J. Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol Pharmacol. 1997;52(5):846–860. doi: 10.1124/mol.52.5.846. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki H, Takei M, Nakahata T, Fukamachi H. Inhibitory effect of adenosine on degranulation of human cultured mast cells upon cross-linking of Fc epsilon RI. Biochem Biophys Res Commun. 1998;242(3):697–702. doi: 10.1006/bbrc.1997.8040. [DOI] [PubMed] [Google Scholar]
- 21.Osipchuk Y, Cahalan M. Cell-to-cell spread of calcium signals mediated by ATP receptors in mast cells. Nature. 1992;359(6392):241–244. doi: 10.1038/359241a0. [DOI] [PubMed] [Google Scholar]
- 22.Marquardt DL, Gruber HE, Wasserman SI. Adenosine release from stimulated mast cells. Proc Natl Acad Sci U S A. 1984;81(19):6192–6196. doi: 10.1073/pnas.81.19.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirshenbaum AS, Akin C, Wu Y, Rottem M, Goff JP, Beaven MA, Rao VK, Metcalfe DD. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leuk Res. 2003;27(8):677–682. doi: 10.1016/S0145-2126(02)00343-0. [DOI] [PubMed] [Google Scholar]
- 24.Kambayashi T, Okumura M, Baker RG, Hsu CJ, Baumgart T, Zhang W, Koretzky GA Independent and cooperative roles of adaptor molecules in proximal signaling during FcepsilonRI-mediated mast cell activation. Mol Cell Biol 30(17) [DOI] [PMC free article] [PubMed]
- 25.Li A, Leung CT, Peterson-Yantorno K, Mitchell CH, Civan MM. Pathways for ATP release by bovine ciliary epithelial cells, the initial step in purinergic regulation of aqueous humor inflow. Am J Physiol Cell Physiol. 2010;299(6):C1308–C1317. doi: 10.1152/ajpcell.00333.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HO, Ji XD, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. 2-Substitution of N6-benzyladenosine-5'-uronamides enhances selectivity for A3 adenosine receptors. J Med Chem. 1994;37(21):3614–3621. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev. 2006;5:1–18. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kecskes M, Kumar TS, Yoo L, Gao ZG, Jacobson KA. Novel Alexa Fluor-488 labeled antagonist of the A(2A) adenosine receptor: Application to a fluorescence polarization-based receptor binding assay. Biochem Pharmacol. 2010;80(4):506–511. doi: 10.1016/j.bcp.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim YC, Ji X, Melman N, Linden J, Jacobson KA. Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A(2B) adenosine receptors. J Med Chem. 2000;43(6):1165–1172. doi: 10.1021/jm990421v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang BT, Haltiwanger B. Adenosine A2a and A2b receptors in cultured fetal chick heart cells. High- and low-affinity coupling to stimulation of myocyte contractility and cAMP accumulation. Circ Res. 1995;76(2):242–251. doi: 10.1161/01.RES.76.2.242. [DOI] [PubMed] [Google Scholar]
- 31.Li A, Leung CT, Peterson-Yantorno K, Stamer WD, Civan MM. Cytoskeletal dependence of adenosine triphosphate release by human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2011;52(11):7996–8005. doi: 10.1167/iovs.11-8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li A, Banerjee J, Peterson-Yantorno K, Stamer WD, Leung CT, Civan MM. Effects of cardiotonic steroids on trabecular meshwork cells: search for mediator of ouabain-enhanced outflow facility. Exp Eye Res. 2012;96(1):4–12. doi: 10.1016/j.exer.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li A, Leung CT, Peterson-Yantorno K, Stamer WD, Mitchell CH, Civan MM. Mechanisms of ATP release by human trabecular meshwork cells, the enabling step in purinergic regulation of aqueous humor outflow. J Cell Physiol. 2012;227:172–182. doi: 10.1002/jcp.22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Möller C, Xiang Z, Nilsson G (2003) Activation of mast cells by immunoglobulin E-receptor cross-linkage, but not through adenosine receptors, induces A1 expression and promotes survival. Clin Exp Allergy 33(8):1135–1140 [DOI] [PubMed]
- 35.Marquardt DL, Walker LL, Heinemann S. Cloning of two adenosine receptor subtypes from mouse bone marrow-derived mast cells. J Immunol. 1994;152(9):4508–4515. [PubMed] [Google Scholar]
- 36.Reeves JJ, Jones CA, Sheehan MJ, Vardey CJ, Whelan CJ. Adenosine A3 receptors promote degranulation of rat mast cells both in vitro and in vivo. Inflamm Res. 1997;46(5):180–184. doi: 10.1007/s000110050169. [DOI] [PubMed] [Google Scholar]
- 37.Hua X, Chason KD, Patel JY, Naselsky WC, Tilley SL. IL-4 amplifies the pro-inflammatory effect of adenosine in human mast cells by changing expression levels of adenosine receptors. PLoS One. 2011;6(9):e24947. doi: 10.1371/journal.pone.0024947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forsythe P, McGarvey LP, Heaney LG, MacMahon J, Ennis M. Adenosine induces histamine release from human bronchoalveolar lavage mast cells. Clin Sci (Lond) 1999;96(4):349–355. doi: 10.1042/CS19980228. [DOI] [PubMed] [Google Scholar]
- 39.Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, Hoogsteden HC, Luttmann W, Ferrari D, Di Virgilio F, Virchow JC, Jr, Lambrecht BN. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nature Medicine. 2007;13(8):913–919. doi: 10.1038/nm1617. [DOI] [PubMed] [Google Scholar]
- 40.Livingston M, Heaney LG, Ennis M. Adenosine, inflammation and asthma–a review. Inflamm Res. 2004;53(5):171–178. doi: 10.1007/s00011-004-1248-2. [DOI] [PubMed] [Google Scholar]
- 41.Hughes PJ, Holgate ST, Church MK. Adenosine inhibits and potentiates IgE-dependent histamine release from human lung mast cells by an A2-purinoceptor mediated mechanism. Biochem Pharmacol. 1984;33(23):3847–3852. doi: 10.1016/0006-2952(84)90050-9. [DOI] [PubMed] [Google Scholar]
- 42.Zhong H, Chunn JL, Volmer JB, Fozard JR, Blackburn MR. Adenosine-mediated mast cell degranulation in adenosine deaminase-deficient mice. J Pharmacol Exp Ther. 2001;298(2):433–440. [PubMed] [Google Scholar]
- 43.Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis. 1993;148(1):91–97. doi: 10.1164/ajrccm/148.1.91. [DOI] [PubMed] [Google Scholar]
- 44.Shin Y, Daly JW, Jacobson KA. Activation of phosphoinositide breakdown and elevation of intracellular calcium in a rat RBL-2H3 mast cell line by adenosine analogs: involvement of A(3)-adenosine receptors? Drug Dev Res. 1996;39(1):36–46. doi: 10.1002/(SICI)1098-2299(19960901)39:1<36::AID-DDR5>3.0.CO;2-L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fredholm BB, Irenius E, Kull B, Schulte G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem Pharmacol. 2001;61(4):443–448. doi: 10.1016/S0006-2952(00)00570-0. [DOI] [PubMed] [Google Scholar]
- 46.Epperson SA, Brunton LL, Ramirez-Sanchez I, Villarreal F. Adenosine receptors and second messenger signaling pathways in rat cardiac fibroblasts. Am J Physiol Cell Physiol. 2009;296(5):C1171–C1177. doi: 10.1152/ajpcell.00290.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53(4):527–552. [PMC free article] [PubMed] [Google Scholar]
- 48.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE, International Union of Basic and Clinical Pharmacology (2011) Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev 63(1):1–34 [DOI] [PMC free article] [PubMed]