

Summary
Recent research has shown: i) that Toll-like receptor (TLR) agonists drive hematopoietic stem and progenitor cells (HSPCs) to proliferate and differentiate along the myeloid lineage in vitro, and ii) that direct TLR-mediated stimulation of HSPCs also promotes macrophage differentiation in vivo following infection. These new insights demonstrate that TLR signaling in HSPCs, in addition to other TLR-dependent mechanisms, can contribute to HSPC expansion and myeloid differentiation after infection. Evidence is therefore mounting that direct TLR-induced programming of hematopoiesis plays a key role in host defense by rapidly replenishing the innate immune system with the cells needed to deal with pathogens.
Keywords: hematopoiesis, HSPCs, infection, TLRs
Introduction
Throughout life, leukocytes arise from a common ancestor in the mammalian bone marrow, the hematopoietic stem cell (HSC), which is functionally defined by its durable capacity for self-renewal and ability to produce all types of blood cells (Figure 1, reviewed in [1, 2]). During homeostasis, the process of HSC self-renewal, as well as the production of lineage-committed progenitors, is tightly controlled to maintain daily blood cell production. Many cytokines, cell-cell interactions and transcription factors “fine-tune” the proliferation of hematopoietic stem and progenitor cells (HSPCs) and their differentiation into mature myeloid and lymphoid cells (reviewed in [3]).
Figure 1. The mouse hematopoietic tree.
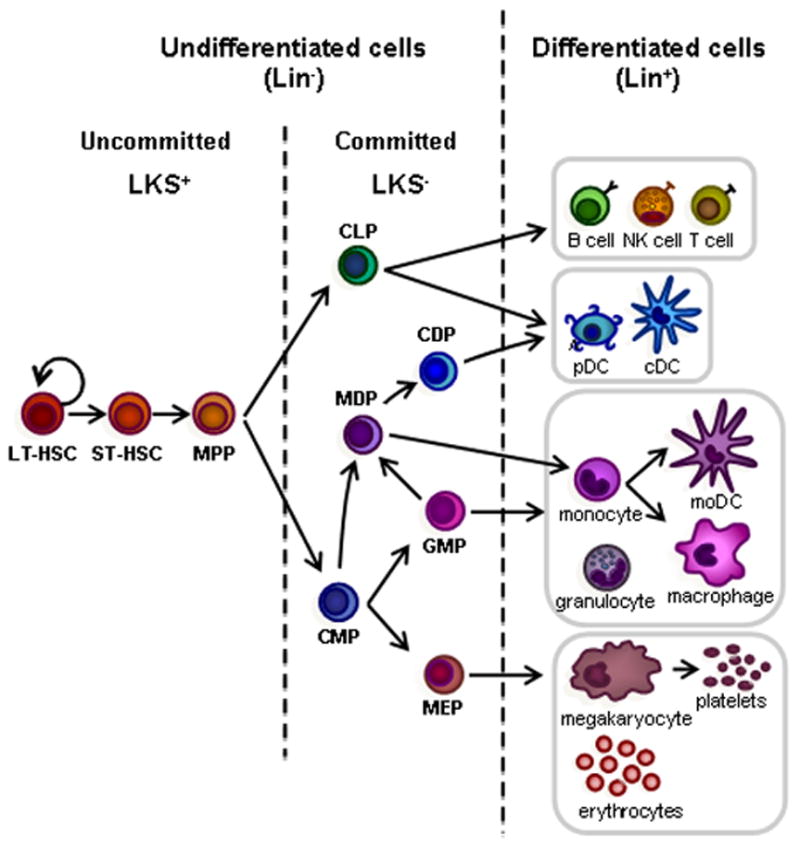
Hematopoiesis is initiated in the bone marrow by normally quiescent long-term hematopoietic stem cells (LT-HSCs), which have the capacity for self-renewal and give rise to proliferating short-term HSCs (ST-HSCs). ST-HSCs produce multipotent progenitors (MPPs), which give rise to progenitors committed to specific hematopoietic lineages, common lymphoid progenitors (CLPs) and common myeloid progenitors (CMPs). Mouse HSPCs from the bone marrow are defined by their lack of expression of markers of differentiated cells. A cocktail of antibodies specific for differentiated blood cell antigens, termed “lineage markers” (Lin; typically CD5, CD45R (B220), CD11b, Gr-1 (Ly-6G/C), 7–4, and Ter-119) can be used to eliminate mature hematopoietic lineages. The remaining Lin− cells can then be enriched for specific stem/progenitor populations. Sorting Lin− c-Kit+ Sca-1+ (LKS+) cells enriches for hematopoietic-reconstituting activity (i.e. HSCs). However, only 10% LKS+ cells are bona fide long-term reconstituting (LT)-HSCs; the LKS+ population also includes short-term (ST)-HSCs and MPPs. The Lin− c-Kit+ Sca-1− (LKS−) fraction contains oligopotent lineage-committed progenitors. CLPs generate all classes of lymphocytes, and CMPs give rise to either megakaryocyte-erythrocyte progenitors (MEPs) or granulocyte-monocyte progenitors (GMPs). Dendritic cell (DC) potential is retained in both CMPs and CLPs. In addition to producing GMPs, CMPs also give rise to monocyte and DC progenitors (MDPs), which can generate monocytes, macrophages, classical DCs (cDCs) and plasmacytoid DCs (pDCs). MDPs lie upstream of the common DC progenitors (CDPs), which are DC-restricted, giving rise to pDCs and cDCs. Monocytes can further differentiate into macrophages or monocyte-derived DCs (moDCs).
Upon infection, or during other forms of immunological stress, there is an increased demand for leukocytes to assist in combating the infection, to replace cells killed by invading microbes or consumed during the immune response, and to increase immune surveillance. The adaptive immune system meets this demand by clonal expansion of T and B cells. In contrast, although there are some reports of proliferation of mature macrophages [4], the increased supply of most innate immune cells, which have a limited lifespan and must be regularly replenished, is achieved by “emergency myelopoiesis” [5, 6]. Acute infection usually triggers the mobilization of myeloid cells, in particular neutrophils and monocytes, from the bone marrow to infected tissues. This is accompanied by the proliferation and differentiation of HSPCs in the bone marrow to maintain the supply of myeloid cells. During most bacterial, viral and fungal infections, myelopoiesis therefore becomes the predominant form of cellular production, with the development of other lineages (lymphoid and erythroid) inhibited; myelopoiesis is also commonly accompanied by alterations in the cellular composition and/or functional characteristics of bone marrow HSPCs [5, 6]. In fact, inflammatory cytokines secreted during infection-induced emergency myelopoiesis reduce the expression of growth and retention factors for lymphopoiesis, and bone marrow lymphocytes are therefore mobilized to secondary lymphoid organs [6]. Emergency myelopoiesis may consist of granulopoiesis (especially neutrophil production), monopoiesis (generation of monocytes and macrophages) or both, depending on the specific microbe as well as the route and severity of infection.
Several cytokines and transcription factors have been implicated in emergency myelopoiesis, although the molecular mechanisms underlying its regulation have not been clearly defined yet. In many cases it is not even yet clear which cells are responsible for instructing the emergency response. Moreover, HSPCs appear to respond to both “pull” and “push” signals (reviewed in [7]). “Pull” signals are exerted on HSPCs by the differentiation of more committed progenitors and the mobilization of differentiated cells from the bone marrow to infected tissues, which induces HSPCs to replace those cells. Myelopoiesis can also be driven by “push” signals, such as myelopoietic factors produced by differentiated cells of hematopoietic (e.g. tissue macrophages) or non-hematopoietic (e.g. epithelial cells) origin that sense the infection. For example, in mice chronically infected with Mycobacterium avium, increased HSC proliferation has been shown to be part of the primary immune response, rather than a compensatory response to progenitor depletion as it occurs in the absence of peripheral cytopenia [7, 8]. Several cytokines have been shown to induce myeloid cell production by HSPCs, including type I and II interferons, TNF-α and IL-6 [5, 7, 9, 10]. In this review we will focus on a new paradigm that has emerged over the past decade: the delivery of myelopoiesis-inducing “push” signals by microbial components directly sensed by HSPCs.
Differentiated innate immune cells such as macrophages and neutrophils recognize characteristic molecular signatures of microbes using pattern recognition receptors (PRRs). Known PRRs include Toll-like receptors (TLRs), Retinoic acid-inducible gene I-like receptors (RLRs), Nucleotide-binding oligomerization domain-like receptors (NLRs) and C-type lectin-like receptors (CLRs) [11]. TLRs, The best characterized PRRs, signal via recruitment of intracellular Toll/IL-1R (TIR) domain-containing adaptors (MyD88, TIRAP, TRIF, TRAM) that interact with the cytoplasmic TIR domains of TLRs to trigger expression of inflammatory cytokines and chemokines [12]. By the early 2000s, a role for TLRs in differentiated myeloid cells was already well established [13], but little was known about the timing of the acquisition of functional TLRs during myeloid differentiation in the bone marrow, and whether these receptors influence hematopoietic development. Studies indicated that TLR signaling can promote terminal differentiation. For example, Hayashi et al. showed that signaling through TLR4 and TLR2 promotes B-cell maturation [14], and Krutzik et al. showed that TLR activation triggers the rapid differentiation of human monocytes into macrophages and dendritic cells [15]. Other studies suggested that TLR signaling influences hematopoiesis at earlier stages. For example, Ueda et al. [16] demonstrated that lipopolysaccharide (LPS) rapidly and profoundly affects bone marrow hematopoiesis by promoting granulopoiesis over lymphopoiesis. However, it was unclear from these studies whether TLR agonists could influence hematopoiesis by targeting HSPCs directly, or by acting indirectly via differentiated cells such as macrophages and neutrophils.
New perspectives on emergency myelopoiesis came in 2006 when reports began to emerge demonstrating that murine and human HSPCs express functional PRRs, including TLRs, and that TLR/PRR signals provoke cell cycle entry and myeloid differentiation [17–19]. Subsequent studies focused on determining whether direct recognition of microbial components by HSPCs induces myelopoiesis in vivo [20, 21]. The idea that PRRs on HSPCs play a role in the selection of innate immune populations during the early stages of infection sits outside the current dogma but is gaining momentum in the literature. In this review we will examine the in vitro and in vivo evidence that TLRs on HSPCs directly sense microbial components and induce emergency myelopoiesis, and discuss the likely contribution of this mechanism to the control of blood cell production in response to microbial challenge, and immunity against infection.
TLR-dependent emergency myelopoiesis during bacterial, viral and fungal infection
HSPC expansion and a bias towards myelopoiesis after infection have been described in several mouse models of bacterial, viral and fungal infection (reviewed in [5]), although the contribution of TLR signaling to this phenomenon was previously not unequivocally demonstrated. For example, the mouse bone marrow Lin− c-Kit+ Sca-1+ (LKS+) population, which comprises HSCs and progenitors (see Figure 1), expands rapidly and is mobilized into the circulation following Escherichia coli bacteremia in Balb/c mice [22]. In addition to stimulating differentiation along the myeloid lineage, infectious agents can induce lymphoid progenitors to produce dendritic cells (DCs). For instance, purified common lymphoid progenitors (CLPs) from HSV-1-infected mice are biased towards DC differentiation in ex vivo cultures [23]. Similarly, CLPs from mice treated with the TLR9 ligand CpG ODN have a limited ability to generate B-lineage cells, but an augmented competence to generate DCs [23].
Infection studies using TLR-deficient mice have perhaps not surprisingly revealed defects in HSPC mobilization and emergency myelopoiesis. CLPs from TLR-deficient mice, for example, are not primed to become DCs during HSV-1 infection [23]. Similarly, vaccinia virus infection induces an increase in LKS+ cell numbers, with an associated decrease in common myeloid progenitors (CMPs) and an increase in the number of later stage myeloid precursors and differentiated myeloid cells; these responses all require MyD88 [24]. Mycobacterial infection also triggers TLR2/MyD88-dependent amplification of the LKS+ population, as well as granulocyte-monocyte progenitors (GMPs), in a murine model [25]. Moreover, we have shown that the bone marrow LKS+ cell population expands rapidly following C. albicans fungemia in a TLR2-dependent manner [26]. In contrast, Scumpia et al. [27] described that this expansion following bacterial infection occurs in the absence of TLR signaling, although the interpretation of the in vivo results is difficult as MyD88−/− mice are more susceptible to most infections; therefore, possible differences between control and knockout mice during infection may be masked by different tissue invasion by the microorganism.
It should be noted that most findings on the expansion of specific cell types, such as LKS positivity following infection, are based on phenotypic characterization, and the phenotype does not necessarily correlate with functionality of HSPCs as stem cells markers are likely to be affected by infection. For instance, lineage-restricted progenitors, which are normally Sca-1−, have been reported to upregulate Sca-1 expression upon infection and/or inflammation and are then found within the LKS+ fraction, with the consequent reduction of myeloid progenitor fraction. Therefore, it is important to validate the HSC status post-infection by using multiple phenotypic criteria as well as functional studies [5, 28].
TLR-dependent alterations in hematopoiesis during infection could be explained in at least two ways: (i) HSPC expansion could be an indirect effect of cytokines or growth factors produced by differentiated hematopoietic or non-hematopoietic cells detecting microbes, or (ii) microbes or microbial components might directly induce HSPC proliferation. These possibilities are not mutually exclusive, and both could involve TLR-mediated recognition of microbes or microbe-derived ligands.
TLR expression by HSPCs and in vitro myeloid differentiation in response to TLR ligands
PRR expression by HSPCs and a role for PRRs in emergency myelopoiesis were first reported in 2006. Nagai et al. [17] demonstrated that highly purified murine hematopoietic stem cells (long-term LKS+ Flk2− and short-term LKS+ Flk2+ HSCs), as well as lineage-restricted progenitors (CLPs, CMPs, GMPs and megakaryocyte-erythrocyte progenitors (MEPs)) (see Figure 1 for HSPC definitions and surface markers), express TLR4 (and its associated accessory molecules MD-2 and CD14) and/or TLR2. They also showed that upon in vitro exposure to LPS (a TLR4 agonist) and Pam3CSK4 (synthetic version of bacterial lipopeptide, detected by TLR1/TLR2 heterodimers), wild type but not MyD88-deficient HSCs enter cell cycle and acquire myeloid lineage markers. Myeloid progenitors stimulated with the TLR ligands produced monocytes and/or macrophages, while TLR agonist-stimulated lymphoid progenitors produced DCs. Accordingly, TLR-mediated signaling in HSPCs causes changes in the expression of transcription factors consistent with increased myeloid differentiation. These data indicated that TLR ligands can act as cues for HSPC proliferation and differentiation [17].
Also in 2006, Sioud et al. reported that human HSPCs (CD34+ cells) express TLR4 and TLR7/8, and that signaling though TLR7/8 induces their differentiation along the myeloid lineage [18]. Kim et al. had previously shown that human CD34+ cells constitutively express TLR9, and that exposure of the cells to its ligand CpG ODN induces IL-8 expression via MAP kinase signaling [29]. De Luca et al. subsequently reported the expression of TLR1, 2, 3, 4, and 6 on human CD34+ cells, and that the TLR1/2 agonist Pam3CSK4 instructs commitment of human HSCs to a myeloid cell fate, by modifying the transcriptional network [19].
Different TLRs have now been shown to induce the production of specific myeloid subsets by mouse and human HSPCs (summarized in Table 1). For instance, while TLR7/8 ligands induce the differentiation of CD34+ cells to produce CD11c+ CD14− DCs, TLR2 ligands instruct the differentiation of CD11c+ CD14+ monocytes [30]. The expression of other PRRs by HSPCs has also been described. For example, the Nod-like receptor Nod2 is expressed by human CD34+ cells, and stimulation of Nod2 with muramyl dipeptide is sufficient to trigger differentiation to CD11c+ myeloid cells [31].
Table 1.
Myeloid cell differentiation induced by ligation of HSPC PRRs.
| Stimulus – PRR agonist | PRR targeted | Species | Context | HSPC subpopulation | Cells produced | References |
|---|---|---|---|---|---|---|
| Pam3CSK4 | TLR2 | mouse | in vitro | LT-HSC, CMP, GMP | macrophages/neutrophils | [17] |
| TLR2 | mouse | in vitro | CLP | DCs | [17] | |
| TLR2 | human | in vitro | CD34+ | CD11c+ CD14+ monocytes | [19, 30] | |
| TLR2 | mouse | in vivo | LKS+, Lin− | macrophages | [21] | |
|
| ||||||
| LPSa) | TLR4 (MyD88) | mouse | in vitro | LT-HSC, CMP, GMP | macrophages | [17] |
| TLR4 | mouse | in vitro | CLP | DCs | [17] | |
| TLR4 | mouse | in vivo | LKS+, Lin− | macrophages | [21] | |
|
| ||||||
| CpG DNA | TLR9 (MyD88) | mouse | ex vivo | CLP | DCs | [23] |
| mouse | in vivo | LKS+, Lin− | macrophages | [21] | ||
|
| ||||||
| R848 | TLR7/8 | human | in vitro | CD34+ | CD11c+ CD14− DCs | [30] |
|
| ||||||
| curdlan | Dectin-1 | mouse | in vitro | Lin− | moDCs | [26] |
|
| ||||||
| MDPb) | Nod2 | human | in vitro | CD34+ | CD11c+ cells | [31] |
|
| ||||||
| Stimulus – infectious agent | PRR targeted | Species | Context | HSPC subpopulation | Cells produced | |
|
| ||||||
| Candida albicans | TLR2 | mouse | in vitro | LT-HSC, LKS+, CMP, GMP | macrophages/neutrophils | [41, 42] |
| TLR2, Dectin-1 | mouse | in vitro | Lin− | moDCs | [26] | |
| TLR2 | mouse | in vivo | Lin− | macrophages | [20] | |
|
| ||||||
| HSV-1c) | TLR9 | mouse | ex vivo | CLP | DCs | [23] |
LPS – lipopolysaccharide,
MDP – muramyl dipeptide
HSV-1– herpes simplex virus-
The involvement of TLRs in the recognition of Candida albicans, the most frequent cause of opportunistic fungal infections, has been widely studied. Mature phagocytic cells recognize the pathogen through a variety of PRRs, including TLRs and the CLR Dectin-1 [32–34]. TLR2 has been shown to be the most important TLR for the detection of both the yeast and hyphal forms of C. albicans, triggering MyD88-dependent cytokine secretion [35–37]; the involvement of TLR4 in C. albicans recognition has also been demonstrated [32, 38, 39]. Dectin-1, a phagocytic receptor that recognizes β-glucan in the cell wall of C. albicans, also collaborates with TLR2 in eliciting proinflammatory cytokines [39,40].
In a study of the interaction between C. albicans and murine HSPCs, it was shown that inactivated yeast and hyphae induce LKS+ cells to proliferate and differentiate towards the myeloid lineage in a TLR2/MyD88-dependent manner [41, 42]. Challenge of LT-HSCs (LKS+ CD105+ Sca1+) with C. albicans yeast also induces their proliferation as well as the upregulation of myeloid progenitor markers (CD34 and Fc R) through a TLR2/MyD88-dependent signaling pathway. TLR2/MyD88 signaling also promotes, upon challenge with yeast or Pam2CSK4, the differentiation of CMPs and GMPs into cells with a morphology of mature myeloid cells expressing CD11b, F4/80 and Gr-1. These myeloid-like cells display functional properties, as they are able to (i) phagocytose C. albicans yeast and (ii) produce proinflammatory cytokines upon stimulation [42].
The specific myeloid subsets that are produced following in vitro exposure of mouse HSPCs (Lin− cells) to C. albicans have been also determined. Inactivated C. albicans yeast induced the differentiation of monocyte-derived dendritic cells (moDCs) (CD11bhigh CD11c+ Ly6C+ F4/80+) via TLR2/MyD88- and Dectin-1-dependent pathways. Interestingly, the response to C. albicans yeast was more similar to the response to curdlan (a pure Dectin-1 ligand) than to Pam2CSK4 (a pure TLR2/TLR6 ligand), as Pam2CSK4 promoted differentiation to macrophages (CD11bhigh CD11clo Ly6C+ F4/80hi) rather than moDCs [26], indicating that Dectin-1 plays a key role in the response to C. albicans. Dectin-1 is not expressed on the most primitive stem cells, the “Side Population” cells (SP cells), but a subset of Lin− cells express detectable levels of Dectin-1 [26], indicating that it is turned on in differentiating progenitors prior to the acquisition of lineage markers. The moDCs generated in vitro, in response to inactivated yeasts, are functional as they have acquired the capability to secrete TNF-α and have fungicidal activity, and therefore could participate in innate immunity against C. albicans. All these data strongly support the notion that TLR signaling programs early progenitors to generate functional mature cells to deal with the fungal pathogen (Figure 2).
Figure 2. C. albicans directly stimulates HSPCs to induce myelopoiesis.
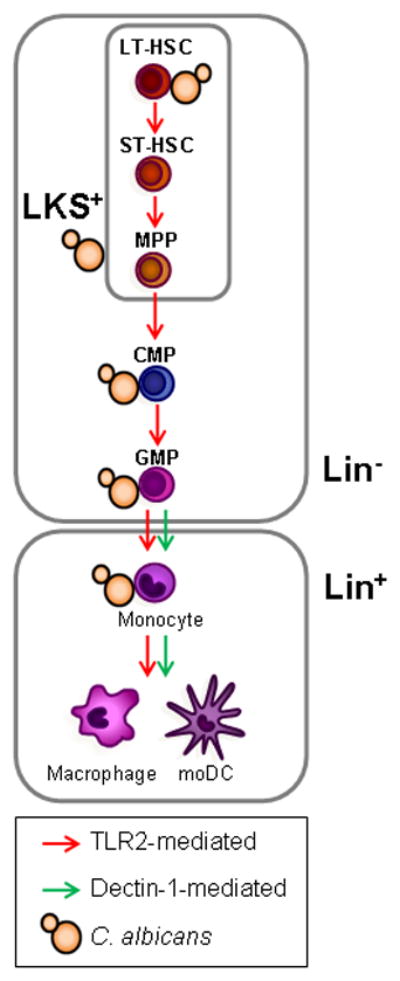
C. albicans interacts in vitro with mouse HSPCs from the most quiescent HSCs (LT-HSCs) to the lineage-committed progenitors (CMPs and GMPs, top), inducing the differentiation of these cells towards the myeloid lineage in a TLR2-dependent manner. C. albicans also induces TLR2- and Dectin-1-dependent production of moDCs by Lin− HSPCs in vitro, and TLR2-dependent macrophage production by transplanted LKS+ and Lin− cells upon C. albicans infection in vivo (bottom). Dectin-1 activation also promotes monocyte differentiation to macrophages and moDCs (bottom).
Does direct stimulation of TLRs on HSPCs induce emergency myelopoiesis in vivo?
Direct in vivo interaction of pathogens and/or their components with TLRs on HSPCs during infection is more difficult to demonstrate since, as noted above, HSPCs in an intact mouse could also respond to other stimuli, such as inflammatory cytokines generated by differentiated cells responding to the infection e.g. TLR-expressing tissue macrophages or epithelial cells [12, 38, 43]. For instance, it is well established that cytokines such as interferons (IFN-α, IFN-β and IFN-γ) and TNF-α play an essential role in HSPC proliferation in response to infection [7, 8, 44]. However, it has been recently shown that IFN-γ impairs proliferation of HSCs in mice by acting as a negative modulator of HSC self-renewal [28], therefore the role of IFN-γ in quiescent HSCs remains to be clearly established. In addition, certain endogenous “danger signals” (such as degradation products of extracellular matrix and heat shock proteins) generated as a result of tissue damage during infection may also be recognized by TLRs on mature cells [12] and/or HSPCs, and could also contribute to inflammation and HSPC activation.
A new experimental approach to address whether TLR agonists can stimulate HSPCs in vivo has been recently used. Purified Lin− or LKS+ cells from the bone marrow of B6Ly5.1 mice (CD45.1+) were transplanted into TLR2−/−, TLR4−/− or MyD88−/− mice (CD45.2+), which were then injected with pure ligands for TLR2, TLR4 or TLR9 (Pam3CSK4, LPS and CpG ODN respectively). Recipient mouse cells are not capable of recognizing or responding to the injected TLR ligands, therefore, any responses observed in the transplanted cells must be due to direct recognition of the agonists by TLRs expressed by the donor HSPCs. Transplanted HSPCs were detected in the bone marrow and spleen of recipient mice and, in response to TLR ligand injection, these cells differentiated preferentially into macrophages, demonstrating unequivocally that HSPCs can respond directly to TLR agonists in vivo, and that the engagement of these receptors induces macrophage differentiation [21] (Figure 2).
A similar in vivo transplantation approach was used to study the effect of C. albicans infection on HSPCs [20]. Transplanted Lin− cells were detected in the spleen and bone marrow of recipient mice, and they differentiated preferentially to macrophages in response to both live and inactivated yeast. Macrophage generation was dependent on TLR2, but independent of TLR4 (Figure 2). These results indicate that TLR-mediated recognition of C. albicans by HSPCs help to replace and/or to increase cells that constitute the first line of defense against the fungus, and suggest that TLR-mediated signaling leads to programming of early progenitors to rapidly replenish the innate immune system and generate the mature cells most urgently needed to deal with the pathogen.
Direct microbial detection by HSPCs of course requires co-localization. HSPCs can be found as resident or migratory populations in uninfected and infected tissues [45, 46], where microbes could induce them to differentiate by extramedullary hematopoiesis. HSPCs located in infected tissues are more likely to have an opportunity to directly detect microbial components than the majority of HSPCs, which reside in the bone marrow. However, HSPCs in the heavily vascularized bone marrow may also be exposed to circulating microbial components, or even to intact microbes following bone marrow invasion during systemic infection. We have previously detected fungal cells in the bone marrow of mice with invasive candidiasis, albeit at lower numbers than in peripheral tissues, but theoretically at sufficient levels to induce measurable activation of HSPCs [26, 42].
Concluding remarks and future directions
The concept of microbial components directly stimulating HSPCs to trigger the rapid generation of myeloid cells to boost the immune response against the infection is certainly attractive. The studies described above demonstrate that TLR signaling in HSPCs does induce myelopoiesis, and that TLR agonists can target HSPCs to directly drive myelopoiesis in vivo, but it remains to be seen how important this mechanism is in the context of an intact organism. It is likely that the hematopoietic response to infection is mediated in large part by the indirect effects of inflammatory mediators produced following TLR-mediated microbial detection by differentiated cells (hematopoietic and non-hematopoietic). However, the findings described above shift the paradigm of microbial detection exclusively by differentiated cells, and demand a reexamination of the role of TLRs in immune responses to include specific evaluation of their involvement in instructing immune cell development following direct detection of microbes and their components by HSPCs.
HSPC activation certainly can occur in response to many stimuli, including growth and differentiation factors, inflammatory cytokines, and microbial components, as well as potentially to endogenous “danger signals” produced during infection or tissue damage. Each of these stimuli may have a relatively greater or lesser impact under specific physiological conditions (during homeostasis, or upon emergency myelopoiesis during inflammation or infection). It will therefore be extremely important to determine how HSPCs integrate multiple signals, either by independent and/or partially overlapping pathways, to orchestrate the differentiation of specific hematopoietic populations under normal physiologic and pathophysiologic conditions. For instance, it has been reported that TLR signaling can influence GM-CSF-driven DC production by bone marrow progenitors in vitro, and that different TLRs have distinct effects. Ligands for TLR4 and TLR9 drive the production of pDCs, whereas influenza viruses and TLR3 ligands reduce DC production but increase neutrophil generation [47].
The functional properties of the myeloid cells produced also likely depend on the specific molecular composition of the pathogen (i.e. the combination of PRRs triggered) and the nature of the other myelopoietic signals the HSPCs receive. This might permit fine-tuning of emergency myelopoiesis to tailor the response to more effectively deal with a specific infection. Conversely, it is possible that some pathogens have evolved mechanisms to modulate HSPC responses in order to evade the immune system. Examination of the function of the myeloid cells produced by HSPCs following TLR ligation is therefore also critical. Indeed, in vitro TLR ligation on HSPCs has been reported to modulate their chemokine receptor expression, and consequently favors HSPC migration to inflammatory/infection sites, indicating that TLRs also regulate HSPC trafficking [6, 48]. Moreover, we recently showed that macrophages produced by HSPCs exposed to the TLR2 agonist Pam3CSK4 either prior to or during differentiation (in vitro and using an in vivo transplantation approach as described above) exhibit reduced inflammatory cytokine and reactive oxygen responses [49]. Further investigation is needed to determine how this impacts the macrophages’ migratory and anti-microbial capacity, and thus their ability to effectively combat infection. It is possible that their reduced inflammatory responsiveness is beneficial in protecting the host from collateral damage that could otherwise result from the presence of large numbers of inflammatory cells. Alternatively, suppression of macrophage responsiveness by targeting TLRs on the HSPCs from which they are produced could be an immune evasion strategy employed by invading organisms.
Future studies will also be required to dissect the mechanisms underlying the specification of myeloid differentiation and function. One key question will be whether TLR signal transduction pathways in HSPCs are similar to those in differentiated cells such as macrophages and neutrophils. It is likely that TLR signaling pathways in HSPCs are at least partially overlapping with differentiated cells, but since TLR signaling in HSPCs uniquely controls myeloid differentiation, it is possible that HSPC TLRs may induce distinct signals in these cells e.g. to activate transcription factors and induce chromatin modifications that specify myeloid cell fate choice. Our studies on the functional consequences of exposure of HSPCs to Pam3CSK4, showed that exposed HSPCs produce soluble factors that can act in a paracrine manner to influence the function of macrophages produced by unexposed HSPCs [49]. The identity of these factors is not currently known, but candidates include several cytokines known to be induced by TLRs in differentiated cells, such as type I and II interferons, TNF-α and IL-6, which have previously been reported to have myelopoietic properties [5, 7, 9, 10]. Thus it is possible that myeloid differentiation may be specified by TLRs in HSPCs without the activation of unique signal transduction pathways.
The answers to all these questions will provide new insights into the role of TLRs in host-pathogen interactions, emergency myelopoiesis and the development of immunity against infection, which may reveal novel targets for anti-microbial intervention.
Acknowledgments
Research in the M.L. Gil laboratory is supported by Grants SAF2010-18256 (Ministerio de Economía y Competitividad, Spain) and ACOMP/2013/168 (Generalitat Valenciana, Valencia, Spain). H.S. Goodridge received a Scientist Development Grant from the American Heart Association and an R21 (AI082379) from the NIH.
Footnotes
Conflict of interest
The authors declare no financial or commercial conflict of interest.
References
- 1.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmid MA, Kingston D, Boddupalli S, Manz MG. Instructive cytokine signals in dendritic cell lineage commitment. Immunol Rev. 2010;234:32–44. doi: 10.1111/j.0105-2896.2009.00877.x. [DOI] [PubMed] [Google Scholar]
- 3.Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity. 2007;26:726–740. doi: 10.1016/j.immuni.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011;32:57–65. doi: 10.1016/j.it.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119:2991–3002. doi: 10.1182/blood-2011-12-380113. [DOI] [PubMed] [Google Scholar]
- 7.King KY, Goodell MA. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol. 2011;11:685–692. doi: 10.1038/nri3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Granick JL, Simon SI, Borjesson DL. Hematopoietic stem and progenitor cells as effectors in innate immunity. Bone Marrow Res. 2012;2012:165107. doi: 10.1155/2012/165107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buechler MB, Teal TH, Elkon KB, Hamerman JA. Cutting edge: Type I IFN drives emergency myelopoiesis and peripheral myeloid expansion during chronic TLR7 signaling. J Immunol. 2013;190:886–891. doi: 10.4049/jimmunol.1202739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 13.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21 :335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi EA, Akira S, Nobrega A. Role of TLR in B cell development: signaling through TLR4 promotes B cell maturation and is inhibited by TLR2. J Immunol. 2005;174:6639–6647. doi: 10.4049/jimmunol.174.11.6639. [DOI] [PubMed] [Google Scholar]
- 15.Krutzik SR, Tan B, Li H, Ochoa MT, Liu PT, Sharfstein SE, Graeber TG, et al. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med. 2005;11:653–660. doi: 10.1038/nm1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, Kincade PW. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sioud M, Floisand Y, Forfang L, Lund-Johansen F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. J Mol Biol. 2006;364:945–954. doi: 10.1016/j.jmb.2006.09.054. [DOI] [PubMed] [Google Scholar]
- 19.De Luca K, Frances-Duvert V, Asensio MJ, Ihsani R, Debien E, Taillardet M, Verhoeyen E, et al. The TLR1/2 agonist PAM(3)CSK(4) instructs commitment of human hematopoietic stem cells to a myeloid cell fate. Leukemia. 2009;23:2063–2074. doi: 10.1038/leu.2009.155. [DOI] [PubMed] [Google Scholar]
- 20.Megías J, Maneu V, Salvador P, Gozalbo D, Gil ML. Candida albicans stimulates in vivo differentiation of haematopoietic stem and progenitor cells towards macrophages by a TLR2-dependent signaling. Cell Microbiol. 2013;15:1143–1153. doi: 10.1111/cmi.12104. [DOI] [PubMed] [Google Scholar]
- 21.Megías J, Yáñez A, Moriano S, O’Connor JE, Gozalbo D, Gil ML. Direct Toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages. Stem Cells. 2012;30:1486–1495. doi: 10.1002/stem.1110. [DOI] [PubMed] [Google Scholar]
- 22.Zhang P, Nelson S, Bagby GJ, Siggins R, Shellito JE, Welsh DA. The lineage-c-Kit+Sca-1+ cell response to Escherichia coli bacteremia in Balb/c mice. Stem Cells. 2008;26:1778–1786. doi: 10.1634/stemcells.2007-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welner RS, Pelayo R, Nagai Y, Garrett KP, Wuest TR, Carr DJ, Borghesi L, et al. Lymphoid precursors are directed to produce dendritic cells as a result of TLR9 ligation during herpes infection. Blood. 2008;112:3753–3761. doi: 10.1182/blood-2008-04-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh P, Yao Y, Weliver A, Broxmeyer H, Hong S, Chang C. Vaccinia virus infection modulates the hematopoietic cell compartments in the bone marrow. Stem Cells. 2008;26:1009–1016. doi: 10.1634/stemcells.2007-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi HH, Kim KK, Kim KD, Kim HJ, Jo EK, Song CH. Effects of mycobacterial infection on proliferation of hematopoietic precursor cells. Microbes Infect. 2011;13:1252–1260. doi: 10.1016/j.micinf.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Yáñez A, Megías J, O’Connor JE, Gozalbo D, Gil ML. Candida albicans induces selective development of macrophages and monocyte derived dendritic cells by a TLR2 dependent signaling. PLoS One. 2011;6:e24761. doi: 10.1371/journal.pone.0024761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scumpia PO, Kelly-Scumpia KM, Delano MJ, Weinsteim JS, Cuenca AG, Al-Quran S, Bovio I, et al. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol. 2010;184:2247–2251. doi: 10.4049/jimmunol.0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Bruin AM, Demirel O, Hooibrink B, Brandts CH, Nolte MA. Interferon-γ impairs proliferation of hematopoietic stem cells in mice. Blood. 2013;121:3578–3585. doi: 10.1182/blood-2012-05-432906. [DOI] [PubMed] [Google Scholar]
- 29.Kim JM, Kim NI, Oh YK, Kim YJ, Youn J, Ahn MJ. CpG oligodeoxynucleotides induce IL-8 expression in CD34+ cells via mitogen-activated protein kinase-dependent and NF-kappaB-independent pathways. Int Immunol. 2005;17 :1525–1531. doi: 10.1093/intimm/dxh345. [DOI] [PubMed] [Google Scholar]
- 30.Sioud M, Floisand Y. TLR agonists induce the differentiation of human bone marrow CD34+ progenitors into CD11c+ CD80/86+ DC capable of inducing a Th1-type response. Eur J Immunol. 2007;37:2834–2846. doi: 10.1002/eji.200737112. [DOI] [PubMed] [Google Scholar]
- 31.Sioud M, Floisand Y. NOD2/CARD15 on bone marrow CD34+ hematopoietic cells mediates induction of cytokines and cell differentiation. J Leukoc Biol. 2009;85:939–946. doi: 10.1189/jlb.1008650. [DOI] [PubMed] [Google Scholar]
- 32.Romani L. Immunity to fungal infections. Nat Rev Immunol. 2011;11:275–288. doi: 10.1038/nri2939. [DOI] [PubMed] [Google Scholar]
- 33.Jouault T, Sarazin A, Martinez-Esparza M, Fradin C, Sendid B, Poulain D. Host responses to a versatile commensal: PAMPs and PRRs interplay leading to tolerance or infection by Candida albicans. Cell Microbiol. 2009;11:1007–1015. doi: 10.1111/j.1462-5822.2009.01318.x. [DOI] [PubMed] [Google Scholar]
- 34.Hardison SE, Brown GD. C-type lectin receptors orchestrate antifungal immunity. Nat Immunol. 2012;13:817–822. doi: 10.1038/ni.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gil ML, Gozalbo D. TLR2, but not TLR4, triggers cytokine production by murine cells in response to Candida albicans yeasts and hyphae. Microbes Infect. 2006;8:2299–2304. doi: 10.1016/j.micinf.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 36.Villamón E, Gozalbo D, Roig P, Murciano C, O’Connor JE, Fradelizi D, Gil ML. Myeloid differentiation factor 88 (MyD88) is required for murine resistance to Candida albicans and is critically involved in Candida-induced production of cytokines. Eur Cytokine Netw. 2004;15:263–271. [PubMed] [Google Scholar]
- 37.Villamón E, Gozalbo D, Roig P, O’Connor JE, Fradelizi D, Gil ML. Toll-like receptor-2 is essential in murine defenses against Candida albicans infections. Microbes Infect. 2004;6:1–7. doi: 10.1016/j.micinf.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 38.Gil ML, Gozalbo D. Role of Toll-like receptors in systemic Candida albicans infections. Front Biosci. 2009;14:570–582. doi: 10.2741/3263. [DOI] [PubMed] [Google Scholar]
- 39.Goodridge H, Underhill D. Fungal Recognition by TLR2 and Dectin-1. Handb Exp Pharmacol. 2008:87–109. doi: 10.1007/978-3-540-72167-3_5. [DOI] [PubMed] [Google Scholar]
- 40.Vautier S, MacCallum DM, Brown GD. C-type lectin receptors and cytokines in fungal immunity. Cytokine. 2012;58:89–99. doi: 10.1016/j.cyto.2011.08.031. [DOI] [PubMed] [Google Scholar]
- 41.Yáñez A, Murciano C, O’Connor JE, Gozalbo D, Gil ML. Candida albicans triggers proliferation and differentiation of hematopoietic stem and progenitor cells by a MyD88-dependent signaling. Microbes Infect. 2009;11:531–535. doi: 10.1016/j.micinf.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 42.Yáñez A, Flores A, Murciano C, O’Connor JE, Gozalbo D, Gil ML. Signaling through TLR2/MyD88 induces differentiation of murine bone marrow stem and progenitor cells to functional phagocytes in response to Candida albicans. Cell Microbiol. 2010;12:114–128. doi: 10.1111/j.1462-5822.2009.01382.x. [DOI] [PubMed] [Google Scholar]
- 43.Gozalbo D, Gil ML. IFN-gamma in Candida albicans infections. Front Biosci. 2009;14:1970–1978. doi: 10.2741/3356. [DOI] [PubMed] [Google Scholar]
- 44.MacNamara KC, Oduro K, Martin O, Jones DD, McLaughlin M, Choi K, Borjesson DL, Winslow GM. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN-γ signaling. J Immunol. 2011;186:1032–1043. doi: 10.4049/jimmunol.1001893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Massberg S, Schaerli P, Knezevic-Maramica I, Köllnberger M, Tubo N, Moseman EA, Huff IV, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mazo IB, Massberg S, von Andrian UH. Hematopoietic stem and progenitor cell trafficking. Trends Immunol. 2011;32:493–503. doi: 10.1016/j.it.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Downes JE, Marshall-Clarke S. Innate immune stimuli modulate bone marrow-derived dendritic cell production in vitro by toll-like receptor-dependent and -independent mechanisms. Immunology. 2010;131:513–524. doi: 10.1111/j.1365-2567.2010.03324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmid MA, Takizawa H, Baumjohann DR, Saito Y, Manz MG. Bone marrow dendritic cell progenitors sense pathogens via Toll-like receptors and subsequently migrate to inflamed lymph nodes. Blood. 2011;118:4829–4840. doi: 10.1182/blood-2011-03-344960. [DOI] [PubMed] [Google Scholar]
- 49.Yáñez A, Hassanzadeh-Kiabi N, Ng MY, Megias J, Subramanian A, Liu GY, Underhill DM, et al. Detection of a TLR2 agonist by hematopoietic stem and progenitor cells (HSPCs) impacts the function of the macrophages they produce. Eur J Immunol. 2013;43:2114–2125. doi: 10.1002/eji.201343403. [DOI] [PMC free article] [PubMed] [Google Scholar]